
Characterization of the Endophytic Mycobiome in Cowpea (Vigna unguiculata) from a Single Location Using Illumina Sequencing



Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Sampling
2.2. Illumina Sequencing of the Metagenomic DNA
2.3. Cluster Analysis
2.4. Phylogenetic Analysis
3. Results
3.1. Illumina Sequencing of the Metagenomic DNA
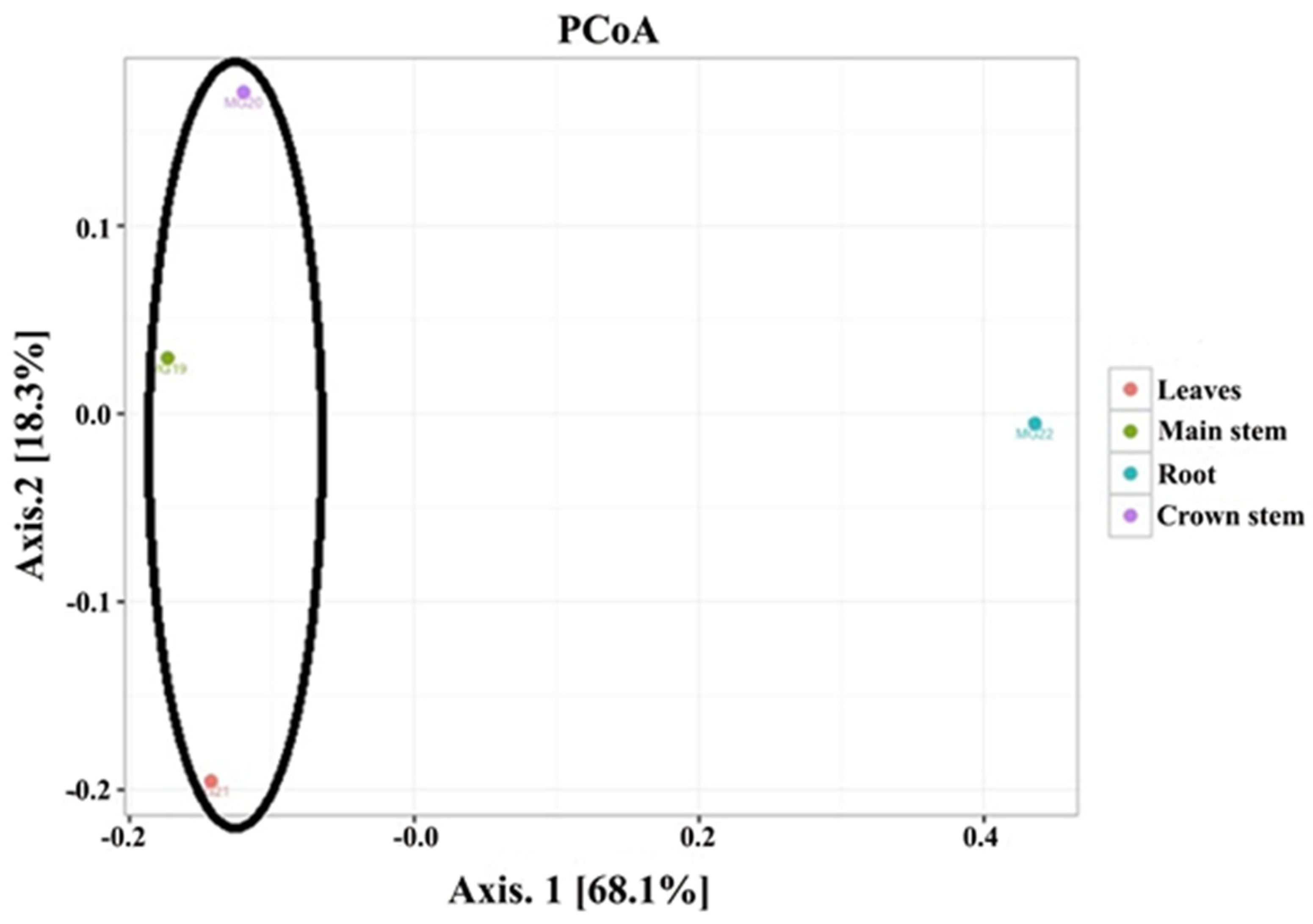
3.2. Cluster Analysis
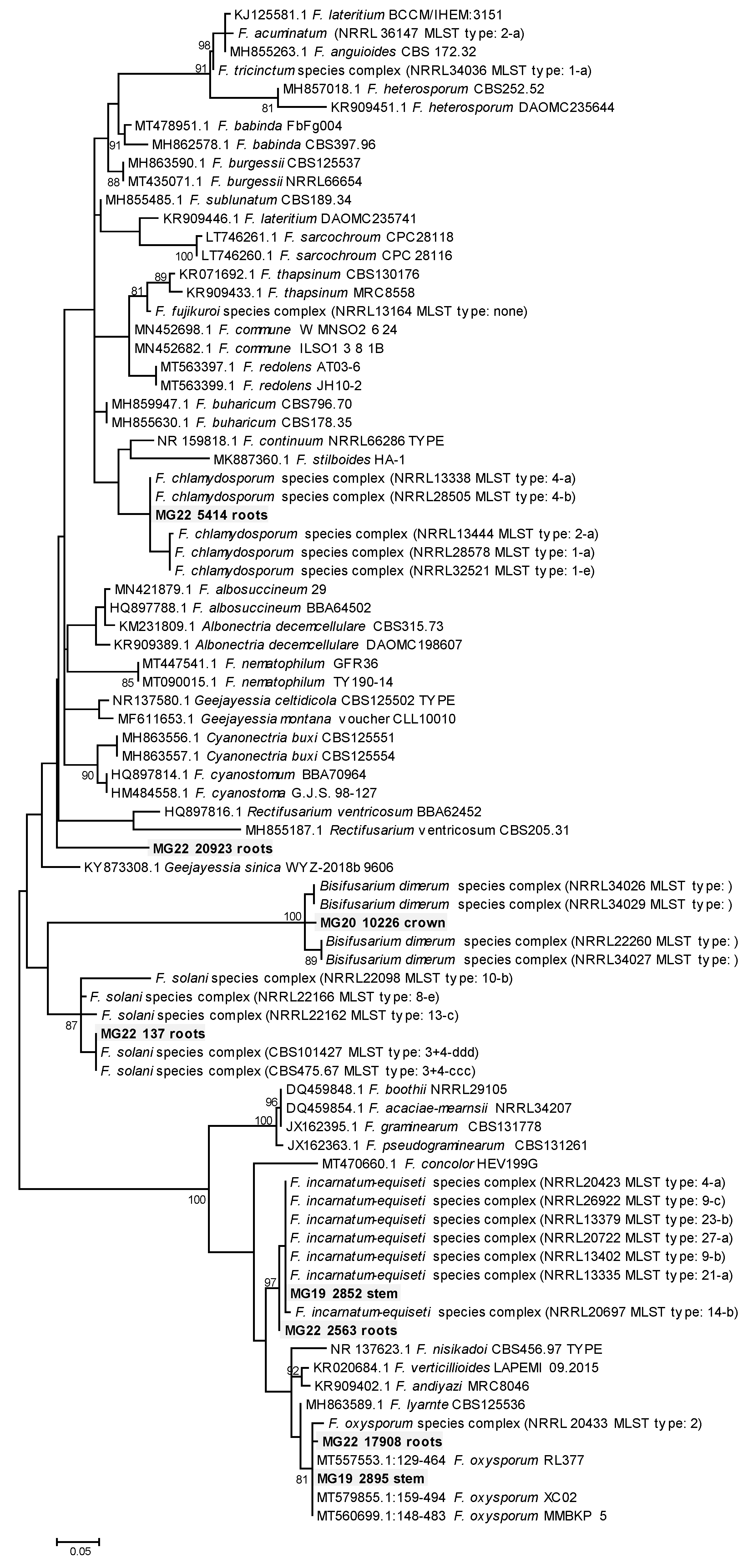
3.3. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Langyintuo, A.S.; Lowenberg-DeBoer, J.; Faye, M.; Lambert, D.; Ibro, G.; Moussa, B.; Kergna, A.; Kushwaha, S.; Musa, S.; Ntoukam, G. Cowpea supply and demand in West and Central Africa. Field Crops Res. 2003, 82, 215–231. [Google Scholar] [CrossRef]
- Singh, B.B.; Hartmann, P.; Fatokun, C.; Tamo, M.; Tarawali, S.; Ortiz, R. Recent progress on cowpea improvement. Chron. Hortic. 2003, 43, 8–12. [Google Scholar]
- Ehlers, J.D.; Hall, A.E. Cowpea (Vigna unguiculata L Walp). Field Crops Res. 1997, 53, 187–204. [Google Scholar] [CrossRef]
- Hamid, S.; Muzaffar, S.; Wani, I.; Ahmed, M.; Farooq, A.B.; Mohd, M. Physical and cooking characteristics of two cowpea cultivars grown in temperate Indian climate. J. Saudi Soc. Agric. Sci. 2016, 15, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Menendez, C.M.; Hall, A.E.; Gepts, P. A genetic linkage map of cowpea (Vigna unguiculata) developed from a cross between two inbred, domesticated lines. Theor. Appl. Genet. 1997, 95, 1210–1217. [Google Scholar] [CrossRef]
- Quin, F.M. Introduction. In Advances in Cowpea Research; Singh, B.B., Mohan Raj, D.R., Dashiell, K.E., Jackai, L.E.N., Eds.; Sayce Publishing: Devon, UK, 1997; pp. ix–xv. [Google Scholar]
- Davis, D.W.; Oelke, E.A.; Oplinger, E.S.; Doll, J.D.; Hanson, C.V.; Putnam, D.H. Cowpea research, production and utilization. In Field Crops Manual; Bressani, R., Ed.; John Wiley and Sons: Oxford, UK, 1991. [Google Scholar]
- Ba, F.S.; Pasquet, R.S.; Gepts, P. Genetic diversity in cowpea [Vigna unguiculata (L.) Walp.] as revealed by RAPD markers. Genet. Resour. Crop Evol. 2004, 51, 539–550. [Google Scholar] [CrossRef]
- Asiwe, J.A.N. Needs assessment of cowpea production practices, constraints and utilization in South Africa. Afr. J. Biotechnol. 2009, 8, 5383–5388. [Google Scholar]
- Asiwe, J.A.N.; Belane, A.; Dakora, F.D. Evaluation of cowpea breeding lines for nitrogen fixatrion at ARC-Grain Crops Institute, Potchefstroom, South Africa. In Proceedings of the 16th International Congress on Biological Nitrogen Fixation, Big Sky, MT, USA, 14–19 June 2009. [Google Scholar]
