
Alterations in Epiphytic Bacterial Communities during the Occurrence of Green Rot Disease in Saccharina japonica Seedlings

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Physicochemical Analysis
2.3. DNA Extraction and Amplification
2.4. Raw Sequence Processing
2.5. Statistical Analyses
3. Results
3.1. Histopathological Observation
3.2. Physicochemical Characteristics of Seawater
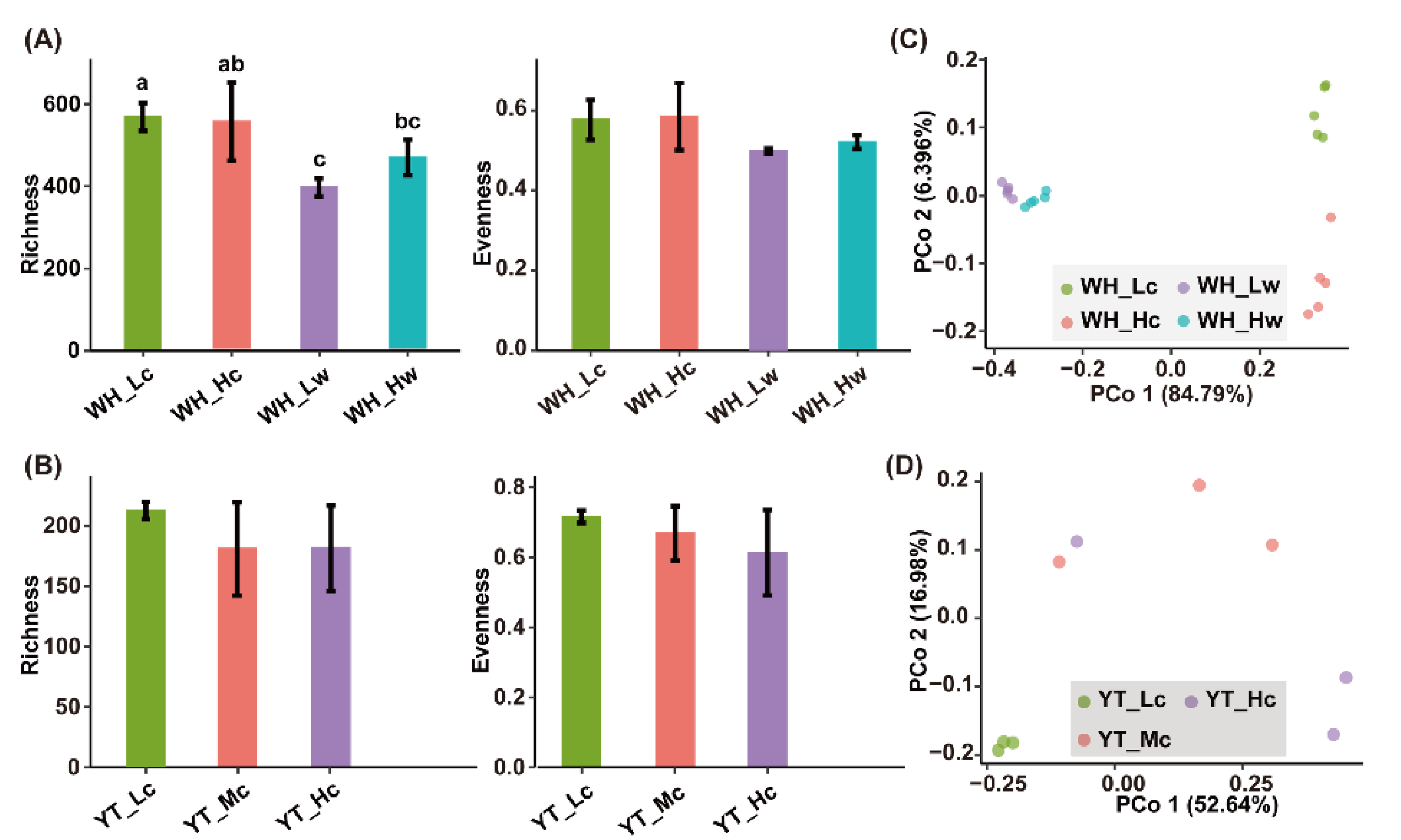
3.3. Bacterial Diversities and Community Compositions
3.4. Significant Differences between Bacterial Communities
3.5. Predicted Bacterial Functions
3.6. Correlation between Bacterial Community Structure and Environmental Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goecke, F.; Labes, A.; Wiese, J.; Imhoff, J.F. Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 2010, 409, 267–299. [Google Scholar] [CrossRef]
- Singh, R.P.; Reddy, C.R.K. Unraveling the functions of the macroalgal microbiome. Front. Microbiol. 2016, 6, 1488. [Google Scholar] [CrossRef] [PubMed]
- Dimitrieva, G.Y.; Crawford, R.L.; Yuksel, G.U. The nature of plant growth-promoting effects of a pseudoalteromonad associated with the marine algae Laminaria japonica and linked to catalase excretion. J. Appl. Microbiol. 2006, 100, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Spoerner, M.; Wichard, T.; Bachhuber, T.; Stratmann, J.; Oertel, W. Growth and thallus morphogenesis of Ulva mutabilis (Chlorophyta) depends on a combination of two bacterial species excreting regulatory factors. J. Phycol. 2012, 48, 1433–1447. [Google Scholar] [CrossRef]
- Yong, J.J.J.Y.; Chew, K.W.; Khoo, K.S.; Show, P.L.; Chang, J. Prospects and development of algal-bacterial biotechnology in environmental management and protection. Biotechnol. Adv. 2021, 47, 107684. [Google Scholar] [CrossRef]
- Dittami, S.M.; Duboscq-Bidot, L.; Perennou, M.; Gobet, A.; Corre, E.; Boyen, C.; Tonon, T. Host–microbe interactions as a driver of acclimation to salinity gradients in brown algal cultures. ISME J. 2016, 10, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Busetti, A.; Maggs, C.A.; Gilmore, B.F. Marine macroalgae and their associated microbiomes as a source of antimicrobial chemical diversity. Eur. J. Phycol. 2017, 52, 452–465. [Google Scholar] [CrossRef]
- Singh, R.P.; Kumari, P.; Reddy, C.R.K. Antimicrobial compounds from seaweeds-associated bacteria and fungi. Appl. Microbiol. Biot. 2015, 99, 1571–1586. [Google Scholar] [CrossRef]
- Campbell, A.H.; Harder, T.; Nielsen, S.; Kjelleberg, S.; Steinberg, P.D. Climate change and disease: Bleaching of a chemically defended seaweed. Global Change Biol. 2011, 17, 2958–2970. [Google Scholar] [CrossRef]
- Fernandes, N.; Case, R.J.; Longford, S.R.; Seyedsayamdost, M.R.; Steinberg, P.D.; Kjelleberg, S.; Thomas, T. Genomes and virulence factors of novel bacterial pathogens causing bleaching disease in the marine red alga Delisea pulchra. PLoS ONE 2011, 6, e27387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zozaya Valdés, E.; Roth Schulze, A.J.; Egan, S.; Thomas, T. Microbial community function in the bleaching disease of the marine macroalgae Delisea pulchra. Environ. Microbiol. 2017, 19, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Moon, K.; Kim, J.; Shim, J.; Klochkova, T.A. A revaluation of algal diseases in Korean Pyropia (Porphyra) sea farms and their economic impact. Algae 2014, 29, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Majzoub, M.E.; Marzinelli, E.M.; Dai, Z.; Thomas, T.; Egan, S. Bacterial controlled mitigation of dysbiosis in a seaweed disease. ISME J. 2021, 16, 378–387. [Google Scholar] [CrossRef]
