miR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Extraction, Reverse Transcription, and qRT-PCR
2.3. Luciferase Reporter Assays and DNA Construction
2.4. Western Blotting Experiments
2.5. Patient Samples
2.6. Proliferation Assays (MTS) and Transfection
2.7. Apoptosis Assays
2.8. Statistical Analysis/Software
3. Results
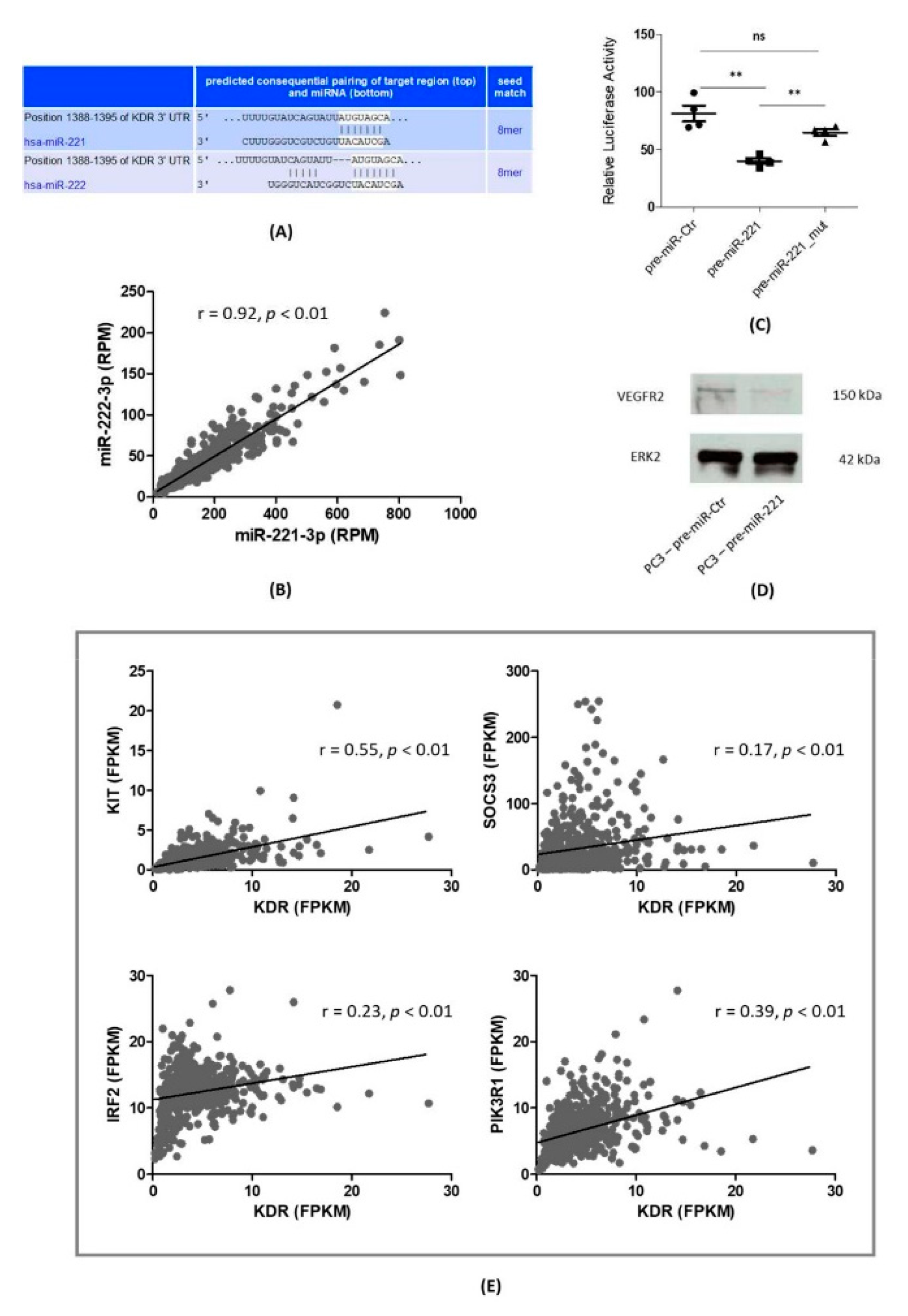
3.1. VEGFR2 as a Direct Target Gene of miR-221-3p in PCa Cells
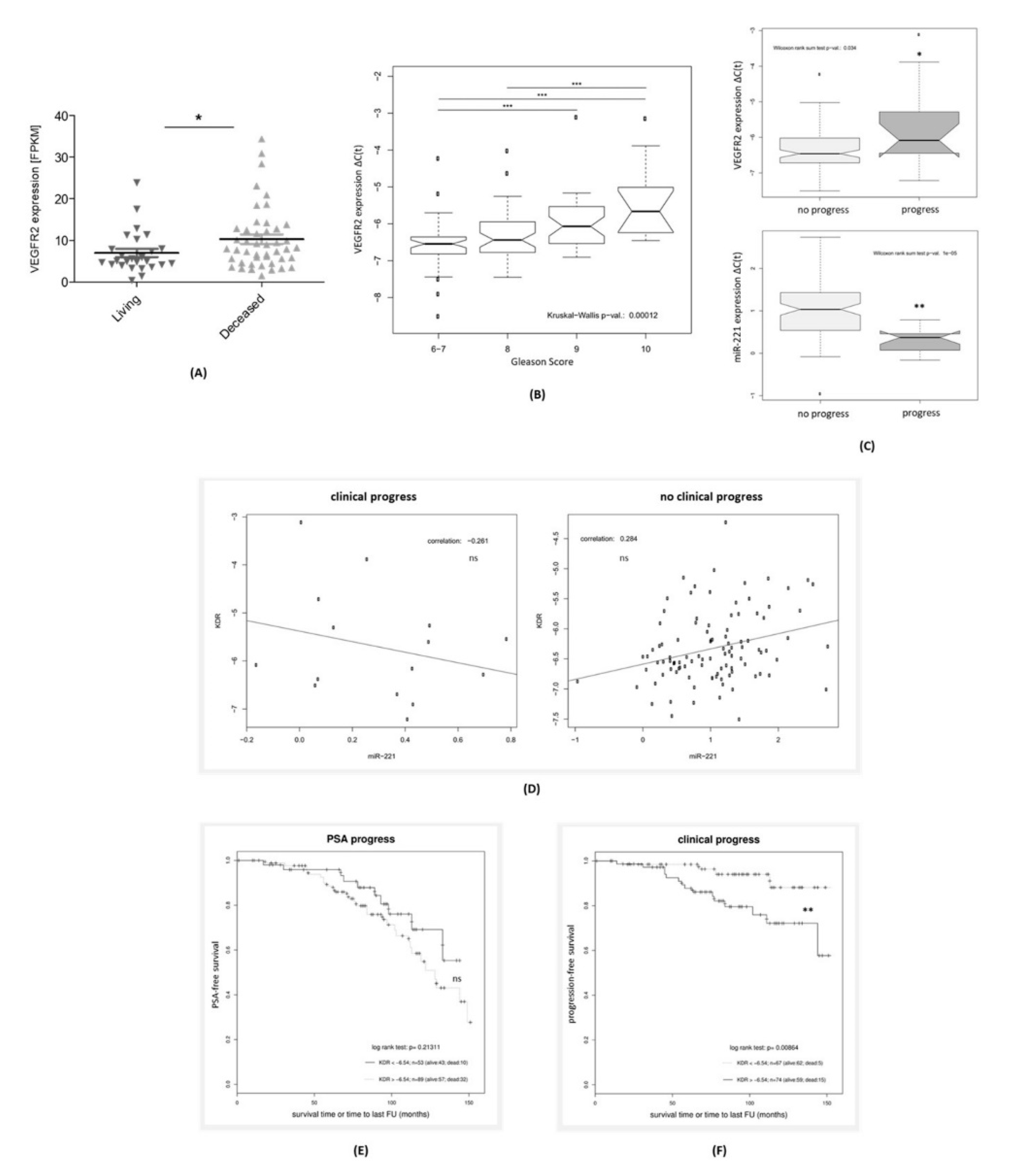
3.2. VEGFR2 Expression in PCa Tissue
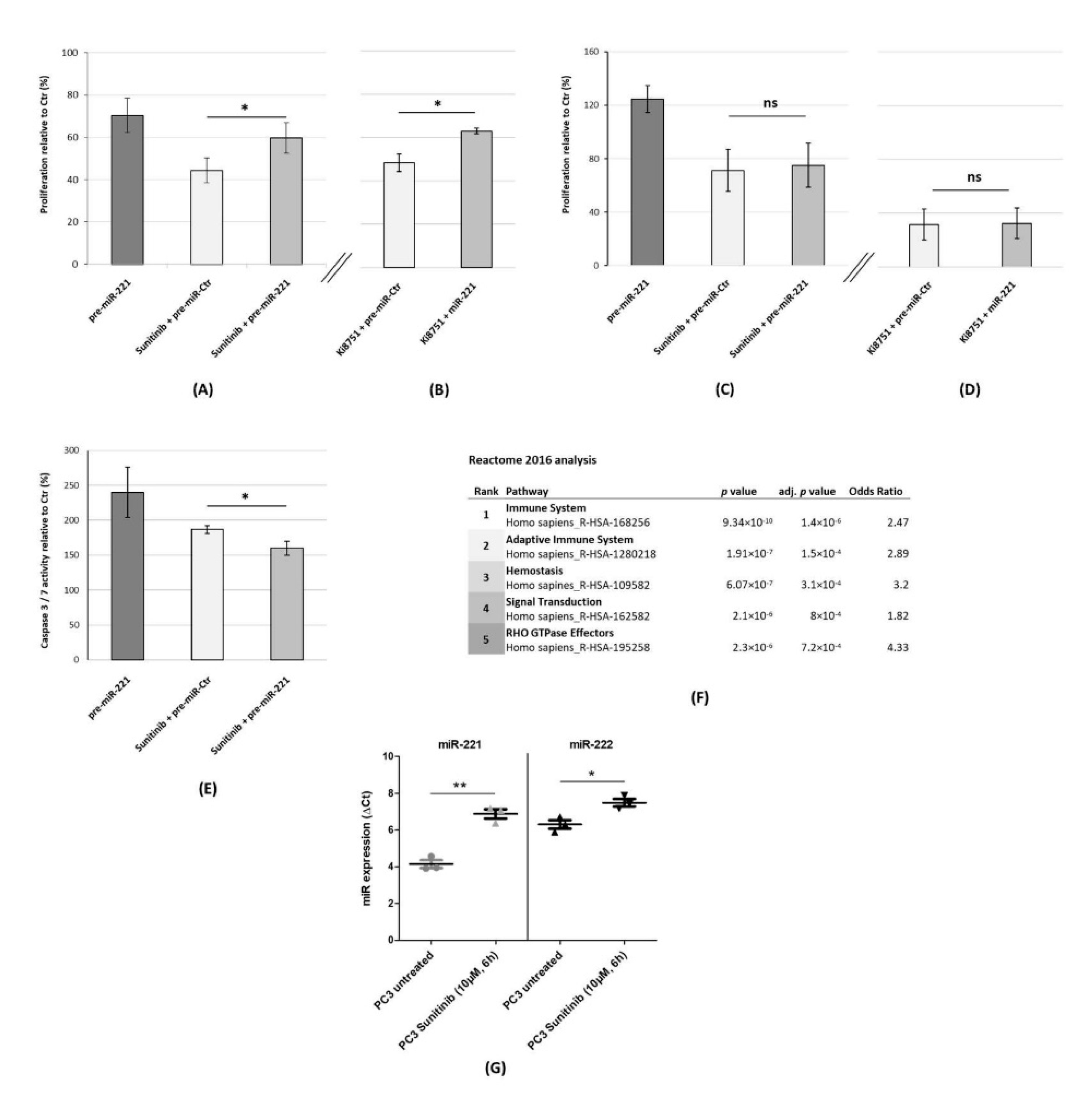
3.3. miR-221-3p Upregulation as an Escape Mechanism from VEGFR2 Inhibition in PC3 Cells
4. Discussion
4.1. miR-221 as a Biomarker Candidate in TKI Therapy
4.2. miR-221 Upregulation as Part of a Sunitinib Escape Mechanism in PCa Cells
4.3. Potential Implications for the Sequence of Anti-Angiogenesis and Immune-Based Approaches
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weidner, N.; Carroll, P.R.; Flax, J.; Blumenfeld, W.; Folkman, J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am. J. Pathol. 1993, 143, 401–409. [Google Scholar]
- Pallares, J.; Rojo, F.; Iriarte, J.; Morote, J.; I Armadans, L.; De Torres, I. Study of microvessel density and the expression of the angiogenic factors VEGF, bFGF and the receptors Flt-1 and FLK-1 in benign, premalignant and malignant prostate tissues. Histol. Histopathol. 2006, 21, 857–865. [Google Scholar] [PubMed]
- Huss, W.J.; Hanrahan, C.F.; Barrios, R.J.; Simons, J.W.; Greenberg, N.M. Angiogenesis and prostate cancer: Identification of a molecular progression switch. Cancer Res. 2001, 61, 2736–2743. [Google Scholar] [PubMed]
- Nordby, Y.; Andersen, S.; Richardsen, E.; Ness, N.; Al-Saad, S.; Melbø-Jørgensen, C.; Patel, H.R.; Donnem, T.; Busund, L.-T.R.; Bremnes, R.M. Stromal expression of VEGF-A and VEGFR-2 in prostate tissue is associated with biochemical and clinical recurrence after radical prostatectomy. Prostate 2015, 75, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Melegh, Z.; Oltean, S. Targeting Angiogenesis in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2676. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Donas, J.; Beuselinck, B.; Inglada-Pérez, L.; Graña-Castro, O.; Schöffski, P.; Wozniak, A.; Bechter, O.; Apellániz-Ruiz, M.; Leandro-Garcia, L.; Esteban, E.; et al. Deep sequencing reveals microRNAs predictive of antiangiogenic drug response. JCI Insight 2016, 1, e86051. [Google Scholar] [CrossRef] [Green Version]
- Khella, H.W.Z.; Butz, H.; Ding, Q.; Rotondo, F.; Evans, K.R.; Kupchak, P.; Dharsee, M.; Latif, A.; Pasic, M.D.; Lianidou, E.; et al. miR-221/222 Are Involved in Response to Sunitinib Treatment in Metastatic Renal Cell Carcinoma. Mol. Ther. 2015, 23, 1748–1758. [Google Scholar] [CrossRef]
- Spahn, M.; Kneitz, S.; Scholz, C.-J.; Nico, S.; Rüdiger, T.; Ströbel, P.; Riedmiller, H.; Kneitz, B.; Stenger, N. Expression of microRNA-221 is progressively reduced in aggressive prostate cancer and metastasis and predicts clinical recurrence. Int. J. Cancer 2009, 127, 394–403. [Google Scholar]
- Gordanpour, A.; Stanimirovic, A.; Nam, R.K.; Moreno, C.S.; Sherman, C.; Sugar, L.; Seth, A. miR-221 Is down-regulated in TMPRSS2: ERG fusion-positive prostate cancer. Anticancer. Res. 2011, 31, 403–410. [Google Scholar]
- Kurul, O.; Ates, F.; Yilmaz, I.; Narli, G.; Yesildal, C.; Senkul, T. The association of let-7c, miR-21, miR-145, miR-182, and miR-221 with clinicopathologic parameters of prostate cancer in patients diagnosed with low-risk disease. Prostate 2019, 79, 1125–1132. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, M.; Han, R.; Zhang, K.; Ding, H.; Liang, C.; Zhang, L. The correlation between microRNA-221/222 cluster overexpression and malignancy: An updated meta-analysis including 2693 patients. Cancer Manag. Res. 2018, 10, 3371–3381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneitz, B.; Krebs, M.; Kalogirou, C.; Schubert, M.; Joniau, S.; Van Poppel, H.; Lerut, E.; Kneitz, S.; Scholz, C.-J.; Ströbel, P.; et al. Survival in Patients with High-Risk Prostate Cancer Is Predicted by miR-221, Which Regulates Proliferation, Apoptosis, and Invasion of Prostate Cancer Cells by Inhibiting IRF2 and SOCS3. Cancer Res. 2014, 74, 2591–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Wang, Q.; Balk, S.; Brown, M.; Lee, G.