CD56bright Natural Killer Cells: A Possible Biomarker of Different Treatments in Multiple Sclerosis


Abstract
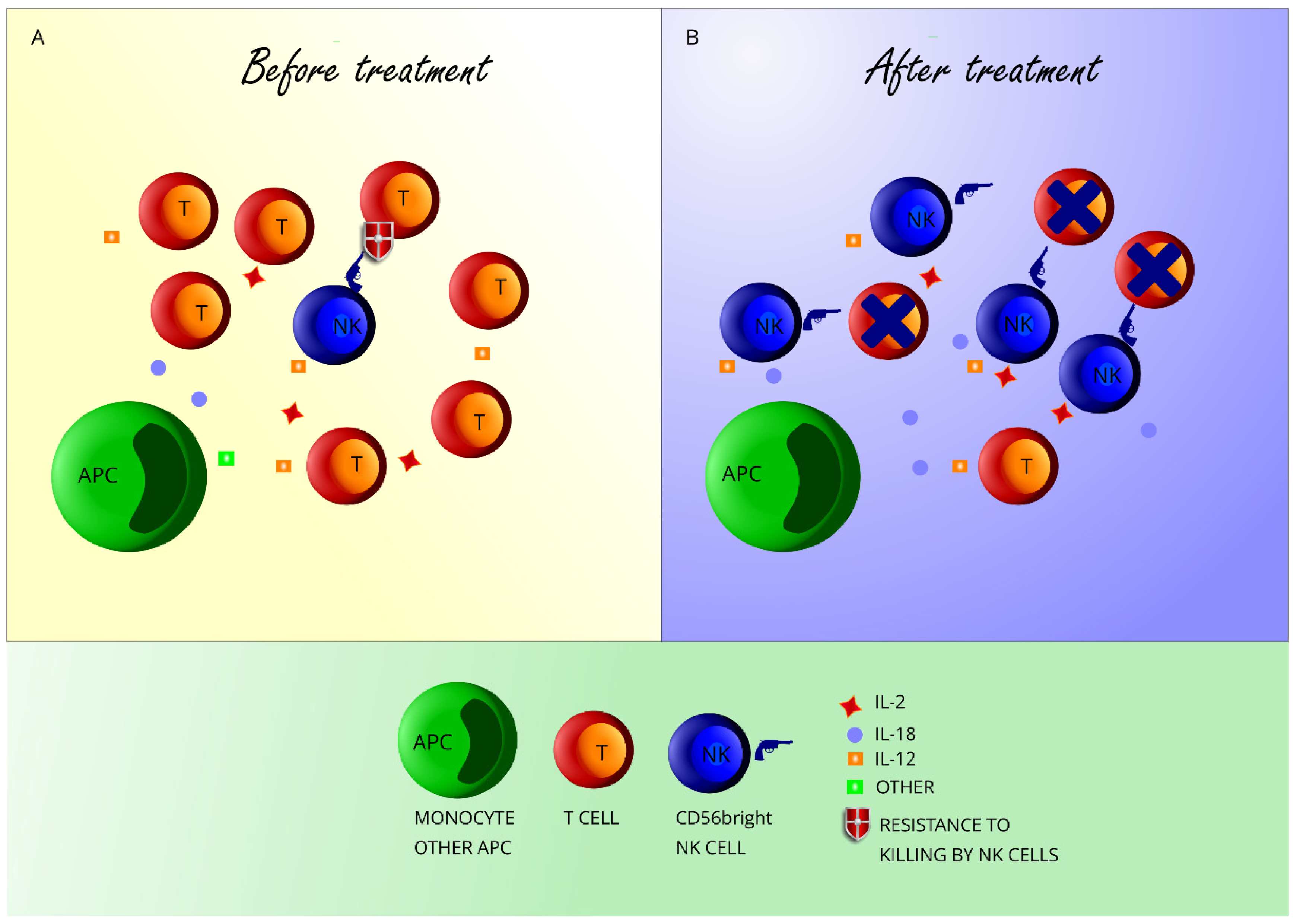
:1. Introduction
2. CD56bright NK Cells: A Regulatory Immune Subset
3. NK Cells in Untreated MS Patients and in Patients Undergoing Disease-Modifying Treatments
3.1. NK Cells in Untreated MS Patients
3.2. NK Cells in Patients Undergoing Disease-Modifying Treatments for MS
3.2.1. Interferon-Beta
3.2.2. Daclizumab
3.2.3. Dimethyl Fumarate
3.2.4. Fingolimod
3.2.5. Alemtuzumab
3.2.6. Autologous Hematopoietic Stem Cell Transplantation
3.3. Clinical Impact of Expansion of NK Cells Upon Treatment
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Burton, J.M.; Freedman, M.S. The Shifting Landscape of Disease-Modifying Therapies for Relapsing Multiple Sclerosis. J. Neuro-Ophthalmol. 2018, 38, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, F.; Laurent, S.; Fink, G.R.; Barnett, M.H.; Hartung, H.P.; Warnke, C. Effects of disease-modifying therapy on peripheral leukocytes in patients with multiple sclerosis. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, G.; Turner, B.; Gnanapavan, S.; Offiah, C.; Schmierer, K.; Marta, M. Is it time to target no evident disease activity (NEDA) in multiple sclerosis? Mult. Scler. Relat. Disord. 2015, 4, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemssen, T.; Akgun, K.; Bruck, W. Molecular biomarkers in multiple sclerosis. J. Neuroinflamm. 2019, 16, 272. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Impellizzieri, D.; Basso, C.; Laroni, A.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B. T-cell trafficking in the central nervous system. Immunol. Rev. 2012, 248, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Kleinewietfeld, M.; Hafler, D.A. Regulatory T cells in autoimmune neuroinflammation. Immunol. Rev. 2014, 259, 231–244. [Google Scholar] [CrossRef]
- Montaldo, E.; Vacca, P.; Vitale, C.; Moretta, F.; Locatelli, F.; Mingari, M.C.; Moretta, L. Human innate lymphoid cells. Immunol. Lett. 2016, 179, 2–8. [Google Scholar] [CrossRef]
- Vitale, M.; Cantoni, C.; Della Chiesa, M.; Ferlazzo, G.; Carlomagno, S.; Pende, D.; Falco, M.; Pessino, A.; Muccio, L.; De Maria, A.; et al. An Historical Overview: The Discovery of How NK Cells Can Kill Enemies, Recruit Defense Troops, and More. Front. Immunol. 2019, 10, 1415. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Wendt, K.; Wilk, E.; Buyny, S.; Buer, J.; Schmidt, R.E.; Jacobs, R. Gene and protein characteristics reflect functional diversity of CD56dim and CD56bright NK cells. J. Leukoc. Biol. 2006, 80, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, C.; Juelke, K.; Falco, M.; Morandi, B.; D’Agostino, A.; Costa, R.; Ratto, G.; Forte, G.; Carrega, P.; Lui, G.; et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J. Immunol. 2007, 178, 4947–4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesce, S.; Squillario, M.; Greppi, M.; Loiacono, F.; Moretta, L.; Moretta, A.; Sivori, S.; Castagnola, P.; Barla, A.; Candiani, S.; et al. New miRNA Signature Heralds Human NK Cell Subsets at Different Maturation Steps: Involvement of miR-146a-5p in the Regulation of KIR Expression. Front. Immunol. 2018, 9, 2360. [Google Scholar] [CrossRef]