- Okereke, G.U.; Egwu, S.E.; Nnabude, P. Effect of cowpea organic residues and fertilizer N on soil fertility, growth and yield of upland rice. In Proceedings of the Eighteenth World Congress Soil Science, Philadelphia, PA, USA, 9–15 July 2006. [Google Scholar]
- Adandonon, A.; Aveling, T.A.S.; Tamo, M. Occurrence and distribution of cowpea damping-off and stem rot and associated fungi in Benin. J. Agric. Sci. 2004, 142, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.B. Advances in Cowpea Research; Sayce Publishing: Devon, UK, 1997. [Google Scholar]
- Singh, S.R.; Rachie, K.O. Cowpea Research, Production and Utilisation; John Wiley and Sons Ltd.: Chichester, UK, 1985. [Google Scholar]
- Aveling, T.A.S.; Adandonon, A. First report of pre- and postemergence damping-off of cowpea caused by Pythium ultimum in South Africa. Plant Dis. 2000, 84, 922. [Google Scholar] [CrossRef]
- Kendrick, J.B. Seed transmission of cowpea wilt. Phytopathology 1931, 21, 979–983. [Google Scholar]
- Kendrick, J.B.; Snyder, W.C. Fusarium yellows of beans. Phytopathology 1942, 32, 1010–1014. [Google Scholar]
- Pio-Ribeiro, G.; Assis-Filho, F.M.; Doenças do, C. Manual de Fitopatologia; Agronômica Ceres: São Paulo, Brazil, 1997. [Google Scholar]
- Kritzinger, Q.; Aveling, T.A.; Marasas, W.F.; Rheeder, J.P.; Van Der Westhuizen, L.; Shephard, G.S. Mycoflora and fumonisin mycotoxins associated with cowpea [Vigna unguiculata (L.) Walp] seeds. J. Agric. Food Chem. 2003, 51, 2188–2192. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E.; Korsten, L.; Aveling, T.A.S. Infection process of Colletotrichum dematium on cowpea stems. Mycol. Res. 1999, 103, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.J.; Thottappilly, G.; Emechebe, A.M.; Singh, B.B. Diseases of cowpea. In In Pathology of Food and Pasture Legumes of Cowpea; Allen, D.J., Lenne, J.M., Eds.; CAB International: Wallingford, UK, 1998; pp. 267–324. [Google Scholar]
- la Grange, N.; Aveling, T.A.S. First Report of Alternaria cassiae on Cowpea. Plant Dis. 1998, 82, 1171. [Google Scholar] [CrossRef]
- Seenappa, M.; Keswani, C.L.; Kundya, T.M. Aspergillus infection and aflatoxin production in some Cowpea (Vigna unguiculata (L) Walp) lines in Tanzania. Mycopathologia 1983, 83, 103–106. [Google Scholar] [CrossRef]
