Angiogenesis and Immunity in Renal Carcinoma: Can We Turn an Unhappy Relationship into a Happy Marriage?

{kind=link}
Abstract
:1. Introduction
2. Mutually Exclusive Features of Clear Cell Renal Carcinoma Microenvironment
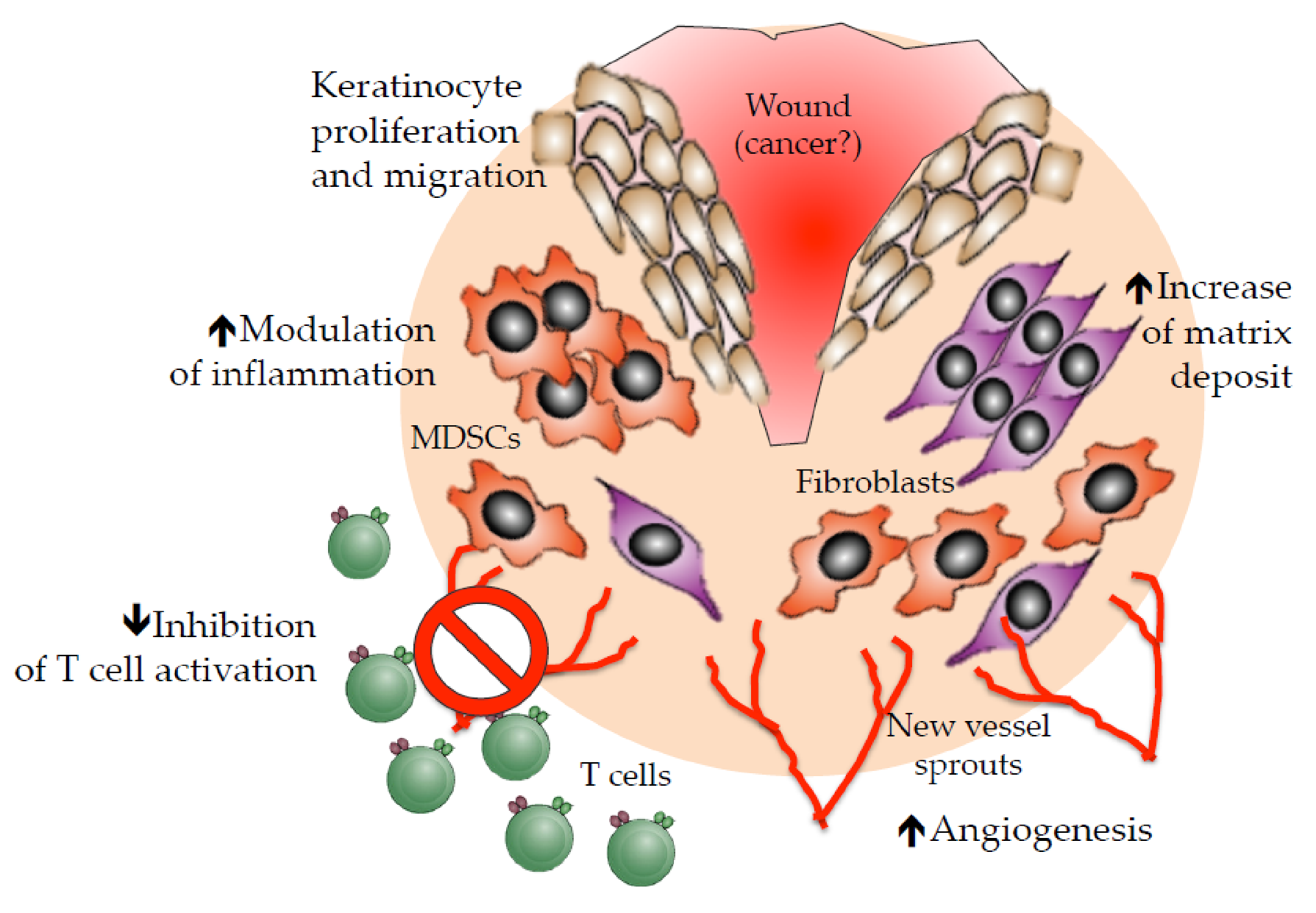
3. Crosstalk between Angiogenesis and Immunity
4. Effects of TKIs on Immunity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Capitanio, U.; Montorsi, F. Renal cancer. Lancet 2016, 387, 894–906. [Google Scholar] [CrossRef]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 1995, 13, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Negrier, S.; Escudier, B.; Lasset, C.; Douillard, J.Y.; Savary, J.; Chevreau, C.; Ravaud, A.; Mercatello, A.; Peny, J.; Mousseau, M.; et al. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. Groupe Francais d’Immunotherapie. N. Engl. J. Med. 1998, 338, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Ratta, R.; Zappasodi, R.; Raggi, D.; Grassi, P.; Verzoni, E.; Necchi, A.; Di Nicola, M.; Salvioni, R.; de Braud, F.; Procopio, G. Immunotherapy advances in uro-genital malignancies. Crit. Rev. Oncol. Hematol. 2016, 105, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Halabi, S.; Rosenberg, J.E.; Stadler, W.M.; Vaena, D.A.; Ou, S.S.; Archer, L.; Atkins, J.N.; Picus, J.; Czaykowski, P.; et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J. Clin. Oncol. 2008, 26, 5422–5428. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; Peltola, K.; et al. METEOR Investigators. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. RECORD-1 Study Group. Phase 3 trial of everolimus for metastatic renal cell carcinoma: Final results and analysis of prognostic factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Global ARCC Trial. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [PubMed] [Green Version]
- Motzer, R.; Escudier, B.; McDermott, D.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. CheckMate 025 Investigators. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Mennitto, A.; Grassi, P.; Ratta, R.; Grassi, P.; Ratta, R.; Verzoni, E.; Prisciandaro, M.; Procopio, G. Nivolumab in the treatment of advanced renal cell carcinoma: Clinical trial evidence and experience. Ther. Adv. Urol. 2016, 8, 319–326. [Google Scholar] [PubMed]
- Calvo, E.; Schmidinger, M.; Heng, D.Y.; Grünwald, V.; Escudier, B. Improvement in survival end points of patients with metastatic renal cell carcinoma through sequential targeted therapy. Cancer Treat. Rev. 2016, 50, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Salgia, N.J.; Dara, Y.; Bergerot, P.; Salgia, M.; Pal, S.K. The Changing Landscape of Management of Metastatic Renal Cell Carcinoma: Current Treatment Options and Future Directions. Curr. Treat. Options Oncol. 2019, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749. [Google Scholar]
- Drake, C.G.; Stein, M.N. The Immunobiology of Kidney Cancer. J. Clin. Oncol. 2018, 36, 3547–3552. [Google Scholar]
