Characterization of Left Ventricular Non-Compaction Cardiomyopathy

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Study Population
2.2. Clinical Evaluation
2.3. Genetic Testing
2.4. Anatomopathological Exam
2.5. Statistical Analysis
3. Results
3.1. Study Population with Genetic and Clinical Evaluation
3.2. Anatomopathological Evaluation
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar]
- Arbustini, E.; Weidemann, F.; Hall, J.L. Left ventricular noncompaction: A distinct cardiomyopathy or a trait shared by different cardiac diseases? J. Am. Coll. Cardiol. 2014, 64, 1840–1850. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Bressan, M.; Hassel, D.; Huisken, J.; Staudt, D.; Kikuchi, K.; Poss, K.D.; Mikawa, T.; Stainier, D.Y. A dual role for ErbB2 signaling in cardiac trabeculation. Development 2010, 137, 3867–3875. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Poss, K.D. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 2012, 484, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Risebro, C.A.; Riley, P.R. Formation of the ventricles. Sci. World J. 2006, 6, 1862–1880. [Google Scholar] [CrossRef]
- Sedmera, D.; Thompson, R.P. Myocyte proliferation in the developing heart. Dev. Dyn. 2011, 240, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Gati, S.; Papadakis, M.; Van Niekerk, N.; Reed, M.; Yeghen, T.; Sharma, S. Increased left ventricular trabeculation in individuals with sickle cell anaemia: Physiology or pathology? Int. J. Cardiol. 2013, 168, 1658–1660. [Google Scholar] [CrossRef]
- Gati, S.; Papadakis, M.; Papamichael, N.D.; Zaidi, A.; Sheikh, N.; Reed, M.; Sharma, R.; Thilaganathan, B.; Sharma, S. Reversible de novo left ventricular trabeculations in pregnant women: Implications for the diagnosis of left ventricular noncompaction in low-risk populations. Circulation 2014, 130, 475–483. [Google Scholar] [CrossRef]
- Gati, S.; Chandra, N.; Bennett, R.L.; Reed, M.; Kervio, G.; Panoulas, V.F.; Ghani, S.; Sheikh, N.; Zaidi, A.; Wilson, M.; et al. Increased left ventricular trabeculation in highly trained athletes: Do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart 2013, 99, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Engberding, R.; Bender, F. Identification of a rare congenital anomaly of the myocardium by two-dimensional echocardiography: Persistence of isolated myocardial sinusoids. Am. J. Cardiol. 1984, 53, 1733–1734. [Google Scholar] [CrossRef]
- Ritter, M.; Oechslin, E.; Sutsch, G.; Attenhofer, C.; Schneider, J.; Jenni, R. Isolated noncompaction of the myocardium in adults. Mayo Clin. Proc. 1997, 72, 26–31. [Google Scholar] [CrossRef]
- Grant, T. An unusual anomaly of the coronary vessels in the malformed heart of a child. Heart 1926, 13, 273–283. [Google Scholar]