- Moss, M.O. Centenary review—Mycotoxins. Mycol. Res. 1996, 100, 513–523. [Google Scholar] [CrossRef]
- Caroll, E.C.; Carroll, F.E. Studies on the incidence of coniferous needle endophytes in the Pacific Northwest. Can. J. Bot. 1978, 56, 3034–3043. [Google Scholar] [CrossRef]
- Petrini, O.; Fisher, P.J. Fungal endophytes in Salicornia perennis. Trans. Br. Mycol. Soc. 1986, 87, 647–651. [Google Scholar] [CrossRef]
- Rodriguez, R.J.; White, J.F., Jr.; Arnold, A.E.; Redman, R.S. Fungal endophytes: Diversity and functional roles. New Phytol. 2009, 182, 314–330. [Google Scholar] [CrossRef]
- Schulz, B.; Boyle, C. The endophytic continuum. Mycol. Res. 2005, 109, 661–686. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.; Flinn, B.S. The use of beneficial microbial endophytes for plant biomass and stress tolerance improvement. Recent Pat. Biotechnol. 2010, 4, 81–95. [Google Scholar] [CrossRef]
- Khan, A.L.; Al-Harrasi, A.; Al-Rawahi, A.; Al-Farsi, Z.; Al-Mamari, A.; Waqas, M.; Asaf, S.; Elyassi, A.; Mabood, F.; Shin, J.H.; et al. Endophytic Fungi from Frankincense Tree Improves Host Growth and Produces Extracellular Enzymes and Indole Acetic Acid. PLoS ONE 2016, 11, e0158207. [Google Scholar] [CrossRef]
- David, V.; Terrat, S.; Herzine, K.; Claisse, O.; Rousseaux, S.; Tourdot-Marechal, R.; Masneuf-Pomarede, I.; Ranjard, L.; Alexandre, H. Highthroughput sequencing of amplicons for monitoring yeast biodiversity in must and during alcoholic fermentation. J. Ind. Microbiol. Biotechnol. 2014, 41, 811–821. [Google Scholar] [CrossRef]
- Cheng, W.X.; Parton, W.J.; Gonzalez-Meler, M.A.; Phillips, R.; Asao, S.; McNickle, G.G.; Brzostek, E.; Jastrow, J.D. Synthesis and modeling perspectives of rhizosphere priming. New Phytol. 2014, 201, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, M.G.; Liu, Y.; Zhang, F.S.; Hodge, A.; Feng, G. Carbon and phosphorus exchange may enable cooperation between an arbuscular mycorrhizal fungus and a phosphate-solubilizing bacterium. New Phytol. 2016, 210, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Gutknecht, J.L.M.; Herman, D.J.; Keck, D.C.; Firestone, M.K.; Cheng, W.X. Rhizosphere priming effects on soil carbon and nitrogen mineralization. Soil Biol. Biochem. 2014, 76, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Beans, C. Core Concept: Probing the phytobiome to advance agriculture. Proc. Natl. Acad. Sci. USA 2017, 114, 8900–8902. [Google Scholar] [CrossRef] [Green Version]