- Şenbabaoğlu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; Van Allen, E.M.; de Velasco, G.; Miao, D.; Ostrovnaya, I.; Drill, E.; Luna, A.; et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Van den Heuvel, C.N.A.M.; van Ewijk, A.; Zeelen, C.; de Bitter, T.; Huynen, M.; Mulders, P.; Oosterwijk, E.; Leenders, W.P.J. Molecular Profiling of Druggable Targets in Clear Cell Renal Cell Carcinoma through Targeted RNA Sequencing. Front. Oncol. 2019, 9, 117. [Google Scholar] [PubMed] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, L.B.; Bergers, G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol. 2015, 36, 240–249. [Google Scholar] [PubMed] [Green Version]
- Atkins, M.B.; McDermott, D.F.; Powles, T.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar]
- Rini, B.I.; Small, E.J. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J. Clin. Oncol. 2005, 23, 1028–1043. [Google Scholar] [CrossRef]
- Na, X.; Wu, G.; Ryan, C.K.; di’Santagnese, P.A.; Messing, E.M. Overproduction of vascular endothelial growth factor related to von Hippel-Lindau tumor suppressor gene mutations and hypoxia-inducible factor-1 alpha expression in renal cell carcinomas. J. Urol. 2003, 170 (2 Pt 1), 588–592. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-cell and NK-cell infiltration into solid tumors: A key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar]
- Azzi, S.; Hebda, J.K.; Gavard, J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, J.; Grankvist, K.; Rasmuson, T.; Bergh, A.; Landberg, G.; Ljungberg, B. Expression of vascular endothelial growth factor protein in human renal cell carcinoma. BJU Int. 2004, 93, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Paradis, V.; Lagha, N.B.; Zeimoura, L.; Blanchet, P.; Eschwege, P.; Ba, N.; Benoît, G.; Jardin, A.; Bedossa, P. Expression of vascular endothelial growth factor in renal cell carcinomas. Virchows Arch. 2000, 436, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009, 29, 881–883. [Google Scholar]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular Endothelial Growth Factor Affects Dendritic Cell Maturation through the Inhibition of Nuclear Factor-κB Activation in Hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Varney, M.L.; Johansson, S.L.; Singh, R.K. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: Role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res. 2005, 15, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Oldenhove, G. Tuning microenvironments: Induction of regulatory T cells by dendritic cells. Immunity 2008, 29, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J. Tregs and rethinking cancer immunotherapy. J. Clin. Investig. 2007, 117, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Finke, J.H.; Rayman, P.A.; Ko, J.S.; Bradley, J.M.; Gendler, S.J.; Cohen, P.A. Modification of the tumor microenvironment as a novel target of renal cell carcinoma therapeutics. Cancer J. 2013, 19, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Toes, R.E.; Ossendorp, F.; Offringa, R.; Melief, C.J. CD4 T cells and their role in antitumor immune responses. J. Exp. Med. 1999, 189, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–627. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune responses to malignancies. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S272–S283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, R.W.; Elkord, E.; Gilham, D.E.; Ramani, V.; Clarke, N.; Stern, P.L.; Hawkins, R.E. Frequency of regulatory T cells in renal cell carcinoma patients and investigation of correlation with survival. Cancer Immunol. Immunother. 2007, 56, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Parihar, J.S.; Tunuguntla, H.S. Role of chemokines in renal cell carcinoma. Rev. Urol. 2014, 16, 118–121. [Google Scholar] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Sevko, A.; Umansky, V. Myeloid-Derived Suppressor Cells Interact with Tumors in Terms of Myelopoiesis, Tumorigenesis and Immunosuppression: Thick as Thieves. J. Cancer 2013, 4, 3–11. [Google Scholar] [CrossRef]
- Fujimura, T.; Mahnke, K.; Enk, A.H. Myeloid derived suppressor cells and their role in tolerance induction in cancer. J. Dermatol. Sci. 2010, 59, 1–6. [Google Scholar] [CrossRef]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [Green Version]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [Green Version]
- Griffioen, A.W.; Damen, C.A.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res. 1996, 56, 1111–1117. [Google Scholar]
- Ribatti, D.; Crivellato, E. Immune cells and angiogenesis. J. Cell. Mol. Med. 2009, 13, 2822–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riboldi, E.; Musso, T.; Moroni, E.; Urbinati, C.; Bernasconi, S.; Rusnati, M.; Adorini, L.; Presta, M.; Sozzani, S. Cutting edge: Proangiogenic properties of alternatively activated dendritic cells. J. Immunol. 2005, 175, 2788–2792. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Ohnishi, T.; Okubo, Y.; Weiler, D.; Abrams, J.S.; Gleich, G.J. Granulocyte/macrophage colony-stimulating factor and interleukin 3 release from human peripheral blood eosinophils and neutrophils. J. Exp. Med. 1991, 174, 745–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, A.; Aloe, L.; Pe’er, J.; Frucht-Pery, J.; Bonini, S.; Bonini, S.; Levi-Schaffer, F. Nerve growth factor is preformed in and activates human peripheral blood eosinophils. J. Allergy Clin. Immunol. 1998, 102, 454–460. [Google Scholar] [CrossRef]
- Hoshino, M.; Takahashi, M.; Aoike, N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J. Allergy Clin. Immunol. 2001, 107, 295–301. [Google Scholar] [CrossRef]
- Sunderkötter, C.; Goebeler, M.; Schulze-Osthoff, K.; Bhardwaj, R.; Sorg, C. Macrophage-derived angiogenesis factors. Pharmacol. Ther. 1991, 51, 195–216. [Google Scholar] [CrossRef]
- Klimp, A.H.; Hollema, H.; Kempinga, C.; van der Zee, A.G.; de Vries, E.G.; Daemen, T. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumor-associated macrophages. Cancer Res. 2001, 61, 7305–7309. [Google Scholar]
- Chavakis, T.; Cines, D.B.; Rhee, J.S.; Liang, O.D.; Schubert, U.; Hammes, H.P.; Higazi, A.A.; Nawroth, P.P.; Preissner, K.T.; Bdeir, K. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): A link between inflammation and angiogenesis. FASEB J. 2004, 18, 1306–1308. [Google Scholar] [CrossRef]