- Pignatelli, R.H.; McMahon, C.J.; Dreyer, W.J.; Denfield, S.W.; Price, J.; Belmont, J.W.; Craigen, W.J.; Wu, J.; El Said, H.; Bezold, L.I.; et al. Clinical characterization of left ventricular noncompaction in children: A relatively common form of cardiomyopathy. Circulation 2003, 108, 2672–2678. [Google Scholar] [CrossRef]
- Jenni, R.; Rojas, J.; Oechslin, E. Isolated noncompaction of the myocardium. N. Engl. J. Med. 1999, 340, 966–967. [Google Scholar] [CrossRef]
- Jenni, R.; Oechslin, E.; Schneider, J.; Attenhofer Jost, C.; Kaufmann, P.A. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart 2001, 86, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.M.; Hinton, R.B.; Czosek, R.; Lorts, A.; Parrott, A.; Shikany, A.R.; Ittenbach, R.F.; Ware, S.M. Genetic testing in pediatric left ventricular noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001735. [Google Scholar] [CrossRef] [Green Version]
- Chin, T.K.; Perloff, J.K.; Williams, R.G.; Jue, K.; Mohrmann, R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation 1990, 82, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Stollberger, C.; Gerecke, B.; Finsterer, J.; Engberding, R. Refinement of echocardiographic criteria for left ventricular noncompaction. Int. J. Cardiol. 2013, 165, 463–467. [Google Scholar] [CrossRef]
- Kohli, S.K.; Pantazis, A.A.; Shah, J.S.; Adeyemi, B.; Jackson, G.; McKenna, W.J.; Sharma, S.; Elliott, P.M. Diagnosis of left-ventricular non-compaction in patients with left-ventricular systolic dysfunction: Time for a reappraisal of diagnostic criteria? Eur. Heart J. 2008, 29, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, G.; Charron, P.; Eicher, J.C.; Giorgi, R.; Donal, E.; Laperche, T.; Boulmier, D.; Pascal, C.; Logeart, D.; Jondeau, G.; et al. Isolated left ventricular non-compaction in adults: Clinical and echocardiographic features in 105 patients. Results from a French registry. Eur. J. Heart Fail. 2011, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Stork, S.; Weidemann, F. Left ventricular noncompaction cardiomyopathy: An overdiagnosed disease. Circulation 2012, 126, 240–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.E.; Hong, Y.J.; Kim, H.K.; Kim, J.A.; Na, J.O.; Yang, D.H.; Kim, Y.J.; Choi, E.Y. The Korean Society of Cardiology and the Korean Society of Radiology. 2014 Korean guidelines for appropriate utilization of cardiovascular magnetic resonance imaging: A joint report of the Korean Society of Cardiology and the Korean Society of Radiology. Korean J. Radiol. 2014, 15, 659–688. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.B.; Jones, K.; Blanch, B.; Puranik, R.; McGeechan, K.; Barratt, A.; Semsarian, C. A systematic review and meta-analysis of the prevalence of left ventricular non-compaction in adults. Eur. Heart J. 2020, 41, 1428–1436. [Google Scholar] [CrossRef]
- Ross, S.B.; McGeechan, K.; Barratt, A.; Semsarian, C. Overdiagnosis of left ventricular non-compaction in adults: The data tells the story. Eur. Heart J. 2019, 40, 3206. [Google Scholar] [CrossRef]