- Wu, C.; Espinoza, D.A.; Koelle, S.J.; Yang, D.; Truitt, L.; Schlums, H.; Lafont, B.A.; Davidson-Moncada, J.K.; Lu, R.; Kaur, A.; et al. Clonal expansion and compartmentalized maintenance of rhesus macaque NK cell subsets. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichocki, F.; Grzywacz, B.; Miller, J.S. Human NK Cell Development: One Road or Many? Front. Immunol. 2019, 10, 2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef] [Green Version]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef] [Green Version]
- Laroni, A.; Gandhi, R.; Beynon, V.; Weiner, H.L. IL-27 imparts immunoregulatory function to human NK cell subsets. PLoS ONE 2011, 6, e26173. [Google Scholar] [CrossRef] [Green Version]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, N.; Odum, N.; Urso, B.; Lanier, L.L.; Spee, P. Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS ONE 2012, 7, e31959. [Google Scholar] [CrossRef] [Green Version]
- Laroni, A.; Armentani, E.; Kerlero de Rosbo, N.; Ivaldi, F.; Marcenaro, E.; Sivori, S.; Gandhi, R.; Weiner, H.L.; Moretta, A.; Mancardi, G.L.; et al. Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J. Autoimmun. 2016, 72, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Runzi, A.; Kuhlmann, T.; Posevitz-Fejfar, A.; Schwab, N.; Schneider-Hohendorf, T.; Herich, S.; Held, K.; Konjevic, M.; et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2973–E2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darlington, P.J.; Stopnicki, B.; Touil, T.; Doucet, J.S.; Fawaz, L.; Roberts, M.E.; Boivin, M.N.; Arbour, N.; Freedman, M.S.; Atkins, H.L.; et al. Natural Killer Cells Regulate Th17 Cells After Autologous Hematopoietic Stem Cell Transplantation for Relapsing Remitting Multiple Sclerosis. Front. Immunol. 2018, 9, 834. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Catalfamo, M.; Reichert-Scrivner, S.; Packer, A.; Cerna, M.; Waldmann, T.A.; McFarland, H.; Henkart, P.A.; Martin, R. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5941–5946. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Wiendl, H.; Marcenaro, E.; Kerlero de Rosbo, N.; Uccelli, A.; Laroni, A. Regulatory Functions of Natural Killer Cells in Multiple Sclerosis. Front. Immunol. 2016, 7, 606. [Google Scholar] [CrossRef] [Green Version]
- Tahrali, I.; Kucuksezer, U.C.; Altintas, A.; Uygunoglu, U.; Akdeniz, N.; Aktas-Cetin, E.; Deniz, G. Dysfunction of CD3−CD16+CD56dim and CD3−CD16−CD56bright NK cell subsets in RR-MS patients. Clin. Immunol. 2018, 193, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.; Alari-Pahissa, E.; Munteis, E.; Vera, A.; Zabalza, A.; Llop, M.; Villarrubia, N.; Costa-García, M.; Álvarez-Lafuente, R.; Villar, L.M.; et al. Adaptive Features of Natural Killer Cells in Multiple Sclerosis. Front. Immunol. 2019, 10, 2403. [Google Scholar] [CrossRef]
- Plantone, D.; Marti, A.; Frisullo, G.; Iorio, R.; Damato, V.; Nociti, V.; Patanella, A.K.; Bianco, A.; Mirabella, M.; Batocchi, A.P. Circulating CD56dim NK cells expressing perforin are increased in progressive multiple sclerosis. J. Neuroimmunol. 2013, 265, 124–127. [Google Scholar] [CrossRef]