- Owen, J.L.; Mohamadzadeh, M. Macrophages and chemokines as mediators of angiogenesis. Front. Physiol. 2013, 4, 159. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Bourbié-Vaudaine, S.; Blanchard, N.; Hivroz, C.; Roméo, P.H. Dendritic cells can turn CD4+ T lymphocytes into vascular endothelial growth factor-carrying cells by intercellular neuropilin-1 transfer. J. Immunol. 2006, 177, 1460–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight 2016, 1, e85974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, A.S.; Schmittnaegel, M.; Rigamonti, N.; Pais-Ferreira, D.; Mueller, P.; Buchi, M.; Ooi, C.H.; Kreuzaler, M.; Hirschmann, P.; Guichard, A.; et al. Optimized antiangiogenic reprogramming of the tumor microenvironment potentiates CD40 immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 541–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwilas, A.R.; Donahue, R.N.; Tsang, K.Y.; Hodge, J.W. Immune consequences of tyrosine kinase inhibitors that synergize with cancer immunotherapy. Cancer Cell Microenviron. 2015, 2, e677. [Google Scholar] [PubMed] [Green Version]
- Adotevi, O.; Pere, H.; Ravel, P.; Haicheur, N.; Badoual, C.; Merillon, N.; Medioni, J.; Peyrard, S.; Roncelin, S.; Verkarre, V.; et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J. Immunother. 2010, 33, 991–998. [Google Scholar] [CrossRef]
- Finke, J.H.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Onishi, H.; Wada, J.; Yamasaki, A.; Tanaka, H.; Nakano, K.; Morisaki, T.; Katano, M. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur. J. Immunol. 2010, 40, 197–203. [Google Scholar] [CrossRef]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.X.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Buart, S.; Van Pelt, J.; Richon, C.; Hasmim, M.; Leleu, N.; Suchorska, W.M.; Jalil, A.; Lecluse, Y.; El Hage, F.; et al. The cooperative induction of hypoxia-inducible factor-1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J. Immunol. 2009, 182, 3510–3521. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.M.; Hilf, N.; Walter, S.; Werth, D.; Brauer, K.M.; Radsak, M.P.; Weinschenk, T.; Singh-Jasuja, H.; Brossart, P. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 2008, 111, 5610–5620. [Google Scholar] [CrossRef] [Green Version]
- Busse, A.; Asemissen, A.M.; Nonnenmacher, A.; Braun, F.; Ochsenreither, S.; Stather, D.; Fusi, A.; Schmittel, A.; Miller, K.; Thiel, E.; et al. Immunomodulatory effects of sorafenib on peripheral immune effector cells in metastatic renal cell carcinoma. Eur. J. Cancer 2011, 47, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Desar, I.M.; Jacobs, J.H.; Hulsbergen-vandeKaa, C.A.; Oyen, W.J.; Mulders, P.F.; van der Graaf, W.T.; Adema, G.J.; van Herpen, C.M.; de Vries, I.J. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int. J. Cancer 2011, 129, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Liu, C.L.; Lee, J.J.; Liu, T.P.; Ko, W.C.; Huang, Y.C.; Wu, C.H.; Chen, Y.J. Sorafenib induces autophagy and suppresses activation of human macrophage. Int. Immunopharmacol. 