- Protonotarios, A.; Elliott, P.M. Left ventricular non-compaction: Have we reached the limits of conventional imaging? Eur. Heart J. 2019, 41, 1437–1438. [Google Scholar] [CrossRef]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left ventricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Hoedemaekers, Y.M.; Caliskan, K.; Michels, M.; Frohn-Mulder, I.; van der Smagt, J.J.; Phefferkorn, J.E.; Wessels, M.W.; ten Cate, F.J.; Sijbrands, E.J.; Dooijes, D.; et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 232–329. [Google Scholar] [CrossRef] [Green Version]
- Lorca, R.; Martín, M.; Gómez, J.; Santamarta, E.; Morís, C.; Reguero, J.J.; Coto, E. Hypertrophic cardiomyopathy and left ventricular non-compaction: Different manifestations of the same cardiomyopathy spectrum? Int. J. Cardiol. 2015, 190, 26–28. [Google Scholar] [CrossRef]
- Van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.; et al. Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, E.N.; Attenhofer Jost, C.H.; Rojas, J.R.; Kaufmann, P.A.; Jenni, R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: A distinct cardiomyopathy with poor prognosis. J. Am. Coll. Cardiol. 2000, 36, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Weiford, B.C.; Subbarao, V.D.; Mulhern, K.M. Noncompaction of the ventricular myocardium. Circulation 2004, 109, 2965–2971. [Google Scholar] [CrossRef] [PubMed]
- Ichida, F.; Hamamichi, Y.; Miyawaki, T.; Ono, Y.; Kamiya, T.; Akagi, T.; Hamada, H.; Hirose, O.; Isobe, T.; Yamada, K.; et al. Clinical features of isolated noncompaction of the ventricular myocardium: Long-term clinical course, hemodynamic properties, and genetic background. J. Am. Coll. Cardiol. 1999, 34, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J.; Stollberger, C.; Towbin, J.A. Left ventricular noncompaction cardiomyopathy: Cardiac, neuromuscular, and genetic factors. Nat. Rev. Cardiol. 2017, 14, 224–237. [Google Scholar] [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Finsterer, J.; Stollberger, C. Are RYR2 exon-3 deletions truly causative for non-compaction? Europace 2014, 16, 1864. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J.; Zarrouk-Mahjoub, S. Lamin A/C mutations do not cause left ventricular hypertrabeculation/noncompaction. Tex. Heart Inst. J. 2015, 42, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Klaassen, S.; Probst, S.; Oechslin, E.; Gerull, B.; Krings, G.; Schuler, P.; Greutmann, M.; Hürlimann, D.; Yegitbasi, M.; Pons, L.; et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008, 117, 2893–2901. [Google Scholar] [CrossRef]
- Wang, C.; Hata, Y.; Hirono, K.; Takasaki, A.; Ozawa, S.W.; Nakaoka, H.; Saito, K.; Miyao, N.; Okabe, M.; Ibuki, K.; et al. A wide and specific spectrum of genetic variants and genotype-phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J. Am. Heart Assoc. 2017, 6, e006210. [Google Scholar] [CrossRef] [PubMed]
