
Metagenomic Insight into the Community Structure and Functional Genes in the Sunflower Rhizosphere Microbiome



Abstract
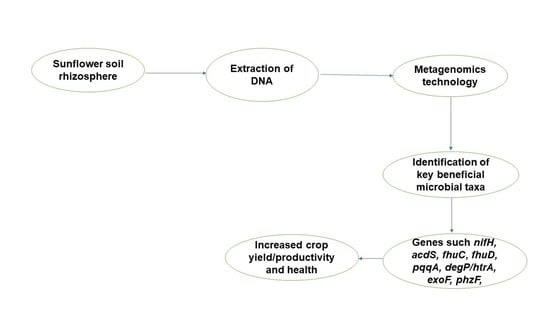
:
1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Analyses
2.2. DNA Extraction from Soil Samples, Sample Preparation, and Sequencing
2.3. Data Processing and Statistical Analysis
3. Results
3.1. Physicochemical Characteristics of the Soil
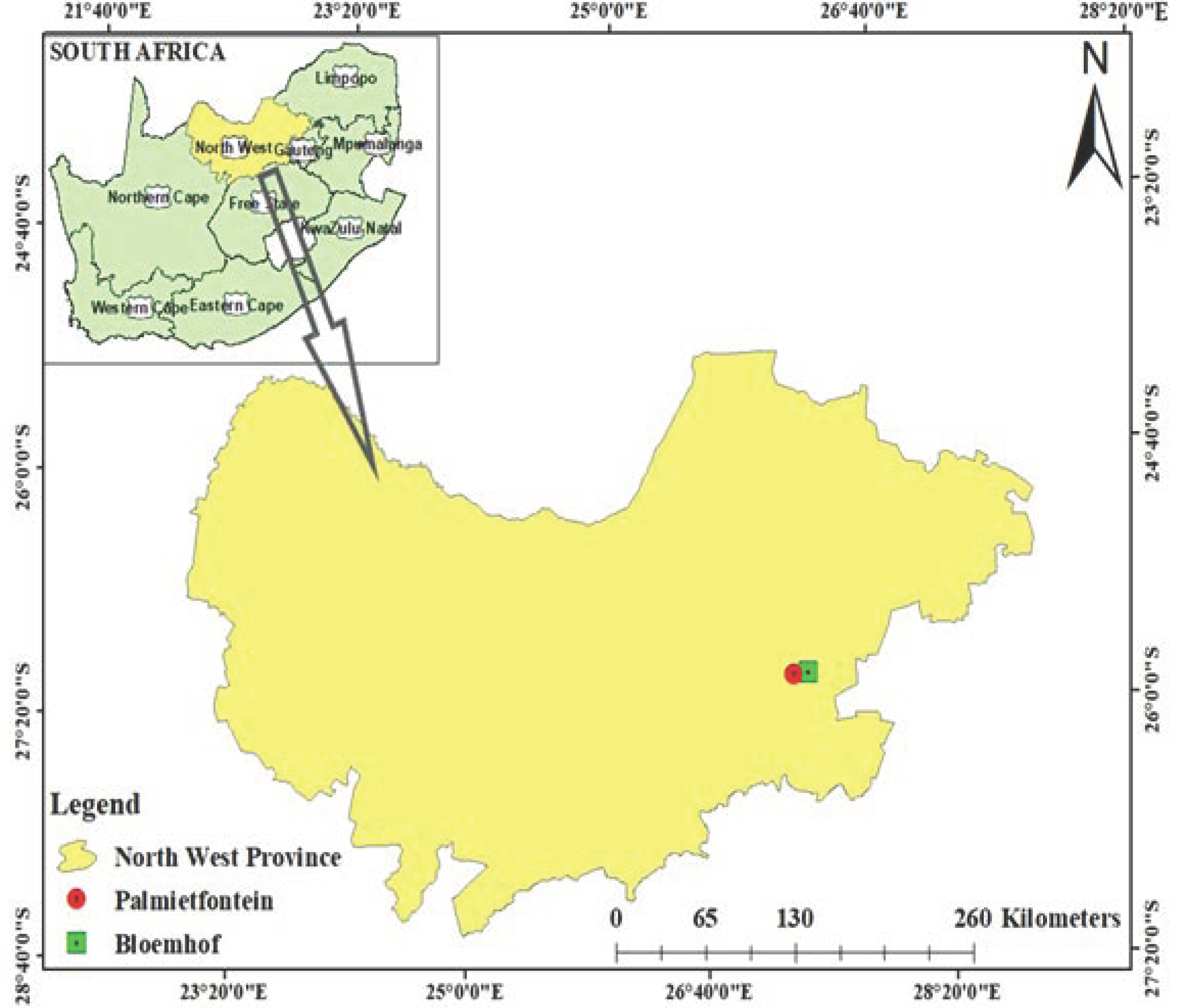
3.2. Genomic Overview of Metagenomic Sequences
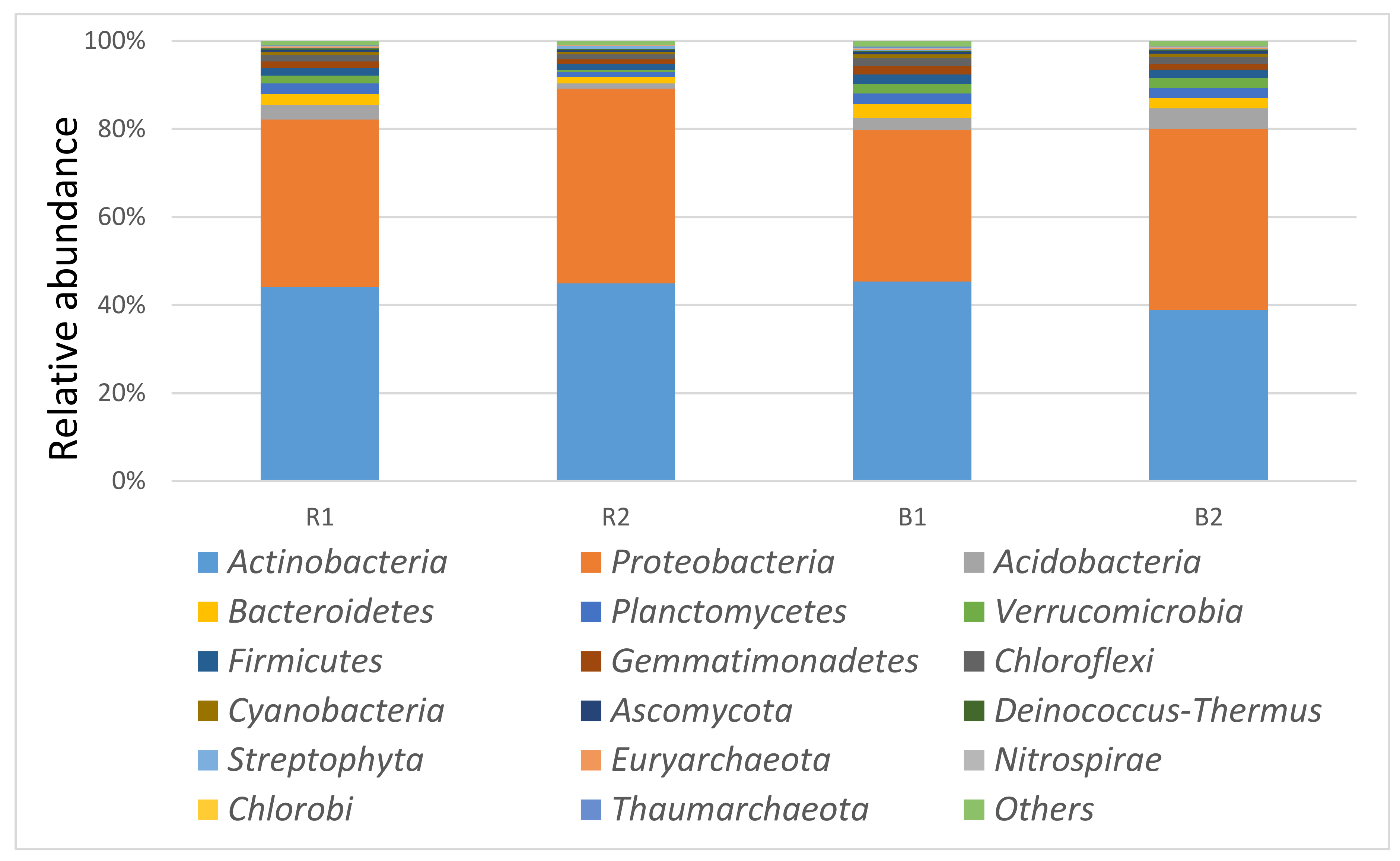
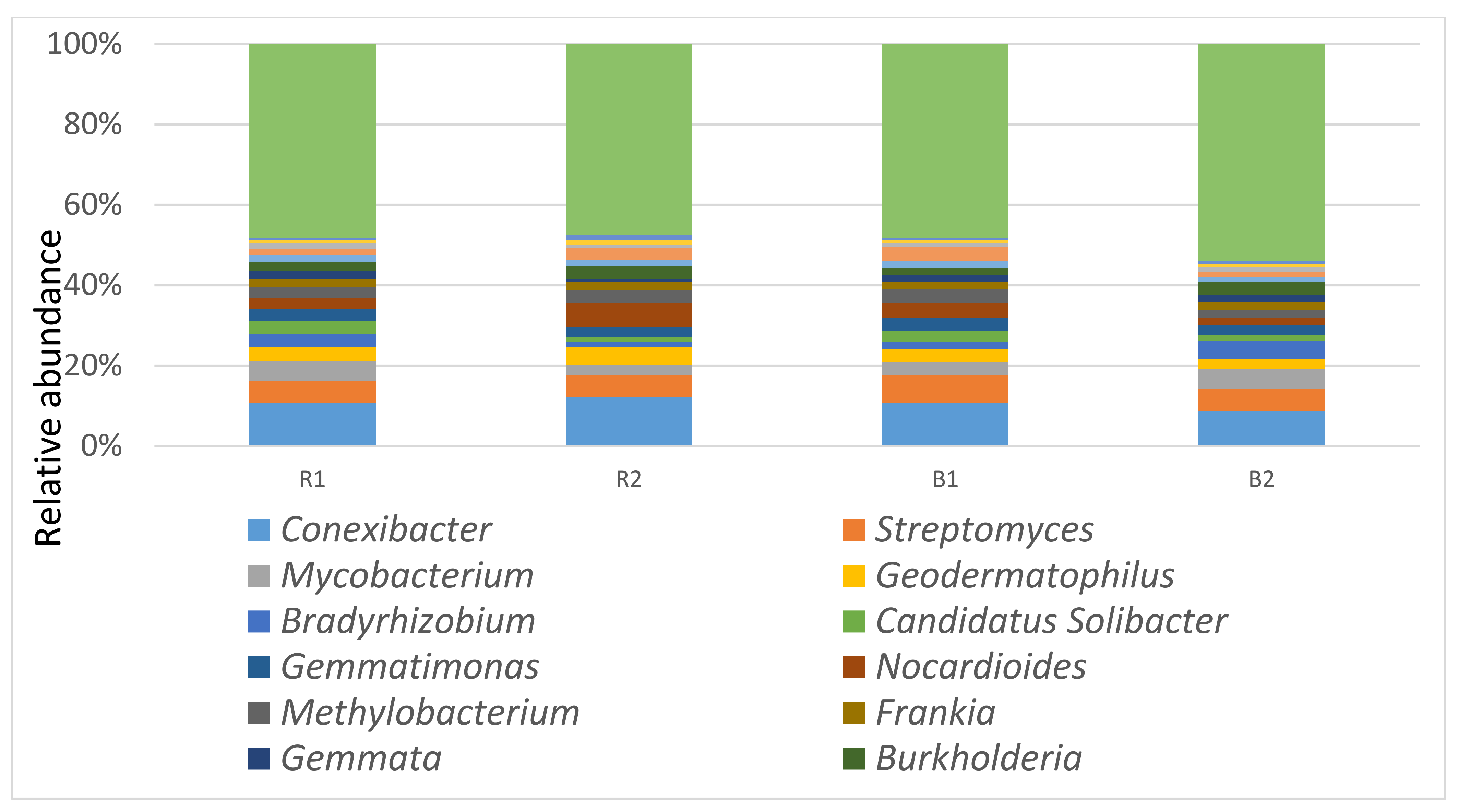
3.3. Taxonomy Diversity and Community Structure
3.4. Microbial Community Diversity Richness and Evenness of Sunflower Rhizosphere R1, R2, and Bulk Soils B1, B2 Examined
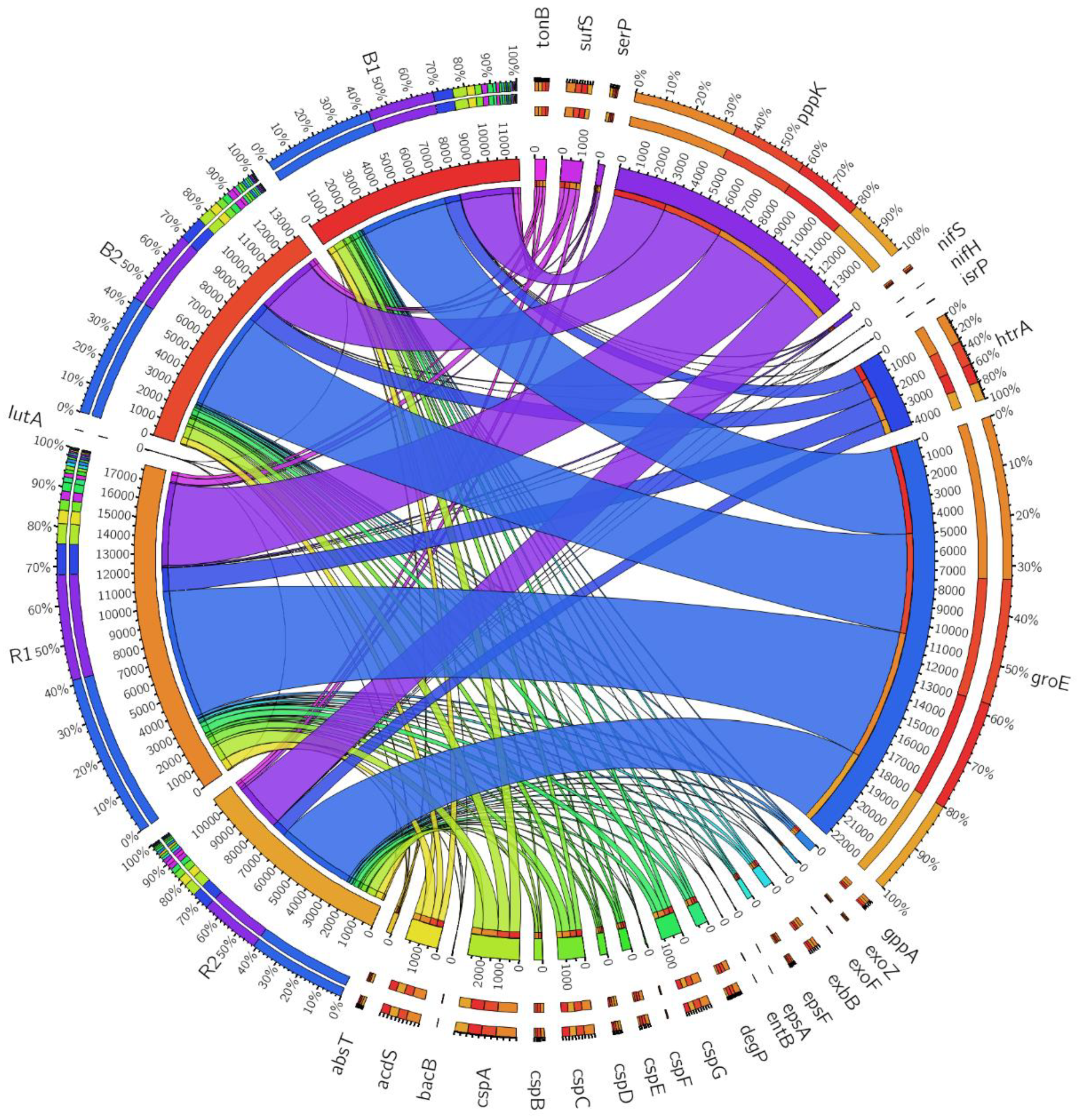
3.5. Diversity Indices of Functional Genes Observed from Sunflower Rhizosphere and Bulk Soils
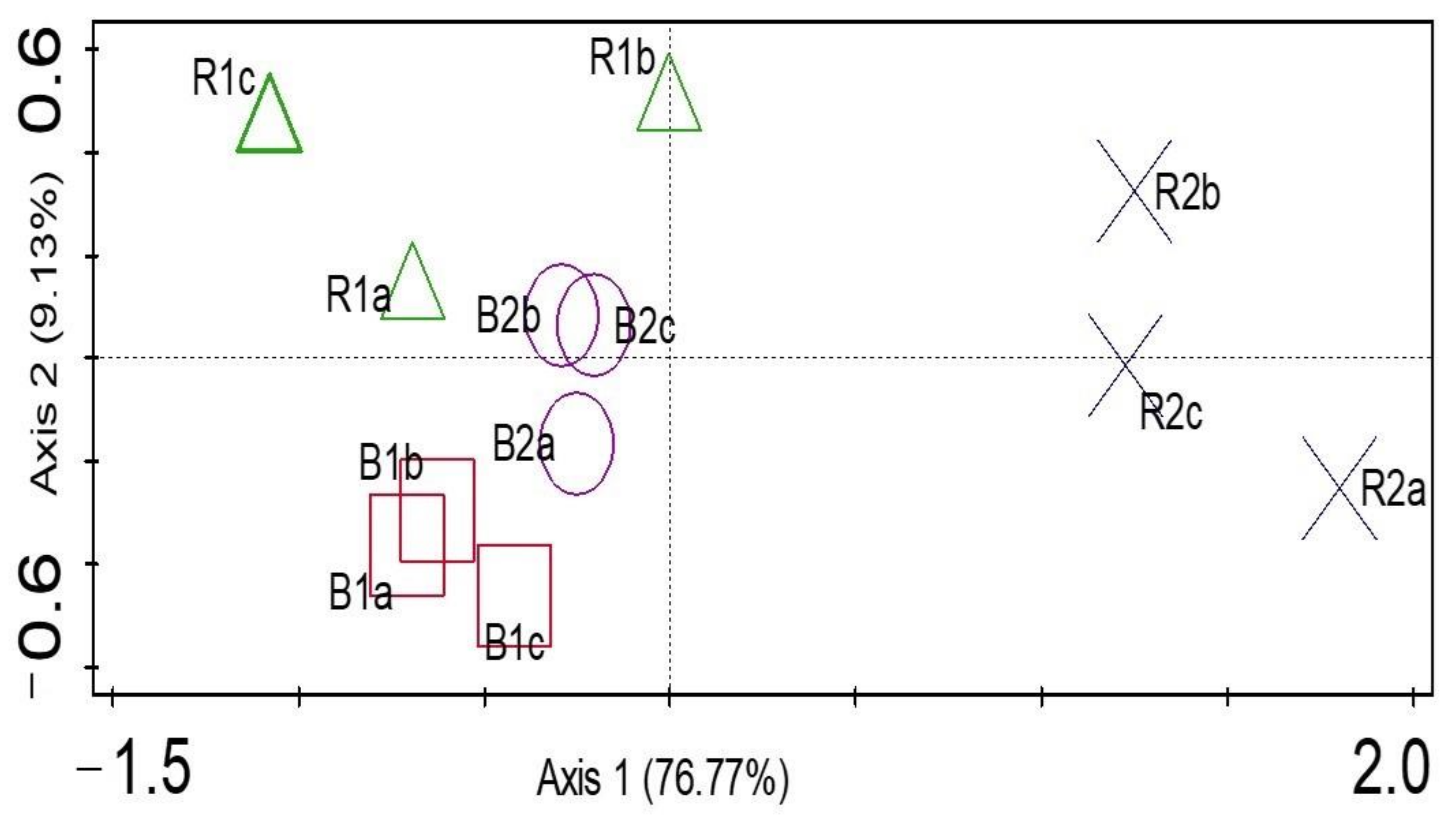
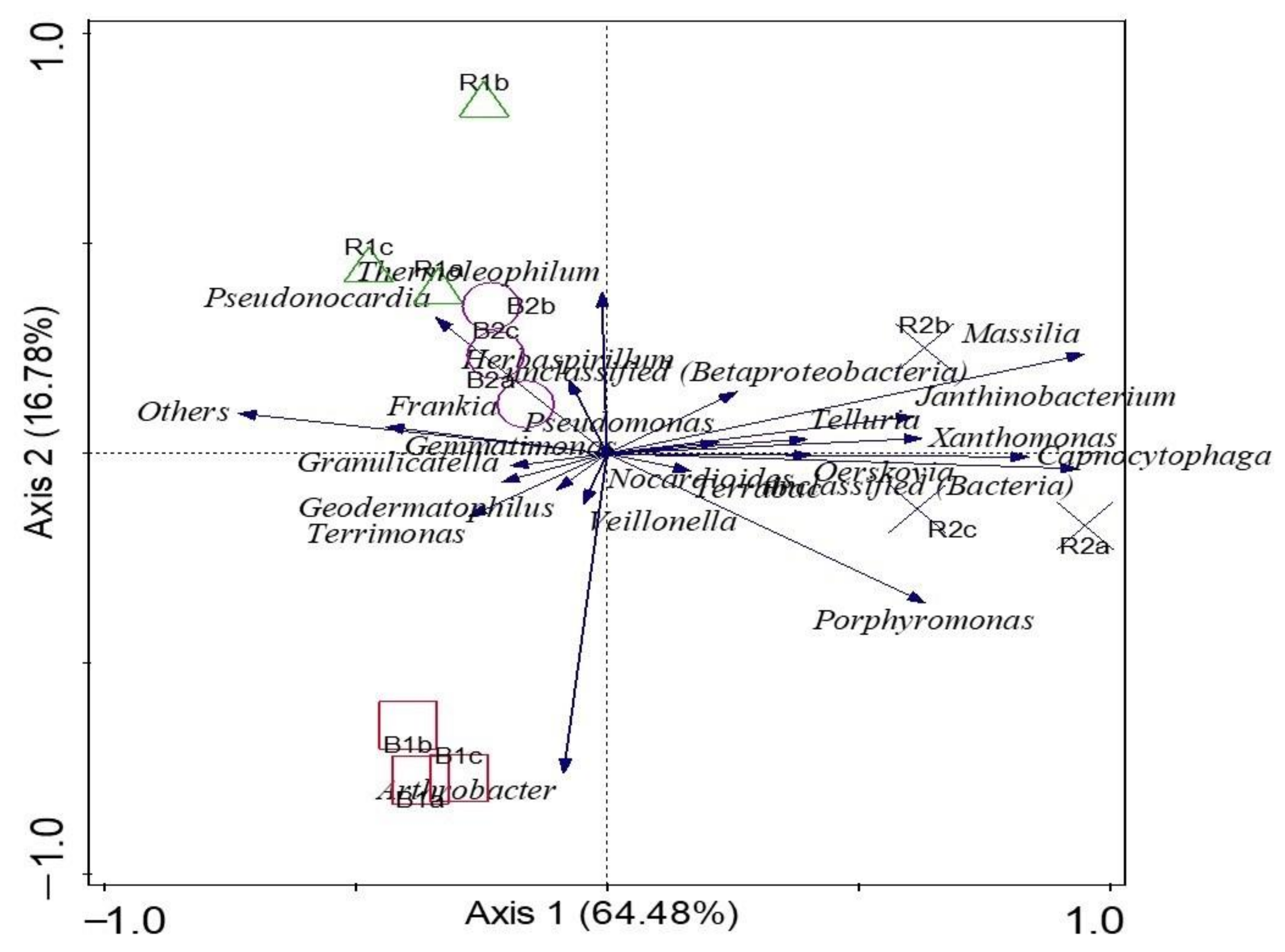
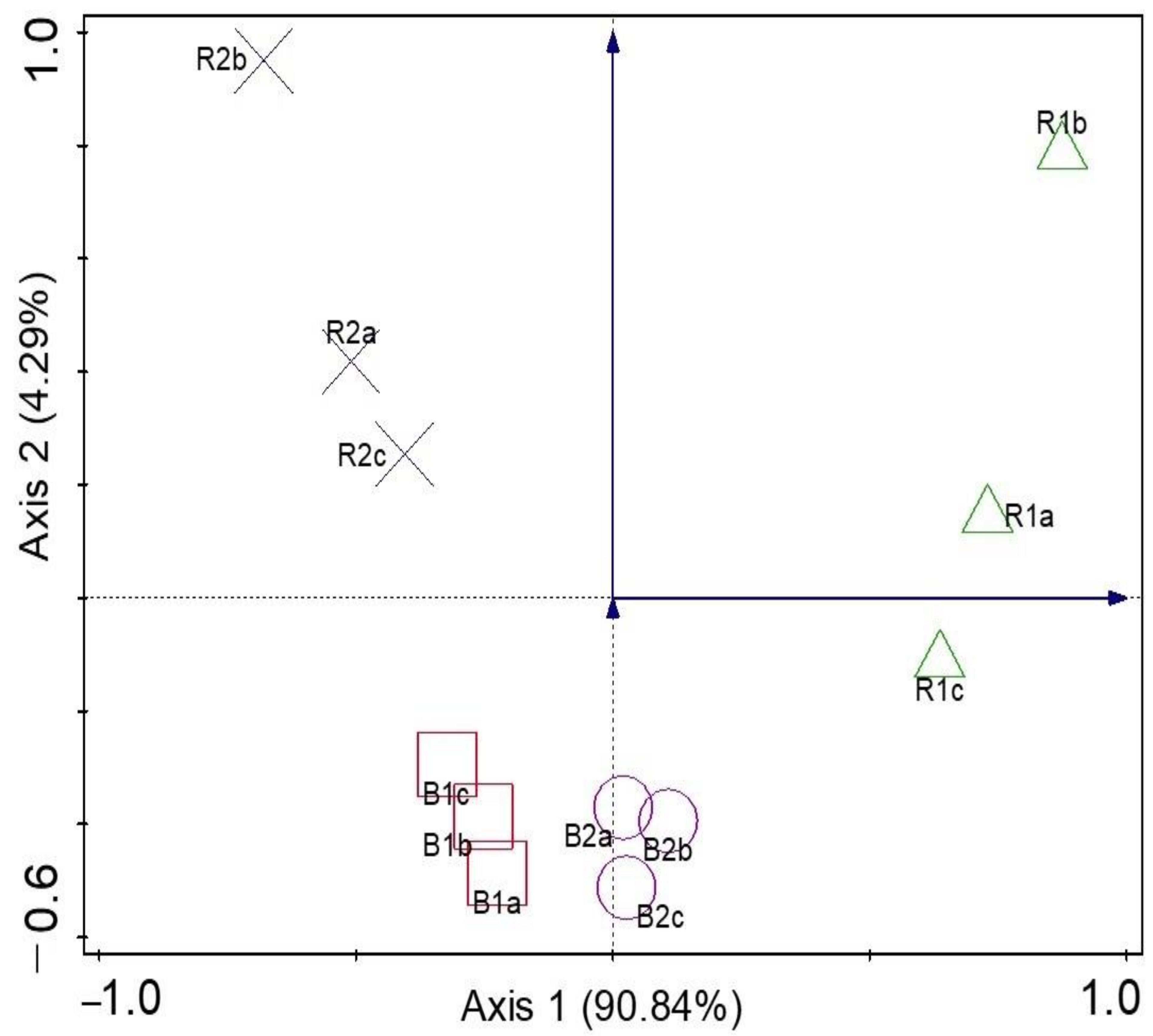
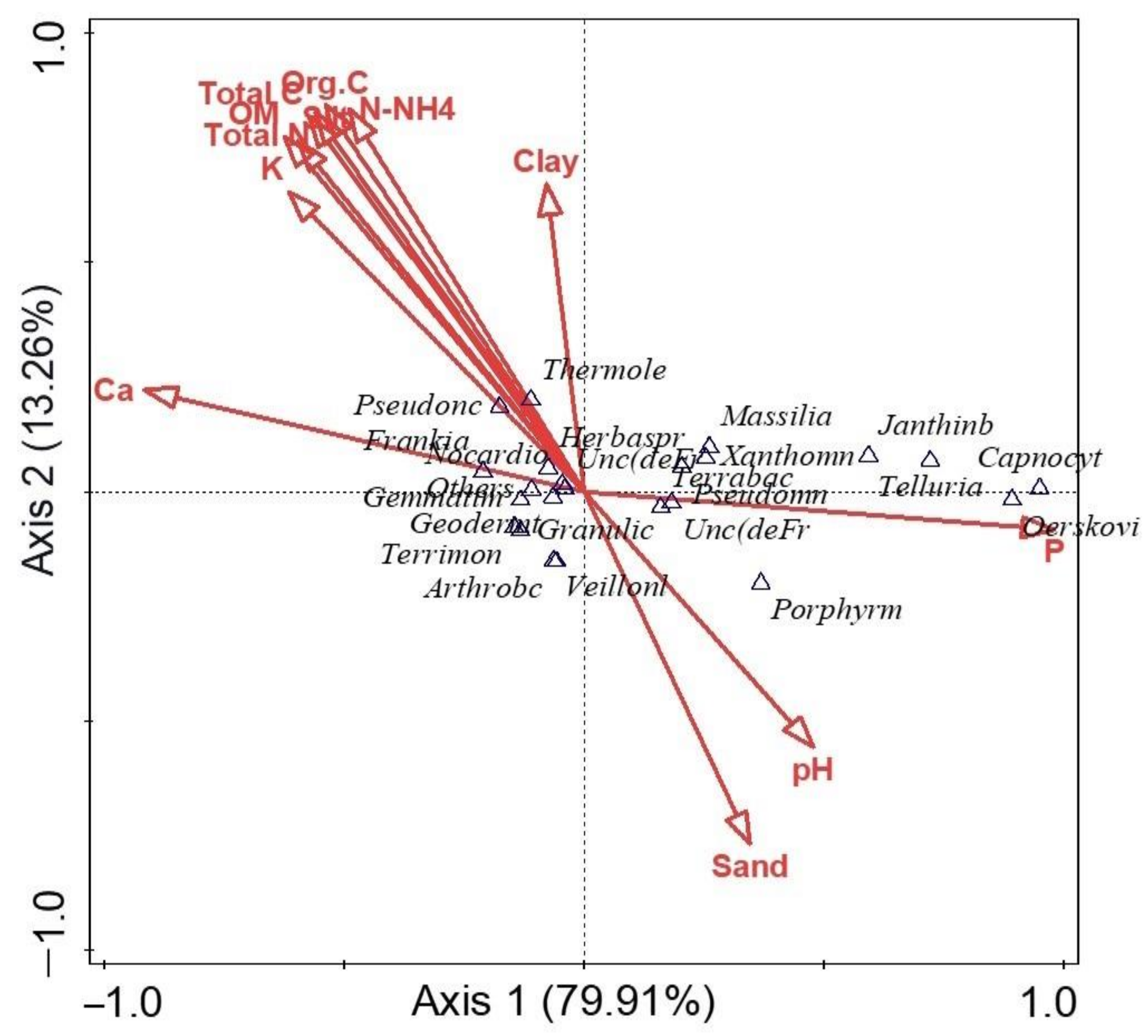
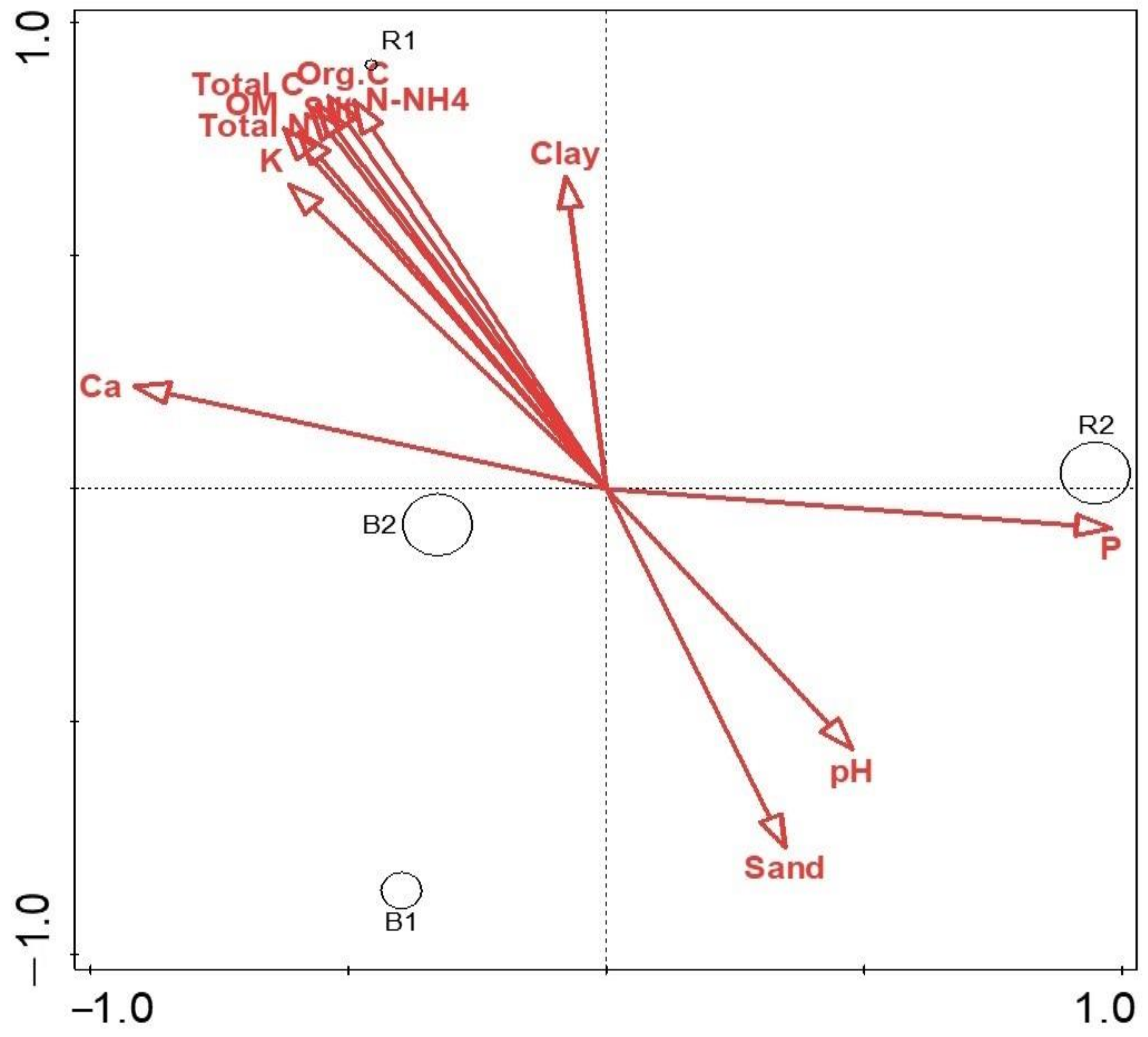
Microbial Diversity and Community Structure Are Influenced by Environmental Variables
3.6. Plant Growth Promoting Properties from Sunflower Rhizosphere and Bulk Soils
3.7. Nitrogen-Fixing Genes
3.7.1. Siderophore-Producing Genes
3.7.2. ACC Deaminase Producing Genes
3.7.3. Exopolysaccharide Producing Genes
3.7.4. Genes Potentially Contributing to Phosphate Solubilisation
3.7.5. High-Temperature Stress Response Genes
3.7.6. Heat and Cold Stress Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendes, L.; Raaijmakers, J.; de Hollander, M.; Mendes, R.; Tsai, S. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 2018, 12, 212. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579. [Google Scholar] [CrossRef]
- Qiao, C.; Penton, C.R.; Xiong, W.; Liu, C.; Wang, R.; Liu, Z.; Xu, X.; Li, R.; Shen, Q. Reshaping the rhizosphere microbiome by bio-organic amendment to enhance crop yield in a maize-cabbage rotation system. Appl. Soil Ecol. 2019, 142, 136–146. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Grube, M.; Köberl, M. The plant microbiome explored: Implications for experimental botany. J. Exp. Bot. 2015, 67, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Durrer, A.; Gumiere, T.; Taketani, R.; da Costa, D.; e Silva, M.; Andreote, F. The drivers underlying biogeographical patterns of bacterial communities in soils under sugarcane cultivation. Appl. Soil Ecol. 2017, 110, 12–20. [Google Scholar] [CrossRef]