- Saha, M.; Weinberger, F. Microbial “gardening” by a seaweed holobiont: Surface metabolites attract protective and deter pathogenic epibacterial settlement. J. Ecol. 2019, 107, 2255–2265. [Google Scholar] [CrossRef]
- Gachon, C.M.M.; Sime-Ngando, T.; Strittmatter, M.; Chambouvet, A.; Kim, G.H. Algal diseases: Spotlight on a black box. Trends Plant Sci. 2010, 15, 633–640. [Google Scholar] [CrossRef]
- Fernandes, N.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Community structure and functional gene profile of bacteria on healthy and diseased thalli of the red seaweed Delisea pulchra. PLoS ONE 2012, 7, e50854. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pang, S.; Shan, T.; Su, L. Changes of microbial community structures associated with seedlings of Saccharina japonica at early stage of outbreak of green rotten disease. J. Appl. Phycol. 2020, 32, 1323–1327. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, H.; Tang, L.; Li, J.; Mao, Y.; Mo, Z. Compositional shifts of bacterial communities associated with Pyropia yezoensis and surrounding seawater co-occurring with red rot disease. Front. Microbiol. 2019, 10, 1666. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Chang, L.; Xiao, L.; Zhang, X.; Han, Q.; Li, N.; Egan, S.; Wang, G. Diversity of the epiphytic bacterial communities associated with commercially cultivated healthy and diseased Saccharina japonica during the harvest season. J. Appl. Phycol. 2020, 32, 2071–2080. [Google Scholar] [CrossRef]
- Brown, S.P.; Cornforth, D.M.; Mideo, N. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends Microbiol. 2012, 20, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Sun, X.; Wang, G.; Xu, P.; Wang, X.; Lin, Z.; Wang, F. Effect of blue light on indoor seedling culture of Saccharina japonica (Phaeophyta). J. Appl. Phycol. 2010, 22, 737–744. [Google Scholar] [CrossRef]
- Wang, G.; Lu, B.; Shuai, L.; Li, D.; Zhang, R. Microbial diseases of nursery and field-cultivated Saccharina japonica (Phaeophyta) in China. Algol. Stud. 2014, 145, 39–51. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, W.; Yan, X.; Duan, D. Distribution and reinfection of alginic acid decomposing bacteria on juvenile Laminaria japonica. Oceanologia Et Limnologia Sinica 2004, 35, 562–567. [Google Scholar]
- Wang, L.; Tang, X.; Wang, M.; Zhang, P. The roles played by alginic acid decomposing bacteria during the time of green decay disease of Laminaria japonica. J. Ocean Univ. Qingdao 2003, 32, 245–248. [Google Scholar]
- Ahmad, R.; Chen, Y.; Zhuang, Y.; Qiu, Q.; Chen, D.; Saha, M.; Wu, H.; Wang, G. Isolation and identification of a pathogenic bacterium, Exiguobacterium oxidotolerans XP-2, from the abnormal diseased mature sporophytes of a commercially cultivated brown seaweed Saccharina japonica. J. Appl. Phycol. 2021, 33, 3239–3249. [Google Scholar] [CrossRef]