- Task Force Members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Yin, L. Non-compact cardiomyopathy or ventricular non-compact syndrome? J. Cardiovasc. Ultrasound. 2014, 22, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, A.; Mont, E.; Kutys, R.; Virmani, R. Left ventricular noncompaction: A pathological study of 14 cases. Hum. Pathol. 2005, 36, 403–411. [Google Scholar] [CrossRef]
- Gómez, J.; Reguero, J.R.; Morís, C.; Martín, M.; Alvarez, V.; Alonso, B.; Iglesias, S.; Coto, E. Mutation analysis of the main hypertrophic cardiomyopathy genes using multiplex amplification and semiconductor next-generation sequencing. Circ. J. 2014, 78, 2963–2971. [Google Scholar] [CrossRef] [Green Version]
- Gómez, J.; Lorca, R.; Reguero, J.R.; Morís, C.; Martín, M.; Tranche, S.; Alonso, B.; Iglesias, S.; Alvarez, V.; Díaz-Molina, B.; et al. Screening of the filamin C gene in a large cohort of hypertrophic cardiomyopathy patients. Circ. Cardiovasc. Genet. 2017, 10, e001584. [Google Scholar] [CrossRef] [Green Version]
- Telenti, A.; Pierce, L.C.; Biggs, W.H.; di Iulio, J.; Wong, E.H.; Fabani, M.M.; Kirkness, E.F.; Moustafa, A.; Shah, N.; Xie, C.; et al. Deep sequencing of 10,000 human genomes. Proc. Natl. Acad. Sci. USA 2016, 113, 11901–11906. [Google Scholar] [CrossRef] [Green Version]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Jacoby, D.; Moon, J.C.; McKenna, W.J. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat. Rev. Cardiol. 2016, 13, 651–675. [Google Scholar] [CrossRef]
- Probst, S.; Oechslin, E.; Schuler, P.; Greutmann, M.; Boyé, P.; Knirsch, W.; Berger, F.; Thierfelder, L.; Jenni, R.; Klaassen, S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ. Cardiovasc. Genet. 2011, 4, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Rigopoulos, A.; Rizos, I.K.; Aggeli, C.; Kloufetos, P.; Papacharalampous, X.; Stefanadis, C.; Toutouzas, P. Isolated left ventricular noncompaction: An unclassified cardiomyopathy with severe prognosis in adults. Cardiology 2002, 98, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Val-Bernal, J.F.; Garijo, M.F.; Rodriguez-Villar, D.; Val, D. Non-compaction of the ventricular myocardium: A cardiomyopathy in search of a pathoanatomical definition. Histol. Histopathol. 2010, 25, 495–503. [Google Scholar] [PubMed]
- Hughes, S.E. The pathology of hypertrophic cardiomyopathy. Histopathology 2004, 44, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Unverferth, D.V.; Baker, P.B.; Pearce, L.I.; Lautman, J.; Roberts, W.C. Regional myocyte hypertrophy and increased interstitial myocardial fibrosis in hypertrophic cardiomyopathy. Am. J. Cardiol. 1987, 59, 932–936. [Google Scholar] [CrossRef]