- Uroz, S.; Oger, P.; Tisserand, E.; Cébron, A.; Turpault, M.; Buée, M.; De Boer, W.; Leveau, J.; Frey-Klett, P. Specific impacts of beech and Norway spruce on the structure and diversity of the rhizosphere and soil microbial communities. Sci. Rep. 2016, 6, 27756. [Google Scholar] [CrossRef] [PubMed]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Oberholster, T.; Vikram, S.; Cowan, D.; Valverde, A. Key microbial taxa in the rhizosphere of sorghum and sunflower grown in crop rotation. Sci. Total Environ. 2018, 624, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Alawiye, T.T.; Babalola, O.O. Bacterial Diversity and Community Structure in Typical Plant Rhizosphere. Diversity 2019, 11, 179. [Google Scholar] [CrossRef] [Green Version]
- Vacheron, J.; Moënne-Loccoz, Y.; Dubost, A.; Gonçalves-Martins, M.; Muller, D.; Prigent-Combaret, C. Fluorescent Pseudomonas strains with only few plant-beneficial properties are favored in the maize rhizosphere. Front. Plant Sci. 2016, 7, 1212. [Google Scholar] [CrossRef] [Green Version]
- Nouioui, I.; Cortés, C.J.; Carro, L.; Castro, J.F.; Gtari, M.; Ghodhbane-Gtari, F.; Klenk, H.-P.; Tisa, L.S.; Sangal, V.; Goodfellow, M. Genomic Insights into plant-growth-promoting potentialities of the genus Frankia. Front. Microbiol. 2019, 10, 1457. [Google Scholar] [CrossRef]
- Bruto, M.; Prigent-Combaret, C.; Muller, D.; Moënne-Loccoz, Y. Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci. Rep. 2014, 4, 6261. [Google Scholar] [CrossRef] [Green Version]
- del Carmen Orozco-Mosqueda, M.; Glick, B.R.; Santoyo, G. ACC deaminase in plant growth-promoting bacteria (PGPB): An efficient mechanism to counter salt stress in crops. Microbiol. Res. 2020, 235, 126439. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Zhang, P.; Trivedi, P.; Riera, N.; Wang, Y.; Liu, X.; Fan, G.; Tang, J.; Coletta-Filho, H.D. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, A.B.; Ounit, R.; Afshinnekoo, E.; Prill, R.J.; Hénaff, E.; Alexander, N.; Minot, S.S.; Danko, D.; Foox, J.; Ahsanuddin, S. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017, 18, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; Van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Enagbonma, B.; Aremu, B.; Babalola, O. Profiling the functional diversity of termite mound soil bacteria as revealed by shotgun sequencing. Genes 2019, 10, 637. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Bing, H.; Fang, L.; Wu, Y.; Yu, J.; Shen, G.; Jiang, M.; Wang, X.; Zhang, X. Diversity patterns of the rhizosphere and bulk soil microbial communities along an altitudinal gradient in an alpine ecosystem of the eastern Tibetan Plateau. Geoderma 2019, 338, 118–127. [Google Scholar] [CrossRef]
- Kettler, T.; Doran, J.; Gilbert, T. Simplified method for soil particle-size determination to accompany soil-quality analyses. Soil Sci. Soc. Am. J. 2001, 65, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Yuan, X.; Lin, H.; Yang, Y.; Li, Z. Differences in soil properties and bacterial communities between the rhizosphere and bulk soil and among different production areas of the medicinal plant Fritillaria thunbergii. Int. J. Mol. Sci. 2011, 12, 3770–3785. [Google Scholar] [CrossRef]