- Leach, J.E.; Triplett, L.R.; Argueso, C.T.; Trivedi, P. Communication in the Phytobiome. Cell 2017, 169, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.W.; Tsai, P.; Anfang, N.; Ross, H.A.; Goddard, M.R. Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ. Microbiol. 2014, 16, 2848–2858. [Google Scholar] [CrossRef] [Green Version]
- Price, L.B.; Liu, C.M.; Melendez, J.H.; Frankel, Y.M.; Engelthaler, D.; Aziz, M.; Bowers, J.; Rattray, R.; Ravel, J.; Kingsley, C.; et al. Community analysis of chronic wound bacteria using 16S rRNA gene-based pyrosequencing: Impact of diabetes and antibiotics on chronic wound microbiota. PLoS ONE 2009, 4, e6462. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Ryberg, M.; Abarenkov, K.; Sjokvist, E.; Kristiansson, E. The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol. Lett. 2009, 296, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinge, T.R.; Cason, E.D.; Valverde, A.; Nyaga, M.; Gryzenhout, M. Endophytic seed mycobiome of six sorghum (Sorghum bicolor) cultivars from commercial seedlots using an Illumina sequencing approach. Mycosphere 2019, 10, 739–756. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Koljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence-Limits on Phylogenies—An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- González, V.; Tello, M.L. The endophytic mycota associated with Vitis vinifera in central Spain. Fungal Divers. 2011, 47, 29–42. [Google Scholar] [CrossRef]
- Kim, D.-H.; Kim, S.-H.; Kwon, S.-W.; Lee, J.-K.; Hong, S.-B. Mycoflora of Soybeans Used for Meju Fermentation. Mycobiology 2013, 41, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Cai, L.; Crous, P.W.; Damm, U. Circumscription of the anthracnose pathogens Colletotrichum lindemuthianum and C. nigrum. Mycologia 2013, 105, 844–860. [Google Scholar] [CrossRef] [Green Version]
- Qin, G.Z.; Tian, S.P. Enhancement of Biocontrol Activity of Cryptococcus laurentii by Silicon and the Possible Mechanisms Involved. Phytopathology 2005, 95, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Li, J.K.; Li, H.; Ji, S.F.; Chen, T.; Tian, S.P.; Qin, G.Z. Enhancement of biocontrol efficacy of Cryptococcus laurentii by cinnamic acid against Penicillium italicum in citrus fruit. Postharvest Biol. Technol. 2019, 149, 42–49. [Google Scholar] [CrossRef]
- Rong, X.; McSpadden Gardener, B.B. Draft Genome Sequence of Cryptococcus flavescens Strain OH182.9_3C, a Biocontrol Agent against Fusarium Head Blight of Wheat. Genome Announc. 2013, 1, e00762-13. [Google Scholar] [CrossRef] [Green Version]