- Takahashi, K.; Miyake, S.; Kondo, T.; Terao, K.; Hatakenaka, M.; Hashimoto, S.; Yamamura, T. Natural killer type 2 bias in remission of multiple sclerosis. J. Clin. Investig. 2001, 107, R23–R29. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Aranami, T.; Endoh, M.; Miyake, S.; Yamamura, T. The regulatory role of natural killer cells in multiple sclerosis. Brain 2004, 127, 1917–1927. [Google Scholar] [CrossRef]
- Saraste, M.; Irjala, H.; Airas, L. Expansion of CD56Bright natural killer cells in the peripheral blood of multiple sclerosis patients treated with interferon-beta. Neurol. Sci. 2007, 28, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, C.E. Interferon-beta: Mechanism of action and dosing issues. Neurology 2007, 68, S8–S11. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.F.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type I IFN Contributes to NK Cell Homeostasis, Activation, and Antitumor Function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef] [PubMed]
- Gill, U.S.; Peppa, D.; Micco, L.; Singh, H.D.; Carey, I.; Foster, G.R.; Maini, M.K.; Kennedy, P.T.F. Interferon Alpha Induces Sustained Changes in NK Cell Responsiveness to Hepatitis B Viral Load Suppression In Vivo. PLoS Pathog. 2016, 12, e1005788. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Radaelli, M.; Soelberg Sorensen, P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet 2017, 389, 1347–1356. [Google Scholar] [CrossRef]
- Vandenbark, A.A.; Huan, J.; Agotsch, M.; La Tocha, D.; Goelz, S.; Offner, H.; Lanker, S.; Bourdette, D. Interferon-beta-1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis. J. Neuroimmunol. 2009, 215, 125–128. [Google Scholar] [CrossRef]
- Martínez-Rodríguez, J.E.; López-Botet, M.; Munteis, E.; Rio, J.; Roquer, J.; Montalban, X.; Comabella, M. Natural killer cell phenotype and clinical response to interferon-beta therapy in multiple sclerosis. Clin. Immunol. 2011, 141, 348–356. [Google Scholar] [CrossRef]
- Lee, A.J.; Chen, B.; Chew, M.V.; Barra, N.G.; Shenouda, M.M.; Nham, T.; van Rooijen, N.; Jordana, M.; Mossman, K.L.; Schreiber, R.D.; et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med. 2017, 214, 1153–1167. [Google Scholar] [CrossRef]
- Jiang, W.; Chai, N.R.; Maric, D.; Bielekova, B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J. Immunol. 2011, 187, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, J.P.; Zhang, Y.; Riester, K.; Tang, M.T.; Efros, L.; Shi, J.; Harris, J.; Vexler, V.; Elkins, J.S. Intermediate-affinity interleukin-2 receptor expression predicts CD56(bright) natural killer cell expansion after daclizumab treatment in the CHOICE study of patients with multiple sclerosis. Mult. Scler. 2011, 17, 1441–1448. [Google Scholar] [CrossRef]
- Martin, J.F.; Perry, J.S.; Jakhete, N.R.; Wang, X.; Bielekova, B. An IL-2 paradox: Blocking CD25 on T cells induces IL-2-driven activation of CD56(bright) NK cells. J. Immunol. 2010, 185, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, A.; Ciccarelli, O. Daclizumab-induced encephalitis in multiple sclerosis. Mult. Scler. 2019, 25, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 or Glatiramer in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef] [Green Version]