- Deke, A.; Adugna, W.; Fite, A. Soil physic-chemical properties in termite mounds and adjacent control soil in Miyo and Yabello Districts of Borana Zone, Southern Ethiopia. Am. J. Agric. For. 2016, 4, 69. [Google Scholar] [CrossRef] [Green Version]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.; Peterson, D.; Biggs, P. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Alvarez, V.; Teal, T.; Schmidt, T. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009, 3, 1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W. BLAT—the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 141. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mazcorro, J.F.; Castillo-Carranza, S.A.; Guard, B.; Gomez-Vazquez, J.P.; Dowd, S.E.; Brigthsmith, D.J. Comprehensive molecular characterization of bacterial communities in feces of pet birds using 16S marker sequencing. Microb. Ecol. 2017, 73, 224–235. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Guo, J.; McCulley, R.; Phillips, T.; McNear, D., Jr. Fungal endophyte and tall fescue cultivar interact to differentially affect bulk and rhizosphere soil processes governing C and N cycling. Soil Biol. Biochem. 2016, 101, 165–174. [Google Scholar] [CrossRef]
- Schlemper, T.R.; Leite, M.F.; Lucheta, A.R.; Shimels, M.; Bouwmeester, H.J.; van Veen, J.A.; Kuramae, E.E. Rhizobacterial community structure differences among sorghum cultivars in different growth stages and soils. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Song, Z.; Yang, X.; Mao, Z.; Nie, X.; Guo, H.; Peng, X. Microbial community analysis of apple rhizosphere around Bohai Gulf. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, N.; Guo, X.; Zhang, Y.; Ye, B. Comparative analysis of bacterial community structure in the rhizosphere of maize by high-throughput pyrosequencing. PLoS ONE 2017, 12, e0178425. [Google Scholar] [CrossRef]
- García-Salamanca, A.; Molina-Henares, M.; van Dillewijn, P.; Solano, J.; Pizarro-Tobías, P.; Roca, A.; Duque, E.; Ramos, J. Bacterial diversity in the rhizosphere of maize and the surrounding carbonate-rich bulk soil. Microb. Biotechnol. 2013, 6, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, W.; Li, S.; Hao, J.; Su, Z.; Sun, M.; Gao, Z.; Zhang, C. Variation of bacterial community diversity in rhizosphere soil of sole-cropped versus intercropped wheat field after harvest. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Mahoney, A.K.; Yin, C.; Hulbert, S.H. Community structure, species variation, and potential functions of rhizosphere-associated bacteria of different winter wheat (Triticum aestivum) cultivars. Front. Plant Sci. 2017, 8, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hol, W.; De Boer, W.; de Hollander, M.; Kuramae, E.; Meisner, A.; van der Putten, W. Context dependency and saturating effects of loss of rare soil microbes on plant productivity. Front. Plant Sci. 2015, 6, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Delgado-Baquerizo, M.; Anderson, I.; Singh, B. Response of soil properties and microbial communities to agriculture: Implications for primary productivity and soil health indicators. Front. Plant Sci. 2016, 7, 990. [Google Scholar] [CrossRef] [Green Version]
- Rousk, J.; Bååth, E.; Brookes, P.; Lauber, C.; Lozupone, C.; Caporaso, J.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340. [Google Scholar] [CrossRef]
- Nelkner, J.; Henke, C.; Lin, T.W.; Pätzold, W.; Hassa, J.; Jaenicke, S.; Grosch, R.; Pühler, A.; Sczyrba, A.; Schlüter, A. Effect of long-term farming practices on agricultural soil microbiome members represented by Metagenomically Assembled Genomes (MAGs) and their predicted plant-beneficial genes. Genes 2019, 10, 424. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Pan, F.; Yao, H. Response of symbiotic and asymbiotic nitrogen-fixing microorganisms to nitrogen fertilizer application. J. Soils Sediments 2019, 19, 1948–1958. [Google Scholar] [CrossRef]
- Zehr, J.P.; Jenkins, B.D.; Short, S.M.; Steward, G.F. Nitrogenase gene diversity and microbial community structure: A cross-system comparison. Environ. Microbiol. 2003, 5, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Díaz, J.F.; Fernández-González, A.J.; Villadas, P.J.; Robles, A.B.; Toro, N.; Fernández-López, M. Metagenomic assessment of the potential microbial nitrogen pathways in the rhizosphere of a Mediterranean forest after a wildfire. Microb. Ecol. 2015, 69, 895–904. [Google Scholar] [CrossRef]
- Sessitsch, A.; Hardoi:m, P.; Döring, J.; Weilharter, A.; Krause, A.; Woyke, T.; Mitter, B.; Hauberg-Lotte, L.; Friedrich, F.; Rahalkar, M. Functional characteristics of an endophyte community colonizing rice roots as revealed by metagenomic analysis. Mol. Plant-Microbe Interact. 2012, 25, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Glick, B.R. Plant growth-promoting bacteria: Mechanisms and applications. Scientifica 2012, 2012, 963401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikava, A.; Volkava, D.; Khramtsova, E. Characteristics of bacterial acdS-gene from the strain Pseudomonas putida B-37 and the creation of a genetic construct for determining its transient expression in the plant cells Nicotiana benthamiana. Proc. Natl. Acad. Sci. Belarus Biol. Ser. 2017, 3, 61–68. [Google Scholar]