2013, 15, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Dikov, M.M.; Ohm, J.E.; Ray, N.; Tchekneva, E.E.; Burlison, J.; Moghanaki, D.; Nadaf, S.; Carbone, D.P. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J. Immunol. 2005, 174, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C. Aspetti clinici di pazopanib: Nuovo farmaco inibitore dell’angiogenesi per il trattamento del carcinoma a cellule renali avanzato. IJPH 2011, 9 (Suppl. S2), 3. [Google Scholar]
- Zizzari, I.G.; Napoletano, C.; Botticelli, A.; Caponnetto, S.; Calabrò, F.; Gelibter, A.; Rughetti, A.; Ruscito, I.; Rahimi, H.; Rossi, E.; et al. TK Inhibitor Pazopanib Primes DCs by Downregulation of the β-Catenin Pathway. Cancer Immunol. Res. 2018, 6, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Verzoni, E.; De Cecco, L.; Dugo, M.; Rinchai, D.; Bedognetti, D.; Grassi, P.; Ratta, R.; Cova, A.; Squarcina, P.; Huber, V.; et al. Broad immunomodulating effect of first-line Pazopanib in metastatic renal cell carcinoma patients. Ann. Oncol. 2017, 28 (Suppl. S5), v295–v329. [Google Scholar] [CrossRef]
- Kumar, R.; Crouthamel, M.C.; Rominger, D.H.; Gontarek, R.R.; Tummino, P.J.; Levin, R.A.; King, A.G. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br. J. Cancer 2009, 101, 1717–1723. [Google Scholar] [CrossRef]
- Verzoni, E.; Ferro, S.; Procopio, G.; Cova, A.; Ratta, R.; Raimondi, A.; Sepe, P.; Squarcina, P.; Lalli, L.V.; Huber, V.; et al. Potent Natural Killer (NK) and myeloid blood cell remodeling by Cabozantinib (Cabo) in pretreated metastatic renal cell carcinoma (mRCC) patients. Ann. Oncol. 2018, 29 (Suppl. S8), viii303–viii331. [Google Scholar] [CrossRef]
- Amemiya, T.; Honma, M.; Kariya, Y.; Ghosh, S.; Kitano, H.; Kurachi, Y.; Fujita, K.I.; Sasaki, Y.; Homma, Y.; Abernethy, D.R.; et al. Elucidation of the molecular mechanisms underlying adverse reactions associated with a kinase inhibitor using systems toxicology. NPJ Syst. Biol. Appl. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracarda, S.; Porta, C.; Sabbatini, R.; Rivoltini, L. Angiogenic and immunological pathways in metastatic renal cell carcinoma: A counteracting paradigm or two faces of the same medal? The GIANUS Review. Crit. Rev. Oncol. Hematol. 2019, 139, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Flörcken, A.; Takvorian, A.; Van Lessen, A.; Singh, A.; Hopfenmüller, W.; Dörken, B.; Pezzutto, A.; Westermann, J. Sorafenib, but not sunitinib, induces regulatory T cells in the peripheral blood of patients with metastatic renal cell carcinoma. Anticancer Drugs 2012, 23, 298–302. [Google Scholar] [CrossRef]
- Stehle, F.; Schulz, K.; Fahldieck, C.; Kalich, J.; Lichtenfels, R.; Riemann, D.; Seliger, B. Reduced immunosuppressive properties of axitinib in comparison with other tyrosine kinase inhibitors. J. Biol. Chem. 2013, 288, 16334–16347. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.; Heishi, T.; Khan, O.F.; Kowalski, P.S.; Incio, J.; Rahbari, N.N.; Chung, E.; Clark, J.W.; Willett, C.G.; Luster, A.D. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J. Clin. Investig. 2017, 127, 3039–3051. [Google Scholar] [CrossRef]