- Freimoser, F.M.; Rueda-Mejia, M.P.; Tilocca, B.; Migheli, Q. Biocontrol yeasts: Mechanisms and applications. World J. Microbiol. Biotechnol. 2019, 35, 154. [Google Scholar] [CrossRef] [Green Version]
- Jensen, D.F.; Knudsen, I.M.B.; Lubeck, M.; Mamarabadi, M.; Hockenhull, J.; Jensen, B. Development of a biocontrol agent for plant disease control with special emphasis on the near commercial fungal antagonist Clonostachys rosea strain ‘IK726’. Australas. Plant Pathol. 2007, 36, 95–101. [Google Scholar] [CrossRef]
- Cota, L.V.; Maffia, L.A.; Mizubuti, E.S.; Macedo, P.E.; Antunes, R.F. Biological control of strawberry grey mould by Clonostachys rosea under field conditions. Phytopathology 2008, 98, S41–S42. [Google Scholar]
- Xue, A.G.; Voldeng, H.D.; Savard, M.E.; Fedak, G.; Tian, X.; Hsiang, T. Biological control of fusarium head blight of wheat with Clonostachys rosea strain ACM941. Can. J. Plant Pathol. 2009, 31, 169–179. [Google Scholar]
- Sun, Z.B.; Li, S.D.; Ren, Q.; Xu, J.L.; Lu, X.; Sun, M.H. Biology and applications of Clonostachys rosea. J. Appl. Microbiol. 2020, 129, 486–495. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xu, D.L.; Chao, L.M.; Liu, H.J.; Bao, Y.Y. Microbial assemblages associated with the rhizosphere and endosphere of an herbage, Leymus chinensis. Microb. Biotechnol. 2020, 13, 1390–1402. [Google Scholar] [CrossRef] [Green Version]
- de Souza, R.S.C.; Okura, V.K.; Armanhi, J.S.L.; Jorrín, B.; Lozano, N.; da Silva, M.J.; González-Guerrero, M.; de Araújo, L.M.; Verza, N.C.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 2016, 6, 28774. [Google Scholar] [CrossRef] [Green Version]
- Eyre, A.W.; Wang, M.Y.; Oh, Y.; Dean, R.A. Identification and Characterization of the Core Rice Seed Microbiome. Phytobiomes J. 2019, 3, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Simonin, M.; Dasilva, C.; Terzi, V.; Ngonkeu, E.L.M.; Diouf, D.; Kane, A.; Béna, G.; Moulin, L. Influence of plant genotype and soil on the wheat rhizosphere microbiome: Evidences for a core microbiome across eight African and European soils. FEMS Microbiol. Ecol. 2020, 96, fiaa067. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; Gordon, T.R. Cryptic fungal infections: The hidden agenda of plant pathogens. Front. Plant Sci. 2014, 5, 506. [Google Scholar] [CrossRef] [Green Version]