- Burke, C.; Kjelleberg, S.; Thomas, T. Selective extraction of bacterial DNA from the surfaces of macroalgae. Appl. Environ. Microbiol. 2009, 75, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 2016, 1, e00009-15. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. Available online: http://www.drive5.com/usearch (accessed on 28 April 2022). [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. Available online: http://qiime.org (accessed on 28 April 2022). [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2014. Available online: https://CRAN.R-project.org (accessed on 28 April 2022).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Community Ecology Package. Available online: https://CRAN.R-project.org/package=vegan (accessed on 28 April 2022).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. Available online: http://huttenhower.sph.harvard.edu/galaxy (accessed on 28 April 2022). [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. Available online: https://github.com/picrust/picrust2 (accessed on 28 April 2022). [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. Available online: https://beikolab.cs.dal.ca/software/STAMP (accessed on 28 April 2022). [CrossRef] [PubMed] [Green Version]
- Egan, S.; Fernandes, N.D.; Kumar, V.; Gardiner, M.; Thomas, T. Bacterial pathogens, virulence mechanism and host defence in marine macroalgae. Environ. Microbiol. 2014, 16, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Egan, S.; Gardiner, M. Microbial dysbiosis: Rethinking disease in marine ecosystems. Front. Microbiol. 2016, 7, 991. [Google Scholar] [CrossRef]
- Bai, L.; Yan, Y.; Wang, S.; Li, J.; Zhang, W.; Yang, H.; Wang, P.; Mo, Z. Etiology analysis of green rot of kelp (Saccharina japonica) seedlings in a nursery farm. J. Anhui Agr. Univ. 2020, 47, 921–926. [Google Scholar]
- Ward, G.M.; Kambey, C.S.B.; Faisan, J.P.; Tan, P.L.; Daumich, C.C.; Matoju, I.; Stentiford, G.D.; Bass, D.; Lim, P.E.; Brodie, J.; et al. Ice-Ice disease: An environmentally and microbiologically driven syndrome in tropical seaweed aquaculture. Rev. Aquacult. 2021, 14, 414–439. [Google Scholar] [CrossRef]
- Shi, W.; Li, M.; Wei, G.; Tian, R.; Li, C.; Wang, B.; Lin, R.; Shi, C.; Chi, X.; Zhou, B.; et al. The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome. Microbiome 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Mallon, C.A.; Elsas, J.D.V.; Salles, J.F. Microbial invasions: The process, patterns, and mechanisms. Trends Microbiol. 2015, 23, 719–729. [Google Scholar] [CrossRef]
- Farhadi, F.; Khameneh, B.; Iranshahi, M.; Iranshahy, M. Antibacterial activity of flavonoids and their structure–activity relationship: An update review. Phytother. Res. 2019, 33, 13–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novaković, M.; Novaković, I.; Cvetković, M.; Sladić, D.; Tešević, V. Antimicrobial activity of the diarylheptanoids from the black and green alder. Braz. J. Bot. 2015, 38, 441–446. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.; Byun, Y.; Park, H. 6-Gingerol reduces Pseudomonas aeruginosa biofilm formation and virulence via quorum sensing inhibition. Sci. Rep. 2015, 5, 8656. [Google Scholar] [CrossRef]