- Igiehon, N.O.; Babalola, O.O.; Aremu, B.R. Genomic insights into plant growth promoting rhizobia capable of enhancing soybean germination under drought stress. BMC Microbiol. 2019, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Shelke, G.M.; Kumar, A.; Jha, P.N. Biochemistry and genetics of ACC deaminase: A weapon to “stress ethylene” produced in plants. Front. Microbiol. 2015, 6, 937. [Google Scholar] [CrossRef]
- Qin, Y.; Druzhinina, I.S.; Pan, X.; Yuan, Z. Microbially mediated plant salt tolerance and microbiome-based solutions for saline agriculture. Biotechnol. Adv. 2016, 34, 1245–1259. [Google Scholar] [CrossRef]
- Viterbo, A.; Landau, U.; Kim, S.; Chernin, L.; Chet, I. Characterization of ACC deaminase from the biocontrol and plant growth-promoting agent Trichoderma asperellum T203. FEMS Microbiol. Lett. 2010, 305, 42–48. [Google Scholar] [CrossRef]
- Thode, S.K.; Rojek, E.; Kozlowski, M.; Ahmad, R.; Haugen, P. Distribution of siderophore gene systems on a Vibrionaceae phylogeny: Database searches, phylogenetic analyses and evolutionary perspectives. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Altomare, C.; Tringovska, I. Beneficial soil microorganisms, an ecological alternative for soil fertility management. In Genetics, Biofuels and Local Farming Systems; Springer: Berlin/Heidelberg, Germany, 2011; pp. 161–214. [Google Scholar] [CrossRef]
- Radzki, W.; Mañero, F.G.; Algar, E.; García, J.L.; García-Villaraco, A.; Solano, B.R. Bacterial siderophores efficiently provide iron to iron-starved tomato plants in hydroponics culture. Antonie Leeuwenhoek 2013, 104, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Thakur, S.; Dhingra, G.; Singh, A.; Pal, M.K.; Harshvardhan, K.; Dubey, R.; Maheshwari, D. Inoculation of siderophore producing rhizobacteria and their consortium for growth enhancement of wheat plant. Biocatal. Agric. Biotechnol. 2018, 15, 264–269. [Google Scholar] [CrossRef]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 2019, 1–12. [Google Scholar] [CrossRef]
- Liu, G.; Kong, Y.; Fan, Y.; Geng, C.; Peng, D.; Sun, M. Whole-genome sequencing of Bacillus velezensis LS69, a strain with a broad inhibitory spectrum against pathogenic bacteria. J. Biotechnol. 2017, 249, 20–24. [Google Scholar] [CrossRef]
- Xu, W.-F.; Ren, H.-S.; Ou, T.; Lei, T.; Wei, J.-H.; Huang, C.-S.; Li, T.; Strobel, G.; Zhou, Z.-Y.; Xie, J. Genomic and functional characterization of the endophytic Bacillus subtilis 7PJ-16 strain, a potential biocontrol agent of mulberry fruit sclerotiniose. Microb. Ecol. 2019, 77, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Singh, P.; Srivastava, A. Synthesis, nature and utility of universal iron chelator–Siderophore: A review. Microbiol. Res. 2018, 212, 103–111. [Google Scholar] [CrossRef]
- Dimkpa, C.; Merten, D.; Svatoš, A.; Büchel, G.; Kothe, E. Siderophores mediate reduced and increased uptake of cadmium by Streptomyces tendae F4 and sunflower (Helianthus annuus), respectively. J. Appl. Microbiol. 2009, 107, 1687–1696. [Google Scholar] [CrossRef]