- Herrera-León, S.; Ramiro, R.; Arroyo, M.; Díez, R.; Usera, M.A.; Echeita, M.A. Blind comparison of traditional serotyping with three multiplex PCRs for the identification of Salmonella serotypes. Res. Microbiol. 2007, 158, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.F.; Summerell, B.A. The Fusarium Laboratory Manual, 1st ed.; Blackwell Publishing Ltd.: Ames, IA, USA, 2006. [Google Scholar]
- Jayasiri, S.C.; Hyde, K.D.; Jones, E.B.G.; Jeewon, R.; Ariyawansa, H.A.; Bhat, J.D.; Camporesi, E.; Kang, J.C. Taxonomy and multigene phylogenetic evaluation of novel species in Boeremia and Epicoccum with new records of Ascochyta and Didymella (Didymellaceae). Mycosphere 2017, 8, 1080–1101. [Google Scholar] [CrossRef]
- Walter, H.; Muggia, L.; Fritscher, M.; Holler, A.; Horvat, D.; Guttenberger, H.; Simon, U.K. Multiple taxa in the Phoma-complex associate with black elder (Sambucus nigra L.). Fungal Biol. 2016, 120, 43–50. [Google Scholar] [CrossRef]
- Bensch, K.; Braun, U.; Groenewald, J.Z.; Crous, P.W. The genus Cladosporium. Stud. Mycol. 2012, 1–379. [Google Scholar] [CrossRef] [Green Version]
- Lodama, K.E. Fumonisin Production by and Biological Control of Fusarium Species Associated with Cowpea Seed; University of Pretoria: Pretoria, South Africa, 2011. [Google Scholar]
- Andrade, O.; Munoz, G.; Galdames, R.; Duran, P.; Honorato, R. Characterization, in vitro culture, and molecular analysis of Thecaphora solani, the causal agent of potato smut. Phytopathology 2004, 94, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Conforto, C.; Cazon, I.; Fernandez, F.D.; Marinelli, A.; Oddino, C.; Rago, A.M. Molecular sequence data of Thecaphora frezii affecting peanut crops in Argentina. Eur. J. Plant Pathol. 2013, 137, 663–666. [Google Scholar] [CrossRef]
- Vasighzadeh, A.; Zafari, D.; Selcuk, F.; Huseyin, E.; Kursat, M.; Lutz, M.; Piatek, M. Discovery of Thecaphora schwarzmaniana on Rheum ribes in Iran and Turkey: Implications for the diversity and phylogeny of leaf smuts on rhubarbs. Mycol. Prog. 2014, 13, 881–892. [Google Scholar] [CrossRef]
- Deng, Q.D.; Yong, M.L.; Li, D.Y.; Lai, C.H.; Chen, H.M.; Fan, J.; Hu, D.W. Survey and examination of the potential alternative hosts of Villosiclava virens, the pathogen of rice false smut, in China. J. Integr. Agric. 2015, 14, 1332–1337. [Google Scholar] [CrossRef]