- Komiya, T.; Huang, C.H.; Neupane, P.; Williamson, S.K.; Chalise, P. Impact of previous anti-angiogenesis treatment in nivolumab-treated advanced non-small cell lung cancer. J. Cancer Metastasis Treat. 2018, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Duléry, R.; Ménard, A.L.; Chantepie, S.; El-Cheikh, J.; François, S.; Delage, J.; Giannotti, F.; Ruggeri, A.; Brissot, E.; Battipaglia, G.; et al. Sequential Conditioning with Thiotepa in T Cell- Replete Hematopoietic Stem Cell Transplantation for the Treatment of Refractory Hematologic Malignancies: Comparison with Matched. Related, Haplo-Mismatched, and Unrelated Donors. Biol. Blood Marrow Transplant. 2018, 24, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Fraccaroli, A.; Prevalsek, D.; Fritsch, S.; Haebe, S.; Bücklein, V.; Schulz, C.; Hubmann, M.; Stemmler, H.J.; Ledderose, G.; Hausmann, A.; et al. Sequential HLA-haploidentical transplantation utilizing post-transplantation cyclophosphamide for GvHD prophylaxis in high-risk and relapsed/refractory AML/MDS. Am. J. Hematol. 2018, 93, 1524–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoellner, A.K.; Fritsch, S.; Prevalsek, D.; Engel, N.; Hubmann, M.; Reibke, R.; Rieger, C.T.; Hellmuth, J.C.; Haas, M.; Mumm, F.; et al. Sequential therapy combining clofarabine and T-cell-replete HLA-haploidentical. haematopoietic SCT is feasible and shows efficacy in the treatment of refractory or relapsed aggressive lymphoma. Bone Marrow Transplant. 2015, 50, 679–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mennitto, A.; Huber, V.; Ratta, R.; Sepe, P.; de Braud, F.; Procopio, G.; Guadalupi, V.; Claps, M.; Stellato, M.; Daveri, E.; et al. Angiogenesis and Immunity in Renal Carcinoma: Can We Turn an Unhappy Relationship into a Happy Marriage? J. Clin. Med. 2020, 9, 930. https://doi.org/10.3390/jcm9040930
Mennitto A, Huber V, Ratta R, Sepe P, de Braud F, Procopio G, Guadalupi V, Claps M, Stellato M, Daveri E, et al. Angiogenesis and Immunity in Renal Carcinoma: Can We Turn an Unhappy Relationship into a Happy Marriage? Journal of Clinical Medicine. 2020; 9(4):930. https://doi.org/10.3390/jcm9040930
Chicago/Turabian StyleMennitto, Alessia, Veronica Huber, Raffaele Ratta, Pierangela Sepe, Filippo de Braud, Giuseppe Procopio, Valentina Guadalupi, Mélanie Claps, Marco Stellato, Elena Daveri, and et al. 2020. "Angiogenesis and Immunity in Renal Carcinoma: Can We Turn an Unhappy Relationship into a Happy Marriage?" Journal of Clinical Medicine 9, no. 4: 930. https://doi.org/10.3390/jcm9040930
APA StyleMennitto, A., Huber, V., Ratta, R., Sepe, P., de Braud, F., Procopio, G., Guadalupi, V., Claps, M., Stellato, M., Daveri, E., Rivoltini, L., & Verzoni, E. (2020). Angiogenesis and Immunity in Renal Carcinoma: Can We Turn an Unhappy Relationship into a Happy Marriage? Journal of Clinical Medicine, 9(4), 930. https://doi.org/10.3390/jcm9040930