- Medina, S.; Villarrubia, N.; Sainz de la Maza, S.; Lifante, J.; Costa-Frossard, L.; Roldan, E.; Picon, C.; Alvarez-Cermeno, J.C.; Villar, L.M. Optimal response to dimethyl fumarate associates in MS with a shift from an inflammatory to a tolerogenic blood cell profile. Mult. Scler. 2018, 24, 1317–1327. [Google Scholar] [CrossRef]
- Smith, M.D.; Calabresi, P.A.; Bhargava, P. Dimethyl fumarate treatment alters NK cell function in multiple sclerosis. Eur. J. Immunol. 2018, 48, 380–383. [Google Scholar] [CrossRef] [Green Version]
- Montes Diaz, G.; Fraussen, J.; Van Wijmeersch, B.; Hupperts, R.; Somers, V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci. Rep. 2018, 8, 8194. [Google Scholar] [CrossRef]
- Marastoni, D.; Buriani, A.; Pisani, A.I.; Crescenzo, F.; Zuco, C.; Fortinguerra, S.; Sorrenti, V.; Marenda, B.; Romualdi, C.; Magliozzi, R.; et al. Increased NK Cell Count in Multiple Sclerosis Patients Treated With Dimethyl Fumarate: A 2-Year Longitudinal Study. Front. Immunol. 2019, 10, 1666. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, B.Z.; Cohen, J.A.; Conway, D.S. Sphingosine 1-Phosphate Receptor Modulators for the Treatment of Multiple Sclerosis. Neurotherapeutics 2017, 14, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Pellkofer, H.L.; Cepok, S.; Korn, T.; Kumpfel, T.; Buck, D.; Hohlfeld, R.; Berthele, A.; Hemmer, B. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology 2011, 76, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Evans, B.L.; Durafourt, B.A.; Blain, M.; Lapierre, Y.; Bar-Or, A.; Antel, J.P. Reduction of the peripheral blood CD56(bright) NK lymphocyte subset in FTY720-treated multiple sclerosis patients. J. Immunol. 2011, 187, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Mehling, M.; Burgener, A.V.; Brinkmann, V.; Bantug, G.R.; Dimeloe, S.; Hoenger, G.; Kappos, L.; Hess, C. Tissue Distribution Dynamics of Human NK Cells Inferred from Peripheral Blood Depletion Kinetics after Sphingosine-1-Phosphate Receptor Blockade. Scand. J. Immunol. 2015, 82, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Ghadiri, M.; Rezk, A.; Li, R.; Evans, A.; Giacomini, P.S.; Barnett, M.H.; Antel, J.; Bar-Or, A. Pre-treatment T-cell subsets associate with fingolimod treatment responsiveness in multiple sclerosis. Sci. Rep. 2020, 10, 356. [Google Scholar] [CrossRef]
- Moreno-Torres, I.; Gonzalez-Garcia, C.; Marconi, M.; Garcia-Grande, A.; Rodriguez-Esparragoza, L.; Elvira, V.; Ramil, E.; Campos-Ruiz, L.; Garcia-Hernandez, R.; Al-Shahrour, F.; et al. Immunophenotype and Transcriptome Profile of Patients With Multiple Sclerosis Treated With Fingolimod: Setting Up a Model for Prediction of Response in a 2-Year Translational Study. Front. Immunol. 2018, 9, 1693. [Google Scholar] [CrossRef] [Green Version]
- Angerer, I.C.; Hecker, M.; Koczan, D.; Roch, L.; Friess, J.; Ruge, A.; Fitzner, B.; Boxberger, N.; Schroder, I.; Flechtner, K.; et al. Transcriptome profiling of peripheral blood immune cell populations in multiple sclerosis patients before and during treatment with a sphingosine-1-phosphate receptor modulator. CNS Neurosci. Ther. 2018, 24, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Al-Jaderi, Z.; Maghazachi, A.A. Effects of vitamin D3, calcipotriol and FTY720 on the expression of surface molecules and cytolytic activities of human natural killer cells and dendritic cells. Toxins 2013, 5, 1932–1947. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.C.; Ahmetspahic, D.; Ruck, T.; Schulte-Mecklenbeck, A.; Schwarte, K.; Jorgens, S.; Scheu, S.; Windhagen, S.; Graefe, B.; Melzer, N.; et al. Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol. (R) Neuroimmunol. Neuroinflamm. 2016, 3, e289. [Google Scholar] [CrossRef] [Green Version]