- Varnava, A.M.; Elliott, P.M.; Sharma, S.; McKenna, W.J.; Davies, M.J. Hypertrophic cardiomyopathy: The interrelation of disarray, fibrosis, and small vessel disease. Heart 2000, 84, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Gerger, D.; Stöllberger, C.; Grassberger, M.; Gerecke, B.; Andresen, H.; Engberding, R.; Finsterer, J. Pathomorphologic findings in left ventricular hypertrabeculation/noncompaction of adults in relation to neuromuscular disorders. Int. J. Cardiol. 2013, 169, 249–253. [Google Scholar] [CrossRef]
- Finsterer, J.; Stollberger, C.; Feichtinger, H. Histological appearance of left ventricular hypertrabeculation/noncompaction. Cardiology 2002, 98, 162–164. [Google Scholar] [CrossRef]
- Ottaviani, G.; Segura, A.M.; Rajapreyar, I.N.; Zhao, B.; Radovancevic, R.; Loyalka, P.; Kar, B.; Gregoric, I.; Buja, L.M. Left ventricular noncompaction cardiomyopathy in end-stage heart failure patients undergoing orthotopic heart transplantation. Cardiovasc. Pathol. 2016, 25, 293–299. [Google Scholar] [CrossRef]
- Jenni, R.; Wyss, C.A.; Oechslin, E.N.; Kaufmann, P.A. Isolated ventricular noncompaction is associated with coronary microcirculatory dysfunction. J. Am. Coll. Cardiol. 2002, 39, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Bleyl, S.B.; Mumford, B.R.; Brown-Harrison, M.C.; Pagotto, L.T.; Carey, J.C.; Pysher, T.J.; Ward, K.; Chin, T.K. Xq28-linked noncompaction of the left ventricular myocardium: Prenatal diagnosis and pathologic analysis of affected individuals. Am. J. Med. Genet. 1997, 72, 257–265. [Google Scholar] [CrossRef]
- Ivan, D.; Flamm, S.D.; Abrams, J.; Kindo, M.; Heck, K.; Frazier, O.H. Isolated ventricular non-compaction in adults with idiopathic cardiomyopathy: Cardiac magnetic resonance and pathologic characterization of the anomaly. J. Heart Lung Transplant. 2005, 24, 781–786. [Google Scholar] [CrossRef] [PubMed]





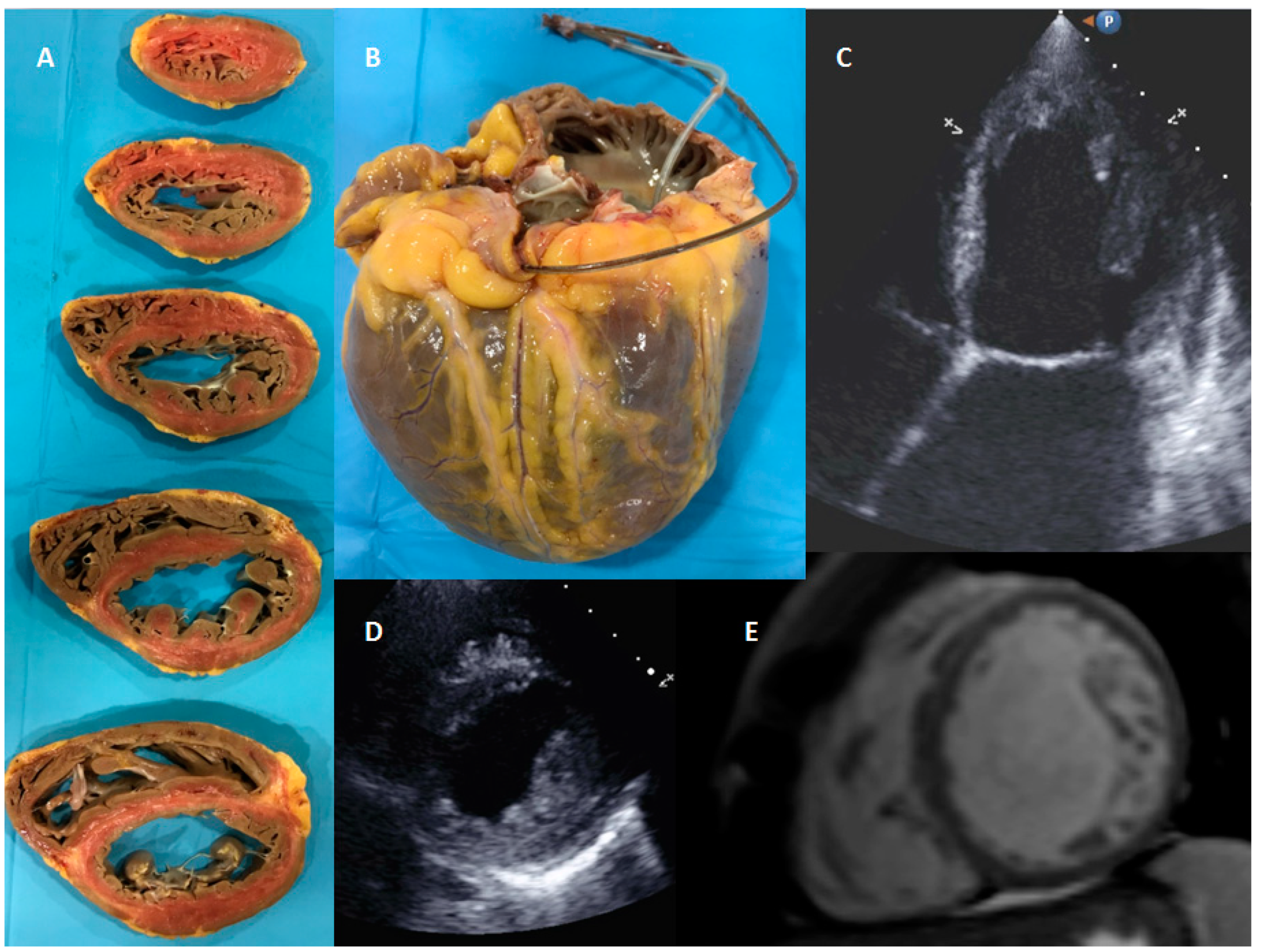{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | GENE | hg38 | NM | PROTEIN | cDNA | FUNCTION | GnomAD Exomes Frequency | HCMG-AMP |
|---|---|---|---|---|---|---|---|---|
| 1 | LMNA | chr1:156134508 | NM_170707 | p.Gln207ArgfsTer273 | c.619delC | Truncating | – | P |
| 2 | LMNA | chr1:156134457 | NM_170707 | p.Arg190Trp | c.568C > T | missense | – | LP |
| 3 | MYBPC3 | chr11:47347891 | NM_000256 | p.Gly263Ter | c.787G > T | Truncating | – | P |
| 4 | MYBPC3 | chr11:47347891 | NM_000256 | p.Gly263Ter | c.787G > T | Truncating | – | P |
| 5 | MYH7 | chr14:23427614 | NM_000257 | p.Leu620Pro | c.1859T > C | missense | – | LP |
| 6 | MYH7 | chr14:23427614 | NM_000257 | p.Leu620Pro | c.1859T > C | missense | – | LP |
| 7 | FLNC | chr7:128848595 | NM_001458 | p.Ala1539Thr | c.4615G > A | missense | – | LP |
| 8 | MYH7 | chr14:23427597 | NM_000257 | p.Gly626Trp | c.1876G > T | missense | – | LP |
| 9 | TTN | chr2: 178553135 | NM_003319 | p.Lys20857ValfsTer7 | c.62569_62570delAA | Truncating | – | LP |
| 9 | FLNC | chr7: 28844249 | NM_001458 | p.Pro1059Ser | c.3175C > T | missense | – | VUS |
| 10 | MYH7 | chr14:23430954 | NM_000257 | p.Arg281Lys | c.842G > C | missense | – | LP |