- Alaylar, B.; Güllüce, M.; Karadayi, M.; Isaoglu, M. Rapid Detection of Phosphate-Solubilizing Bacteria from Agricultural Areas in Erzurum. Curr. Microbiol. 2019, 76, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Enagbonma, B.J.; Babalola, O.O. Unveiling Plant-Beneficial Function as Seen in Bacteria Genes from Termite Mound Soil. J. Soil Sci. Plant Nutr. 2020, 1–10. [Google Scholar] [CrossRef]
- You, M.; Fang, S.; MacDonald, J.; Xu, J.; Yuan, Z.-C. Isolation and characterization of Burkholderia cenocepacia CR318, a phosphate solubilizing bacterium promoting corn growth. Microbiol. Res. 2020, 233, 126395. [Google Scholar] [CrossRef]
- Farhat, M.B.; Farhat, A.; Bejar, W.; Kammoun, R.; Bouchaala, K.; Fourati, A.; Antoun, H.; Bejar, S.; Chouayekh, H. Characterization of the mineral phosphate solubilizing activity of Serratia marcescens CTM 50650 isolated from the phosphate mine of Gafsa. Arch. Microbiol. 2009, 191, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Bharwad, K.; Rajkumar, S. Modulation of PQQ-dependent glucose dehydrogenase (mGDH and sGDH) activity by succinate in phosphate solubilizing plant growth promoting Acinetobacter sp. SK2. 3 Biotech 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Misra, H.; Rajpurohit, Y.; Khairnar, N. Pyrroloquinoline-quinone and its versatile roles in biological processes. J. Biosci. 2012, 37, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, J.J. Enzyme catalysis: The serine proteases. Nat. Educ. 2010, 3, 21. [Google Scholar]
- Vranova, V.; Rejsek, K.; Formanek, P. Proteolytic activity in soil: A review. Appl. Soil Ecol. 2013, 70, 23–32. [Google Scholar] [CrossRef]
- Phillips, R.W.; Roop, R.M. Brucella abortus HtrA functions as an authentic stress response protease but is not required for wild-type virulence in BALB/c mice. Infect. Immun. 2001, 69, 5911–5913. [Google Scholar] [CrossRef] [Green Version]
- Asari, S.Y. Studies on Plant-Microbe Interaction to Improve Stress Tolerance in Plants for Sustainable Agriculture. Ph.D. Thesis, Swedish University of Agricutural Sciences, Uppsala, Sweden, 2015. [Google Scholar]
- Kasim, W.A.; Gaafar, R.M.; Abou-Ali, R.M.; Omar, M.N.; Hewait, H.M. Effect of biofilm forming plant growth promoting rhizobacteria on salinity tolerance in barley. Ann. Agric. Sci. 2016, 61, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Stephan, B.I.; Colombo, R.P.; Silvani, V.A.; Pérgola, M.; Godeas, A.M.; Pardo, A.G.; Bidondo, L.F. Short-term effects of genetically modified potato on arbuscular mycorrhizal fungal communities. J. Soil Sci. Plant Nutr. 2019, 19, 352–356. [Google Scholar] [CrossRef]