-S.M.; Kantoff, P. The role of microRNA-221 and -222 in Androgen-independent Prostate Cancer Cell lines. Cancer Res. 2009, 69, 3356–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Wang, X.; He, H.H.; Sweeney, C.J.; Liu, S.X.; Brown, M.; Balk, S.; Lee, G.-S.; Kantoff, P. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene 2013, 33, 2790–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Du, S.-Y.; Armenia, J.; Qu, F.; Fan, J.; Wang, X.; Fei, T.; Komura, K.; Liu, S.X.; Lee, G.-S.M.; et al. Expression of lncRNA MIR222HG co-transcribed from the miR-221/222 gene promoter facilitates the development of castration-resistant prostate cancer. Oncogenesis 2018, 7, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Dart, D.A.; Koushyar, S.; Lanning, B.E.; Jiang, W. MiR-221 Is Specifically Elevated in PC3 Cells and its Deletion Reduces Adhesion, Motility and Growth. Anticancer. Res. 2019, 39, 5311–5327. [Google Scholar] [CrossRef] [Green Version]
- Krebs, M.; Behrmann, C.; Kalogirou, C.; Sokolakis, I.; Kneitz, S.; Julio, M.K.-D.; Zoni, E.; Rech, A.; Schilling, B.; Kübler, H.; et al. miR-221 Augments TRAIL-Mediated Apoptosis in Prostate Cancer Cells by Inducing Endogenous TRAIL Expression and Targeting the Functional Repressors SOCS3 and PIK3R1. BioMed Res. Int. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- D’Amico, A.V. Biochemical Outcome After Radical Prostatectomy, External Beam Radiation Therapy, or Interstitial Radiation Therapy for Clinically Localized Prostate Cancer. JAMA 1998, 280, 969. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, B. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, 4. [Google Scholar] [CrossRef]