- Carlucci, A.; Raimondo, M.L.; Santos, J.; Phillips, A.J.L. Plectosphaerella species associated with root and collar rots of horticultural crops in southern Italy. Persoonia 2012, 28, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Gullino, M.L.; Gilardi, G.; Garibaldi, A. Seed-Borne Fungal Pathogens of Leafy Vegetable Crops. In Global Perspectives on the Health of Seeds and Plant Propagation Material; Gullino, M., Munkvold, G., Eds.; Springer: Dordrecht, The Netherlands, 2014; Volume 6. [Google Scholar]
- Smith, J.E.; Aveling, T.A.S. Colletotrichum dematium: Causal agent of a new cowpea stem disease in South Africa. Plant Dis. 1997, 81, 832. [Google Scholar] [CrossRef]
- Latunde-Dada, A.O.; O’Connell, R.J.; Nash, C.; Lucas, J.A. Stomata penetration of cow pea (Vigna unguiculata) leaves by Colletotrichum species causing latent anthracnose. Plant Pathol. 1996, 48, 777–785. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemanceau, P.; Blouin, M.; Muller, D.; Moenne-Loccoz, Y. Let the core microbiota be functional. Trends Plant Sci. 2017, 22, 583–595. [Google Scholar] [CrossRef]
- Pancher, M.; Ceol, M.; Corneo, P.E.; Longa, C.M.O.; Yousaf, S.; Pertot, I.; Campisano, A. Fungal endophytic communities in grapevines (Vitis vinifera L.) respond to crop management. Appl. Environ. Microbiol. 2012, 78, 4308–4317. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Total Number of Reads after QC | Total Number of MOTUs |
|---|---|---|
| Main stem | 11,843 | 61 |
| Crown stem | 13,596 | 51 |
| Leaves | 11,417 | 77 |
| Roots | 21,680 | 135 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinge, T.R.; Ghosh, S.; Cason, E.D.; Gryzenhout, M. Characterization of the Endophytic Mycobiome in Cowpea (Vigna unguiculata) from a Single Location Using Illumina Sequencing. Agriculture 2022, 12, 333. https://doi.org/10.3390/agriculture12030333
Kinge TR, Ghosh S, Cason ED, Gryzenhout M. Characterization of the Endophytic Mycobiome in Cowpea (Vigna unguiculata) from a Single Location Using Illumina Sequencing. Agriculture. 2022; 12(3):333. https://doi.org/10.3390/agriculture12030333
Chicago/Turabian StyleKinge, Tonjock Rosemary, Soumya Ghosh, Errol D. Cason, and Marieka Gryzenhout. 2022. "Characterization of the Endophytic Mycobiome in Cowpea (Vigna unguiculata) from a Single Location Using Illumina Sequencing" Agriculture 12, no. 3: 333. https://doi.org/10.3390/agriculture12030333
APA StyleKinge, T. R., Ghosh, S., Cason, E. D., & Gryzenhout, M. (2022). Characterization of the Endophytic Mycobiome in Cowpea (Vigna unguiculata) from a Single Location Using Illumina Sequencing. Agriculture, 12(3), 333. https://doi.org/10.3390/agriculture12030333