- Arseneault, T.; Goyer, C.; Filion, M. Biocontrol of potato common scab is associated with high Pseudomonas fluorescens LBUM223 populations and phenazine-1-carboxylic acid biosynthetic transcript accumulation in the potato geocaulosphere. Phytopathology 2016, 106, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobadi, M.; Taherabadi, S.; Ghobadi, M.-E.; Mohammadi, G.-R.; Jalali-Honarmand, S. Antioxidant capacity, photosynthetic characteristics and water relations of sunflower (Helianthus annuus L.) cultivars in response to drought stress. Ind. Crops Prod. 2013, 50, 29–38. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Values | Sample Locations | |||
|---|---|---|---|---|
| R1 | R2 | B1 | B2 | |
| Organic C (%) | 1.77 ± 0.06 a | 0.49 ± 0.08 b | 0.47 ± 0.21 b | 0.98 ± 0.21 c |
| OM (%) | 1.83 ± 0.41 a | 0.62 ± 0.07 b | 0.70 ± 0.05 b | 1.27 ± 0.54 ab |
| N-NH4 (mg kg−1) | 19.67 ± 2.30 a | 16.23 ± 1.73 a | 15.67 ± 2.79 a | 18.37 ± 1.23 a |
| Total C (%) | 1.60 ± 0.29 a | 0.51 ± 0.09 b | 0.52 ± 0.06 b | 1.01 ± 0.07 c |
| Total N (%) | 0.13 ± 0.03 a | 0.05 ± 0.01 b | 0.05 ± 0.01 b | 0.09 ± 0.02 ab |
| pH (H20) | 6.00 ± 0.26 a | 6.50 ± 0.30 a | 6.60 ± 0.35 a | 5.78 ± 0.59 a |
| P3- (mg kg−1) | 19.33 ± 3.47 a | 74.43 ± 10.69 b | 28.31 ± 2.38 ac | 16.95 ± 2.40 ac |
| Ca2+ (mg kg−1) | 536.00 ± 34.51 a | 246.00 ± 13.89 b | 462.00 ± 24.56 c | 385.00 ± 21.79 d |
| K+ (mg kg−1) | 349.00 ± 32.23 a | 220.00 ± 17.00 b | 223.00 ± 41.33 b | 342.00 ± 32.70 a |
| Sand (%) | 72.00 ± 7.00 a | 80.00 ± 9.64 a | 84.00 ± 2.00 a | 72.00 ± 9.85 a |
| Silt (%) | 6.00 ± 1.50 a | 0.00 ± 0.00 b | 0.00 ± 0.00 b | 4.00 ± 1.00 a |
| Clay (%) | 22.00 ± 4.00 a | 20.00 ± 3.40 a | 16.00 ± 0.00 a | 24.00 ± 3.61 a |
| R1 | R2 | B1 | B2 | p Value | |
|---|---|---|---|---|---|
| Simpson-1-D | 0.6891 | 0.7804 | 0.7137 | 0.6817 | 0.54 |
| Shannon-H | 1.673 | 2.096 | 1.745 | 1.637 | |
| Eveness-e^H/S | 0.2538 | 0.3537 | 0.2604 | 0.2235 |
| R1 | R2 | B1 | B2 | p-Value | |
|---|---|---|---|---|---|
| Simpson-1-D | 0.7774 | 0.7841 | 0.7748 | 0.7755 | 0.76 |
| Shannon-H | 2.0110 | 2.0630 | 2.0040 | 2.0030 | |
| Eveness-e^H/S | 0.2576 | 0.2714 | 0.2649 | 0.2646 |
| Genes | Biological Functions | Aliases | Relative Abundance of Genes | |||
|---|---|---|---|---|---|---|
| R1 | R2 | B1 | B2 | |||
| Nitrogen fixation | Cysteine desulfurase nifS | EC 2.8.1.7 nifS | 94 | 19 | 44 | 69 |
| Cysteine desulfurase sufS | EC 2.8.1.7 sufS | 395 | 211 | 262 | 272 | |
| Nitrogenase (molybdenum-iron) reductase and maturation protein nifH | nifH | 6 | 5 | 1 | 0 | |
| Siderophore | 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase | EC 1.3.1.28 bacillibactin | 8 | 2 | 1 | 3 |
| 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase | EC 1.3.1.28 enterobactin | 4 | 4 | 0 | 3 | |
| ABC Fe3+ siderophore transporter | 73 | 112 | 58 | 43 | ||