| 11 | TTN | chr2: 78557876 | NM_003319 | p.Glu20095Ter | c.60283G > T | Truncating | – | LP |
| 12 | TTN | chr2:178546323 | NM_003319 | p.Arg22605Ter | c.67813C > T | Truncating | 0.00000402 | LP |
| 13 | TTN | chr2:178563588 | NM_003319 | p.Arg18450SerfsTer28 | c.55346_55349dupTTAG | Truncating | – | LP |
| 13 | ACTN2 | chr1: 236717925 | NM_001103 | p.Asp65Ala | c.194A > C | missense | – | VUS |
| 14 | MYH6 | chr14:23862208 | NM_002471.3 | p.Arg1055Gln | c.3164G > A | missense | 0.000123 | VUS |
| 15 | MYH7 | chr14: 23424965 | NM_000257 | p.Pro828Leu | c.2483C > T | missense | – | VUS |
| 16 | RBM20 | chr10:110780815 | NM_001134363 | p.Leu69Pro | c.206T > C | missense | – | VUS |
| 17 | TTN | chr2: 178775139 | NM_003319 | p.Met2145GlyfsTer4 | c.6433_6434delAT | Truncating | – | VUS |
| Patient | Gender | Genetics | LVNC Suspicion in TTE | Reason for Referral | CVA | Tx | FH | Trigger Factors | LVEF Evolution |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | P/LP | Yes | Dyspnoea/arrhythmia | no | yes | yes | no | 2–3 |
| 2 | Male | P/LP | No | EKG | no | yes | yes | no | 2–3 |
| 3 | Female | P/LP | No | CVA | Yes | no | yes | no | 0–2 |
| 4 | Male | P/LP | Yes | Heart murmur | Yes | no | yes | no | 0–3 |
| 5 | Male | P/LP | No | Syncope | no | no | yes | no | 0–1–0 |
| 6 | Female | P/LP | Yes | Family screening | no | no | yes | no | 0 |
| 7 | Female | P/LP | Yes | Dyspnoea/palpitations | no | yes | yes | no | 0–3 |
| 8 | Male | P/LP | Yes | Heart failure | no | no | yes | no | 3–2 |
| 9 | Male | P/LP | No | unknown | no | yes | no | no | 3–2–3 |
| 10 | Female | P/LP | No | Cardiogenic Shock | Yes | no | no | no | 0 |
| 11 | Male | P/LP | Yes | Cardiogenic Shock | no | no | no | no | 3–1–3 |
| 12 | Male | P/LP | Yes | unknown | no | no | no | no | 3–2 |
| 13 | Female | P/LP | No | Dyspnoea | no | no | no | no | 0 |
| 14 | Female | VUS | No | Dyspnoea | no | Yes | yes | no | 2–3 |
| 15 | Female | VUS | Yes | Palpitations/Syncope | no | no | no | no | 0 |
| 16 | Male | VUS | No | EKG | no | no | no | no | 2–3 |
| 17 | Male | VUS | No | Ischemic heart disease | no | no | no | no | 0–3–1 |
| 18 | Female | Negative | No | Dyspnoea | no | no | no | yes | 1–0 |
| 19 | Female | Negative | Yes | Vagal syncope | no | no | yes | yes | 0 |
| 20 | Male | Negative | No | Dyspnoea | no | no | no | no | 3–1 |
| 21 | Male | Negative | Yes | Family screening | no | no | yes | no | 3–1 |
| 22 | Male | Negative | No | Neurological study | Yes | no | no | no | 3–0 |
| 23 | Female | Negative | Yes | Dyspnoea | Yes | no | no | no | 2–0 |
| 24 | Male | Negative | Yes | Heart failure | no | no | no | no | 3–0 |
| 25 | Female | Negative | No | Heart failure | no | no | no | no | 3–0 |
| 26 | Male | Negative | Yes | Heart failure | no | no | no | no | 3–2 |
| 27 | Male | Negative | No | EKG | no | no | no | no | 1–2–1 |
| 28 | Male | Negative | No | EKG | no | no | no | yes | 0 |