- Mattio, L.M.; Catinella, G.; Dallavalle, S.; Pinto, A. Stilbenoids: A natural arsenal against bacterial pathogens. Antibiotics 2020, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Stratil, S.B.; Neulinger, S.C.; Knecht, H.; Friedrichs, A.K.; Wahl, M. Temperature-driven shifts in the epibiotic bacterial community composition of the brown macroalga Fucus vesiculosus. MicrobiologyOpen 2013, 2, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Zozaya-Valdés, E.; Roth-Schulze, A.J.; Thomas, T. Effects of temperature stress and aquarium conditions on the red macroalga Delisea pulchra and its associated microbial community. Front. Microbiol. 2016, 7, 161. [Google Scholar] [CrossRef]
- Zozaya-Valdes, E.; Egan, S.; Thomas, T. A comprehensive analysis of the microbial communities of healthy and diseased marine macroalgae and the detection of known and potential bacterial pathogens. Front. Microbiol. 2015, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Hakamada, Y.; Ohkubo, Y.; Ohashi, S. Purification and characterization of β-Mannanase from Reinekea sp. KIT-YO10 with transglycosylation activity. Biosci. Biotech. Bioch. 2014, 78, 722–728. [Google Scholar] [CrossRef]
- Martin, M.; Barbeyron, T.; Martin, R.; Portetelle, D.; Michel, G.; Vandenbol, M. The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal-polysaccharide-degrading bacteria. Front. Microbiol. 2015, 6, 1487. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Joung, Y.; Joh, K. Lewinella maritima sp. nov., and Lewinella lacunae sp. nov., novel bacteria from marine environments. Int. J. Syst. Evol. Microbiol. 2017, 67, 3603–3609. [Google Scholar] [CrossRef]
- Bengtsson, M.M.; Sjøtun, K.; Storesund, J.E.; Øvreås, J. Utilization of kelp-derived carbon sources by kelp surface-associated bacteria. Aquat. Microb. Ecol. 2011, 62, 191–199. [Google Scholar] [CrossRef]
- Ng, J.C.; Chan, Y.; Tun, H.M.; Leung, F.C.; Shin, P.K.; Chiu, J.M. Pyrosequencing of the bacteria associated with Platygyra carnosus corals with skeletal growth anomalies reveals differences in bacterial community composition in apparently healthy and diseased tissues. Front. Microbiol. 2015, 6, 1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quéré, G.; Intertaglia, L.; Payri, C.; Galand, P.E. Disease specific bacterial communities in a coralline algae of the northwestern Mediterranean Sea: A combined culture dependent and -independent approach. Front. Microbiol. 2019, 10, 1850. [Google Scholar] [CrossRef] [PubMed]
- Ward, G.M.; Faisan, J.P.; Cottier Cook, E.J.; Gachon, C.; Hurtado, A.Q.; Lim, P.E.; Matoju, I.; Msuya, F.E.; Bass, D.; Brodie, J. A review of reported seaweed diseases and pests in aquaculture in Asia. J. World Aquacult. Soc. 2020, 51, 815–828. [Google Scholar] [CrossRef]
- Karimi, E.; Geslain, E.; KleinJan, H.; Tanguy, G.; Legeay, E.; Corre, E.; Dittami, S.M. Genome sequences of 72 bacterial strains isolated from Ectocarpus subulatus: A resource for algal microbiology. Genome Biol. Evol. 2020, 12, 3647–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tang, X.; Mo, Z.; Mao, Y. Metagenome-assembled genomes from Pyropia haitanensis microbiome provide insights into the potential metabolic functions to the seaweed. Front. Microbiol. 2022, 13, 857901. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.M.; Nishimura, M.; Haider, M.N.; Sano, M.; Ijichi, M.; Kogure, K.; Yoshizawa, S. Diversity and composition of microbial communities in an eelgrass (Zostera marina) bed in Tokyo Bay, Japan. Microbes Environ. 2021, 36, ME21037. [Google Scholar] [CrossRef]