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439. [Google Scholar] [CrossRef] [PubMed]
- Wiendl, H.; Carraro, M.; Comi, G.; Izquierdo, G.; Kim, H.J.; Sharrack, B.; Tornatore, C.; Daizadeh, N.; Chung, L.; Jacobs, A.K.; et al. Lymphocyte pharmacodynamics are not associated with autoimmunity or efficacy after alemtuzumab. Neurol. (R) Neuroimmunol. Neuroinflamm. 2019, 7, e635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, M.G.; Moreau, T.; Greenwood, J.; Smith, R.M.; Hale, G.; Isaacs, J.; Waldmann, H.; Lachmann, P.J.; Compston, A. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: Involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J. Clin. Investig. 1996, 98, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Muraro, P.A.; Martin, R.; Mancardi, G.L.; Nicholas, R.; Sormani, M.P.; Saccardi, R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 391–405. [Google Scholar] [CrossRef]
- Dulphy, N.; Haas, P.; Busson, M.; Belhadj, S.; Peffault de Latour, R.; Robin, M.; Carmagnat, M.; Loiseau, P.; Tamouza, R.; Scieux, C.; et al. An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J. Immunol. 2008, 181, 2227–2237. [Google Scholar] [CrossRef] [Green Version]
- Vukicevic, M.; Chalandon, Y.; Helg, C.; Matthes, T.; Dantin, C.; Huard, B.; Chizzolini, C.; Passweg, J.; Roosnek, E. CD56bright NK cells after hematopoietic stem cell transplantation are activated mature NK cells that expand in patients with low numbers of T cells. Eur. J. Immunol. 2010, 40, 3246–3254. [Google Scholar] [CrossRef]
- Lugthart, G.; Goedhart, M.; van Leeuwen, M.M.; Melsen, J.E.; Jol-van der Zijde, C.M.; Vervat, C.; van Ostaijen-ten Dam, M.M.; Jansen-Hoogendijk, A.M.; van Tol, M.J.D.; Lankester, A.C.; et al. Expansion of cytotoxic CD56bright natural killer cells during T-cell deficiency after allogeneic hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 2017, 140, 1466–1469. [Google Scholar] [CrossRef]
- Elkins, J.; Sheridan, J.; Amaravadi, L.; Riester, K.; Selmaj, K.; Bielekova, B.; Parr, E.; Giovannoni, G. CD56(bright) natural killer cells and response to daclizumab HYP in relapsing-remitting MS. Neurol. (R) Neuroimmunol. Neuroinflamm. 2015, 2, e65. [Google Scholar] [CrossRef] [Green Version]
- Caruana, P.; Lemmert, K.; Ribbons, K.; Lea, R.; Lechner-Scott, J. Natural killer cell subpopulations are associated with MRI activity in a relapsing-remitting multiple sclerosis patient cohort from Australia. Mult. Scler. 2017, 23, 1479–1487. [Google Scholar] [CrossRef]
- Laroni, A. Enhancing natural killer cells is beneficial in multiple sclerosis-Yes. Mult. Scler. 2019, 25, 510–512. [Google Scholar] [CrossRef] [Green Version]