- Wang, Z.; Lachmann, A.; Keenan, A.B.; Ma’Ayan, A. L1000FWD: Fireworks visualization of drug-induced transcriptomic signatures. Bioinform. 2018, 34, 2150–2152. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Skanderup, A.J.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, M.D.; Regan, M.M.; Oh, W.; Kaufman, D.S.; Olivier, K.; Michaelson, S.Z.; Spicer, B.; Gurski, C.; Kantoff, P.; Smith, M.R. Phase II study of sunitinib in men with advanced prostate cancer. Ann. Oncol. 2009, 20, 913–920. [Google Scholar] [CrossRef]
- Kuehbacher, A.; Urbich, C.; Dimmeler, S. Targeting microRNA expression to regulate angiogenesis. Trends Pharmacol. Sci. 2008, 29, 12–15. [Google Scholar] [CrossRef]
- Leone, P.; Buonavoglia, A.; Fasano, R.; Solimando, A.G.; De Re, V.; Cicco, S.; Vacca, A.; Racanelli, V.; Re, D. Insights into the Regulation of Tumor Angiogenesis by Micro-RNAs. J. Clin. Med. 2019, 8, 2030. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Chen, J.; Narizhneva, N.V.; Heston, W.; Brainard, J.; Sage, E.H.; Byzova, T.V. Molecular pathway for cancer metastasis to bone. J. Boil. Chem. 2003, 278, 39044–39050. [Google Scholar] [CrossRef] [Green Version]
- A Sidky, Y.; Borden, E.C. Inhibition of angiogenesis by interferons: Effects on tumor- and lymphocyte-induced vascular responses. Cancer Res. 1987, 47, 5155–5161. [Google Scholar] [PubMed]
- Persano, L.; Moserle, L.; Esposito, G.; Bronte, V.; Barbieri, V.; Iafrate, M.; Gardiman, M.P.; Larghero, P.; Pfeffer, U.; Naschberger, E.; et al. Interferon-α counteracts the angiogenic switch and reduces tumor cell proliferation in a spontaneous model of prostatic cancer. Carcinog. 2009, 30, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.-J.; Hwang, J.Y.; Lee, K.-S.; Choi, Y.K.; Choe, J.; Kim, J.-Y.; Moon, H.-E.; Kim, K.-W.; Koh, G.Y.; Lee, H.; et al. TRAIL negatively regulates VEGF-induced angiogenesis via caspase-8-mediated enzymatic and non-enzymatic functions. Angiogenesis 2013, 17, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Pollutri, D.; Patrizi, C.; La Bella, T.; Marinelli, S.; Gardini, A.C.; Marisi, G.; Toaldo, M.B.; Baglioni, M.; Salvatore, V.; et al. In Hepatocellular Carcinoma miR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3–Mediated Apoptosis. Clin. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, P.; Lu, J.; Zhang, H.; Shai, A.; Chun, M.G.; Wang, Y.; Libutti, S.K.; Nakakura, E.K.; Golub, T.R.; Hanahan, U. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genome Res. 2009, 23, 2152–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Yang, M.; Chen, S.; Balk, S.; Pomerantz, M.; Hsieh, C.-L.; Brown, M.; Lee, G.-S.M.; Kantoff, P.W. The altered expression of MiR-221/-222 and MiR-23b/-27b is associated with the development of human castration resistant prostate cancer. Prostate 2012, 72, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Sheehan, K.C.F.; Old, L.J.; Schreiber, R.D. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005, 65, 3447–3453. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.-S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar]
- Vlietstra, R.J.; Van Alewijk, D.C.; Hermans, K.G.; Van Steenbrugge, G.J.; Trapman, J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [Google Scholar]
- E McMenamin, M.; Soung, P.; Perera, S.; Kaplan, I.; Loda, M.; Sellers, W.R. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999, 59, 4291–4296. [Google Scholar]
- Finke, J.H.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib Reverses Type-1 Immune Suppression and Decreases T-Regulatory Cells in Renal Cell Carcinoma Patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.S.; Zea, A.H.; Rini, B.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib Mediates Reversal of Myeloid-Derived Suppressor Cell Accumulation in Renal Cell Carcinoma Patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teleanu, R.; Chircov, C.; Grumezescu, A.; Teleanu, D. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| n | 142 |
|---|---|
| age at surgery | 66 (47–81) |
| initial PSA (µg/L) | 35.71 (20–597) |
| follow-up (months) | 82.5 (1–154) |
| Gleason Score | |
| 6 | n = 3 |
| 7 | n = 45 |
| 8 | n = 46 |
| 9 | n = 36 |
| 10 | n = 12 |
| pT stage | |
| 2a | n = 4 |
| 2b | n = 14 |
| 2c | n = 3 |
| 3a | n = 41 |
| 3b | n = 59 |
| 4 | n = 21 |
| PSA progress | |
| yes (n) | n = 42 |
| no (n) | n = 100 |
| Clinical progress | |
| yes (n) | n = 20 |
| no (n) | n = 122 |