- Staerck, C.; Gastebois, A.; Vandeputte, P.; Calenda, A.; Larcher, G.; Gillmann, L.; Papon, N.; Bouchara, J.; Fleury, M.J.J. Microbial antioxidant defense enzymes. Microb. Pathogenesis 2017, 110, 56–65. [Google Scholar] [CrossRef]
- Bertani, B.; Ruiz, N.; Slauch, J.M. Function and biogenesis of lipopolysaccharides. Ecosal Plus 2018, 8, 1–19. [Google Scholar] [CrossRef]
- Chaban, B.; Hughes, H.V.; Beeby, M. The flagellum in bacterial pathogens: For motility and a whole lot more. Semin. Cell Dev. Biol. 2015, 46, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Matilla, M.A.; Krell, T.S. The effect of bacterial chemotaxis on host infection and pathogenicity. FEMS Microbiol. Rev. 2017, 42, 40–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Senadheera, D.B.; Cvitkovitch, D.G. An intimate link: Two-component signal transduction systems and metal transport systems in bacteria. Future Microbiol. 2014, 9, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, R.; Tiwari, M.; Donelli, G.; Tiwari, V. Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action. Virulence 2018, 9, 522–554. [Google Scholar] [CrossRef] [PubMed]
- The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinf. 2021, 19, 578–583. [CrossRef] [PubMed]
- Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WH_L Tanks | WH_H Tanks | YT tanks | WH_L vs. WH_H p-Values | |
|---|---|---|---|---|
| Temperature (°C) | 7.82 ± 0.26 a | 8.28 ± 0.19 | 7.75 ± 0.05 | 0.006 |
| pH | 8.06 ± 0.02 | 8.03 ± 0.02 | 7.69 ± 0.05 | 0.055 |
| Salinity (‰) | 35.20 ± 0.45 | 34.00 ± 0.00 | 34.10 ± 0.10 | <0.001 |
| NO3−-N (mg/L) | 34.306 ± 2.616 | 34.698 ± 2.736 | 0.720 ± 0.126 | 0.411 |
| PO43−-P (mg/L) | 2.173 ± 0.088 | 2.202 ± 0.024 | 1.288 ± 0.037 | 0.034 |
| NH4+-N (mg/L) | 0.503 ± 0.006 | 0.534 ± 0.058 | 0.004 ± 0.001 | 0.150 |
| NO2−-N (mg/L) | 0.434 ± 0.001 | 0.432 ± 0.002 | 0.031 ± 0.047 | 0.013 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Wang, S.; Li, J.; Liu, F.; Mo, Z. Alterations in Epiphytic Bacterial Communities during the Occurrence of Green Rot Disease in Saccharina japonica Seedlings. J. Mar. Sci. Eng. 2022, 10, 730. https://doi.org/10.3390/jmse10060730
Yan Y, Wang S, Li J, Liu F, Mo Z. Alterations in Epiphytic Bacterial Communities during the Occurrence of Green Rot Disease in Saccharina japonica Seedlings. Journal of Marine Science and Engineering. 2022; 10(6):730. https://doi.org/10.3390/jmse10060730
Chicago/Turabian StyleYan, Yongwei, Shanshan Wang, Jie Li, Fuli Liu, and Zhaolan Mo. 2022. "Alterations in Epiphytic Bacterial Communities during the Occurrence of Green Rot Disease in Saccharina japonica Seedlings" Journal of Marine Science and Engineering 10, no. 6: 730. https://doi.org/10.3390/jmse10060730
APA StyleYan, Y., Wang, S., Li, J., Liu, F., & Mo, Z. (2022). Alterations in Epiphytic Bacterial Communities during the Occurrence of Green Rot Disease in Saccharina japonica Seedlings. Journal of Marine Science and Engineering, 10(6), 730. https://doi.org/10.3390/jmse10060730