| Ferric siderophore transport system, biopolymer transport protein exbB | exbB | 146 | 116 | 110 | 103 | |
| Aerobactin siderophore receptor IutA | IutA | 2 | 7 | 3 | 2 | |
| Iron siderophore receptor protein | 9 | 2 | 6 | 10 | ||
| TonB-dependent siderophore receptor | tonB | 176 | 163 | 135 | 161 | |
| ACC deaminase | 1-aminocyclopropane-1-carboxylate deaminase acdS | EC 3.5.99.7 | 599 | 395 | 371 | 400 |
| Exopolysaccharides | Exopolysaccharide biosynthesis glycosyltransferase epsF | epsF EC 2.4.1.- | 80 | 48 | 74 | 80 |
| Exopolysaccharide production protein exoF precursor | exoF | 17 | 3 | 4 | 2 | |
| Exopolysaccharide production protein exoZ | exoZ | 38 | 45 | 24 | 39 | |
| Exopolysaccharide biosynthesis transcriptional activator epsA | epsA | 4 | 3 | 3 | 3 | |
| High-temperature stress response | Protease/chaperone protein htrA | htrA | 1379 | 798 | 874 | 1100 |
| Serine protease, degP/htrA, do-like | degP/htrA EC 3.4.21.- | 246 | 104 | 148 | 151 | |
| Serine protease | EC 3.4.21.- | 78 | 147 | 67 | 52 | |
| Phosphate solubilization | Polyphosphate kinase | EC 2.7.4.1 | 4725 | 2691 | 2966 | 3388 |
| Phosphatase, ppx/gppA family | ppx/gppA | 145 | 108 | 81 | 106 | |
| Heat and cold stress shock | Cold shock protein cspA | cspA | 898 | 557 | 616 | 653 |
| Cold shock protein cspB | cspB | 166 | 63 | 93 | 128 | |
| Cold shock protein cspC | cspC | 456 | 319 | 312 | 341 | |
| Cold shock protein cspD | cspD | 110 | 116 | 77 | 92 | |
| Cold shock protein cspE | cspE | 167 | 75 | 115 | 120 | |
| Cold shock protein cspF | cspF | 18 | 9 | 15 | 21 | |
| Cold shock protein cspG | cspG | 388 | 232 | 226 | 291 | |
| Heat shock protein 60 family chaperone groEL | groEL | 7318 | 4453 | 4977 | 5603 | |
| Heat shock protein 60 family co-chaperone groES | groES | 1409 | 814 | 1016 | 1134 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alawiye, T.T.; Babalola, O.O. Metagenomic Insight into the Community Structure and Functional Genes in the Sunflower Rhizosphere Microbiome. Agriculture 2021, 11, 167. https://doi.org/10.3390/agriculture11020167
Alawiye TT, Babalola OO. Metagenomic Insight into the Community Structure and Functional Genes in the Sunflower Rhizosphere Microbiome. Agriculture. 2021; 11(2):167. https://doi.org/10.3390/agriculture11020167
Chicago/Turabian StyleAlawiye, Temitayo Tosin, and Olubukola Oluranti Babalola. 2021. "Metagenomic Insight into the Community Structure and Functional Genes in the Sunflower Rhizosphere Microbiome" Agriculture 11, no. 2: 167. https://doi.org/10.3390/agriculture11020167
APA StyleAlawiye, T. T., & Babalola, O. O. (2021). Metagenomic Insight into the Community Structure and Functional Genes in the Sunflower Rhizosphere Microbiome. Agriculture, 11(2), 167. https://doi.org/10.3390/agriculture11020167