- Dubuisson, N.; Baker, D.; Kang, A.S.; Pryce, G.; Marta, M.; Visser, L.H.; Hofmann, W.E.; Gnanapavan, S.; Giovannoni, G.; Schmierer, K. Alemtuzumab depletion failure can occur in multiple sclerosis. Immunology 2018, 154, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielekova, B.; Richert, N.; Herman, M.L.; Ohayon, J.; Waldmann, T.A.; McFarland, H.; Martin, R.; Blevins, G. Intrathecal effects of daclizumab treatment of multiple sclerosis. Neurology 2011, 77, 1877–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| DMT | Effect of DMT on Circulating Total NK Cells | Effect of DMT on Circulating CD56bright NK Cells | Effect of DMT on Circulating CD56dim NK Cells | # of Enrolled Subjects | Reference |
|---|---|---|---|---|---|
| IFN-b | ↓ % NK of PBMCs at 12 months | ↑ % CD56bright of PBMCs at 3 and 12 months ↑ % CD56bright of NK cells at 12 months | ↓ % CD56dim of PBMCs at 12 months | 11 | [31] |
| Unchanged | ↑ % CD56bright of PBMCs at 12 months | 11 | [36] | ||
| ↑ % CD56bright of NK cells at 24 months ↑ % CD56bright of NK compared to untreated ↑Abs No compared to untreated | ↓ % CD56dim of NK compared to untreated ↓ Abs No compared to untreated | 25 (longitudinal) 27 (cross-sectional) | [37] | ||
| DMF | = % NK of PBMCs at 6 months | ↑ % CD56bright of PBMCs at 6 months | 64 | [46] | |
| = % NK of PBMCs at 6 months | ↑ % CD56bright of NK cells at 6 months | 18 | [47] | ||
| ↑ Abs number at 12 months | ↓ Abs number at 12 months | 12 | [48] | ||
| ↑ Abs number at 12 and 24 months | [49] | ||||
| Fingolimod | ↑ NK cells in treated vs. untreated | 20 untreated, 12 treated | [53] | ||
| ↑ % NK cells of PBMCs in treated vs. untreated | ↓ % CD56bright of PBMCs in treated vs. untreated | NK: 5 untreated, 8 treated CD56bright: 8 untreated, 10 treated | [54] | ||
| ↓ Abs number at 6 h | = Abs number at 6 h | 8 | [55] | ||
| = Abs number at 24 months | ↑ Abs number at 24 months ↓ % CD56bright of PBMCs at 24 months | = Abs number at 24 months ↑ % CD56dim of PBMCs at 24 months | 36 | [56] | |
| ↑ % CD56bright of NK at 6 months (responders) | 40 | [57] | |||
| Alemtuzumab | ↑ % CD56bright of PBMCs at 6 months ↑ Abs number at 6 monthsnkg2d | =% CD56dim of PBMCs at 6 months = Abs number at 6 months | 12 | [60] | |
| AHSCT | ↑ Abs Number at 12 and 21 months compared to 3 weeks after treatment ↑ % NK of PBMCs at 3–6–9–12–15–18 months | ↑ % CD56bright of PBMCs at 3-6-9-12 months ↑ % CD56bright of NK cells at 3 and 6 months | ↑ % CD56dim of PBMCs at 3, 6, 9, 12, 15, 18 months | 7 | [23] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laroni, A.; Uccelli, A. CD56bright Natural Killer Cells: A Possible Biomarker of Different Treatments in Multiple Sclerosis. J. Clin. Med. 2020, 9, 1450. https://doi.org/10.3390/jcm9051450
Laroni A, Uccelli A. CD56bright Natural Killer Cells: A Possible Biomarker of Different Treatments in Multiple Sclerosis. Journal of Clinical Medicine. 2020; 9(5):1450. https://doi.org/10.3390/jcm9051450
Chicago/Turabian StyleLaroni, Alice, and Antonio Uccelli. 2020. "CD56bright Natural Killer Cells: A Possible Biomarker of Different Treatments in Multiple Sclerosis" Journal of Clinical Medicine 9, no. 5: 1450. https://doi.org/10.3390/jcm9051450
APA StyleLaroni, A., & Uccelli, A. (2020). CD56bright Natural Killer Cells: A Possible Biomarker of Different Treatments in Multiple Sclerosis. Journal of Clinical Medicine, 9(5), 1450. https://doi.org/10.3390/jcm9051450