| n | 500 |
|---|---|
| age at surgery | 61 (41–78) |
| Gleason Score | |
| 6 | n = 45 |
| 7 | n = 250 |
| 8 | n = 64 |
| 9 | n = 137 |
| 10 | n = 4 |
| pT stage | |
| 2a | n = 13 |
| 2b | n = 10 |
| 2c | n = 165 |
| 3a | n = 159 |
| 3b | n = 136 |
| 4 | n = 10 |
| PSA progress | |
| yes (n) | n = 58 |
| no (n) | n = 373 |
| not assessed (n) | n = 69 |
| Upregulated | Survival Impact | Downregulated | Survival Impact | |
|---|---|---|---|---|
| Glioblastoma multiforme | ns | Cervical squamous cell carcinoma | ns | |
| RCC, clear cell | ** | Endometrial carcinoma | ns | |
| Stomach adenocarcinoma | ns | Lung adenocarcinoma | ns | |
| Lung squamous cell carcinoma | ns | |||
| PCa | ns | |||
| RCC, chromophobe | ns | |||
| RCC, papillary | ** | |||
| Urothelial carcinoma | ns | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krebs, M.; Solimando, A.G.; Kalogirou, C.; Marquardt, A.; Frank, T.; Sokolakis, I.; Hatzichristodoulou, G.; Kneitz, S.; Bargou, R.; Kübler, H.; et al. miR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro. J. Clin. Med. 2020, 9, 670. https://doi.org/10.3390/jcm9030670
Krebs M, Solimando AG, Kalogirou C, Marquardt A, Frank T, Sokolakis I, Hatzichristodoulou G, Kneitz S, Bargou R, Kübler H, et al. miR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro. Journal of Clinical Medicine. 2020; 9(3):670. https://doi.org/10.3390/jcm9030670
Chicago/Turabian StyleKrebs, Markus, Antonio Giovanni Solimando, Charis Kalogirou, André Marquardt, Torsten Frank, Ioannis Sokolakis, Georgios Hatzichristodoulou, Susanne Kneitz, Ralf Bargou, Hubert Kübler, and et al. 2020. "miR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro" Journal of Clinical Medicine 9, no. 3: 670. https://doi.org/10.3390/jcm9030670
APA StyleKrebs, M., Solimando, A. G., Kalogirou, C., Marquardt, A., Frank, T., Sokolakis, I., Hatzichristodoulou, G., Kneitz, S., Bargou, R., Kübler, H., Schilling, B., Spahn, M., & Kneitz, B. (2020). miR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro. Journal of Clinical Medicine, 9(3), 670. https://doi.org/10.3390/jcm9030670