| 29 | Male | Negative | Yes | Palpitations | no | no | no | yes | 0 |
| 30 | Male | Negative | No | EKG | no | no | yes | yes | 0–1 |
| 31 | Female | Negative | Yes | CVA | Yes | no | no | no | 0 |
| 32 | Male | Negative | Yes | Heart murmur | no | no | no | no | 0 |
| 33 | Male | Negative | Yes | CVA | Yes | no | no | no | 0 |
| 34 | Male | Negative | Yes | EKG | no | no | no | no | 0 |
| 35 | Male | Negative | Yes | Syncope | no | no | no | no | 0 |
| 36 | Male | Negative | Yes | unknown | no | no | no | yes | 0 |
| 37 | Female | Negative | Yes | Palpitations | no | no | no | no | 0 |
| 38 | Male | Negative | No | EKG | no | no | no | yes | 0 |
| Group 1 | Group 2 | |
|---|---|---|
| % Men | 61.5% | 75% |
| Possible trigger factors for LVNC | 0% | 33.3% |
| Family history of cardiomyopathy | 61.5% | 15% |
| LVEF evolution | Tendency to worsen | Normal/Tendency to improve |
| Heart transplantation | 30.% | 0% |
| Heart | NC Thickness | C Thickness | LV wall Thickness | NC/C | Fibrosis | NC Cellular Hypertrophy | C Cellular Hypertrophy | Genetic Variants |
|---|---|---|---|---|---|---|---|---|
| 1 | 16 | 8 | 23 | 2 | yes | 3 | 3 | LMNA p.Gln207fs |
| 2 | 16 | 5 | 21 | 3.2 | no | 3 | 2 | LMNA p.Arg190Trp |
| 3 | 16 | 7 | 23 | 2.3 | yes | Not valuable | 1 | FLNC p.Ala1539Thr |
| 4 | 11 | 4 | 15 | 2.7 | no | 3 | 2 | TTN p.K20857VfsdelAA FLNC p.Pro1059Ser |
| 5 | 17 | 5 | 22 | 3.4 | no | 3 | 2 | MYH6 R1055Q |
| 6 | 20 | 5 | 25 | 4 | no | 3 | 2 | Negative |
| 7 | 14 | 3 | 17 | 4.6 | no | 3 | 2 | unavailable |
| 8 | 17 | 7 | 24 | 2.4 | yes | 2 | 2 | unavailable |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorca, R.; Martín, M.; Pascual, I.; Astudillo, A.; Díaz Molina, B.; Cigarrán, H.; Cuesta-Llavona, E.; Avanzas, P.; Rodríguez Reguero, J.J.; Coto, E.; et al. Characterization of Left Ventricular Non-Compaction Cardiomyopathy. J. Clin. Med. 2020, 9, 2524. https://doi.org/10.3390/jcm9082524
Lorca R, Martín M, Pascual I, Astudillo A, Díaz Molina B, Cigarrán H, Cuesta-Llavona E, Avanzas P, Rodríguez Reguero JJ, Coto E, et al. Characterization of Left Ventricular Non-Compaction Cardiomyopathy. Journal of Clinical Medicine. 2020; 9(8):2524. https://doi.org/10.3390/jcm9082524
Chicago/Turabian StyleLorca, Rebeca, María Martín, Isaac Pascual, Aurora Astudillo, Beatriz Díaz Molina, Helena Cigarrán, Elías Cuesta-Llavona, Pablo Avanzas, José Julían Rodríguez Reguero, Eliecer Coto, and et al. 2020. "Characterization of Left Ventricular Non-Compaction Cardiomyopathy" Journal of Clinical Medicine 9, no. 8: 2524. https://doi.org/10.3390/jcm9082524
APA StyleLorca, R., Martín, M., Pascual, I., Astudillo, A., Díaz Molina, B., Cigarrán, H., Cuesta-Llavona, E., Avanzas, P., Rodríguez Reguero, J. J., Coto, E., Morís, C., & Gómez, J. (2020). Characterization of Left Ventricular Non-Compaction Cardiomyopathy. Journal of Clinical Medicine, 9(8), 2524. https://doi.org/10.3390/jcm9082524