
Kinetic Energy Density Functionals Based on a Generalized Screened Coulomb Potential: Linear Response and Future Perspectives



Abstract
:1. Introduction
2. Theory
2.1. Kinetic Energy Potential
2.2. Linear Response of a yGGA Functional
Asymptotic Behavior
3. Linear yGGA Functionals
4. Computational Details
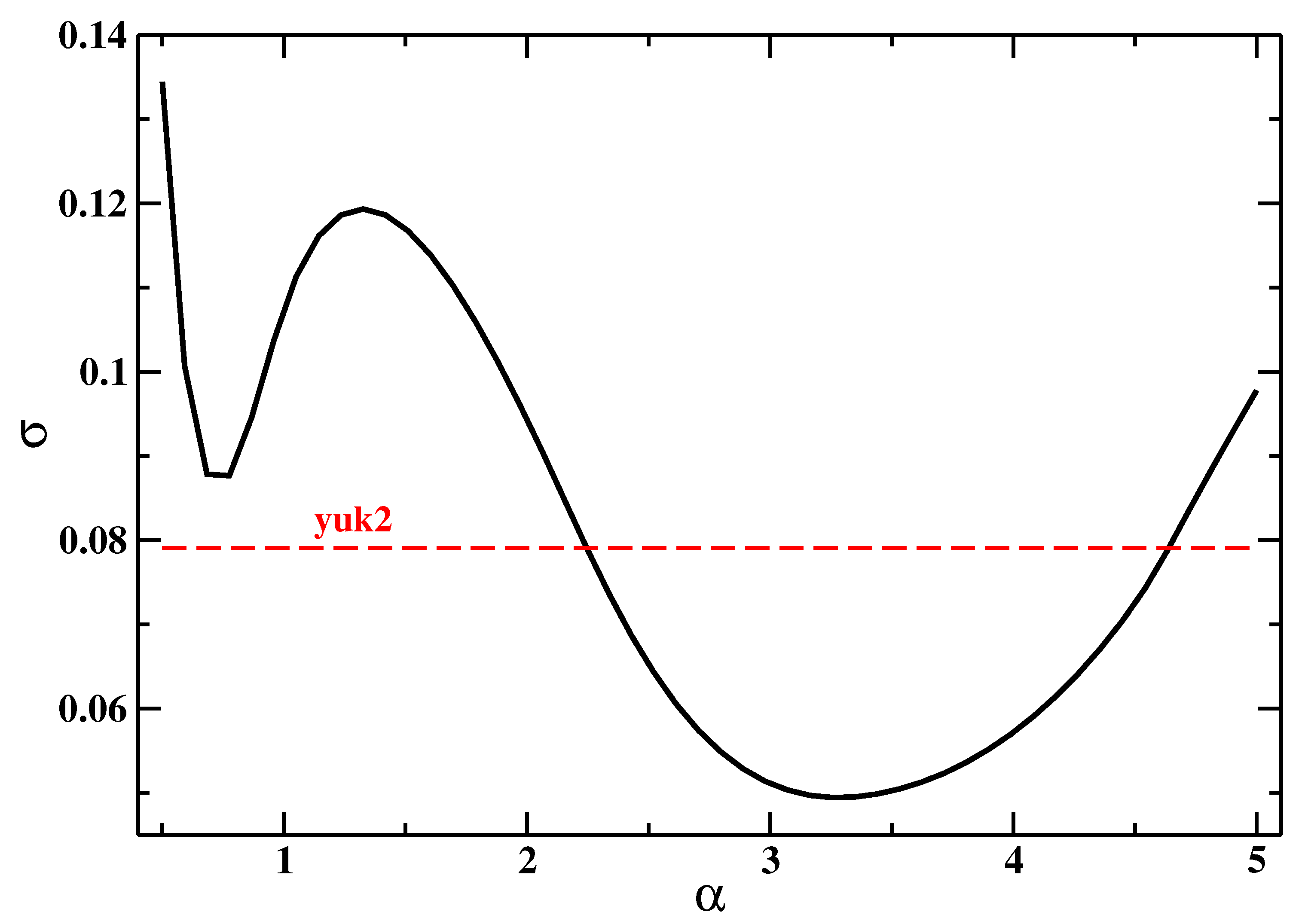
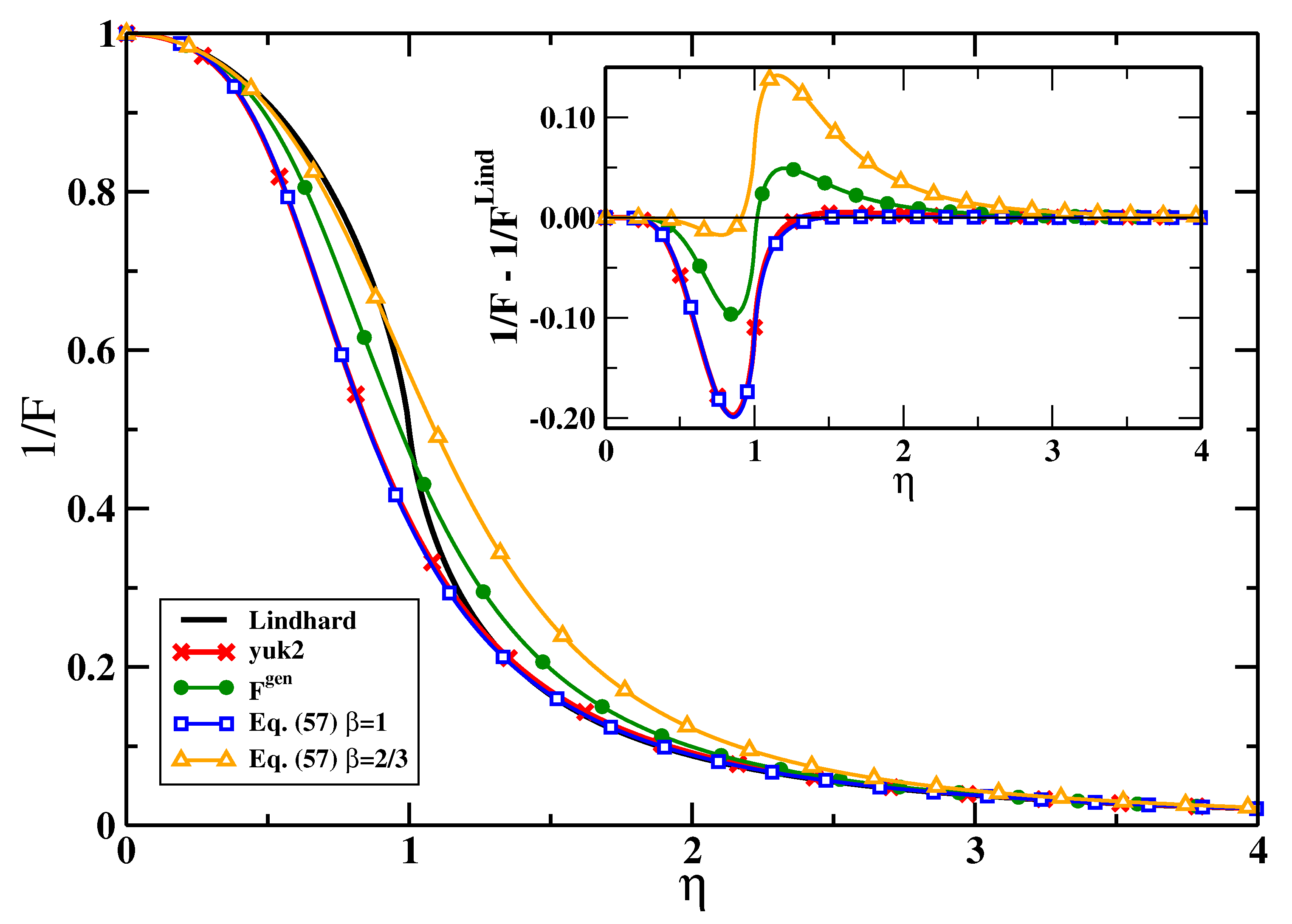
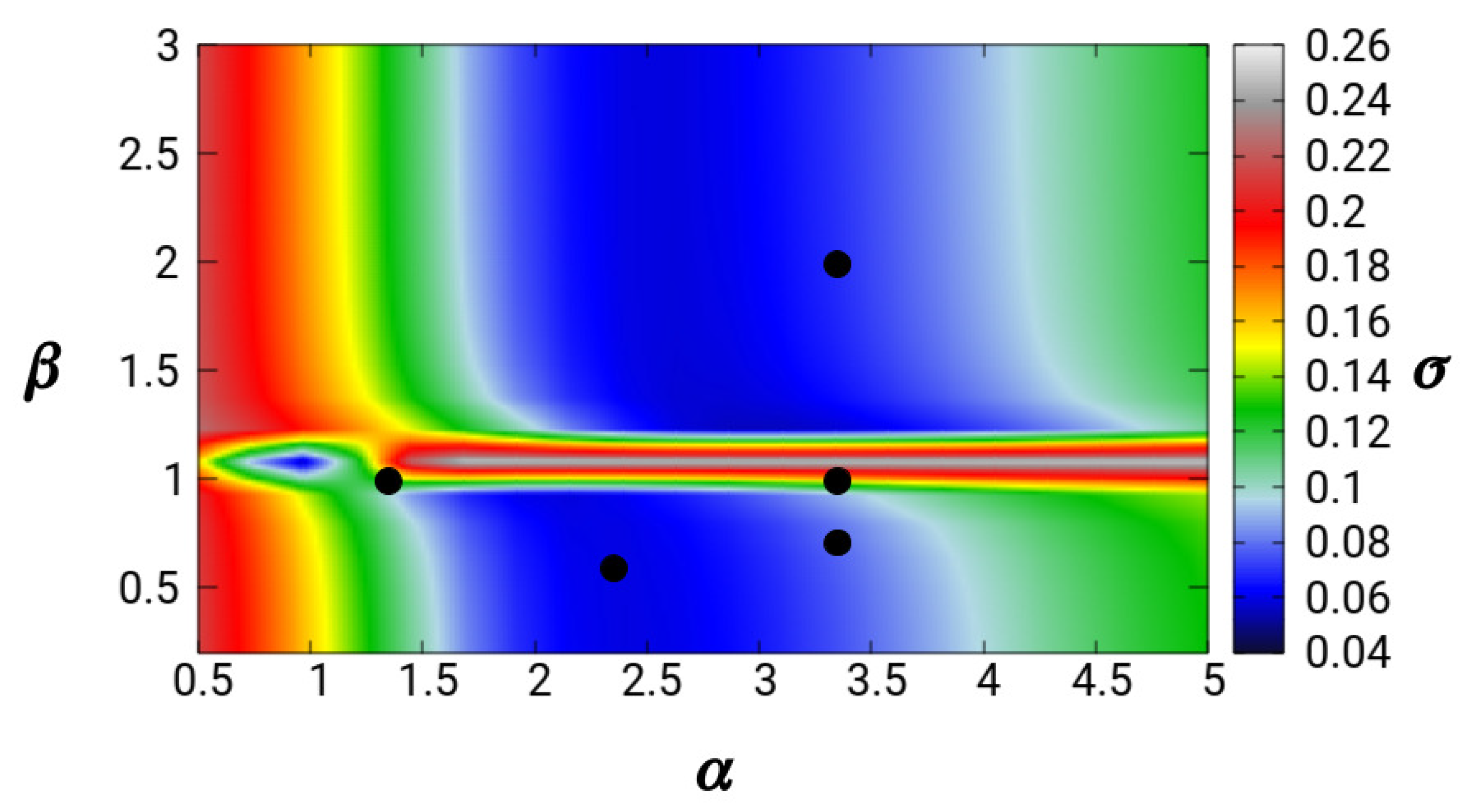
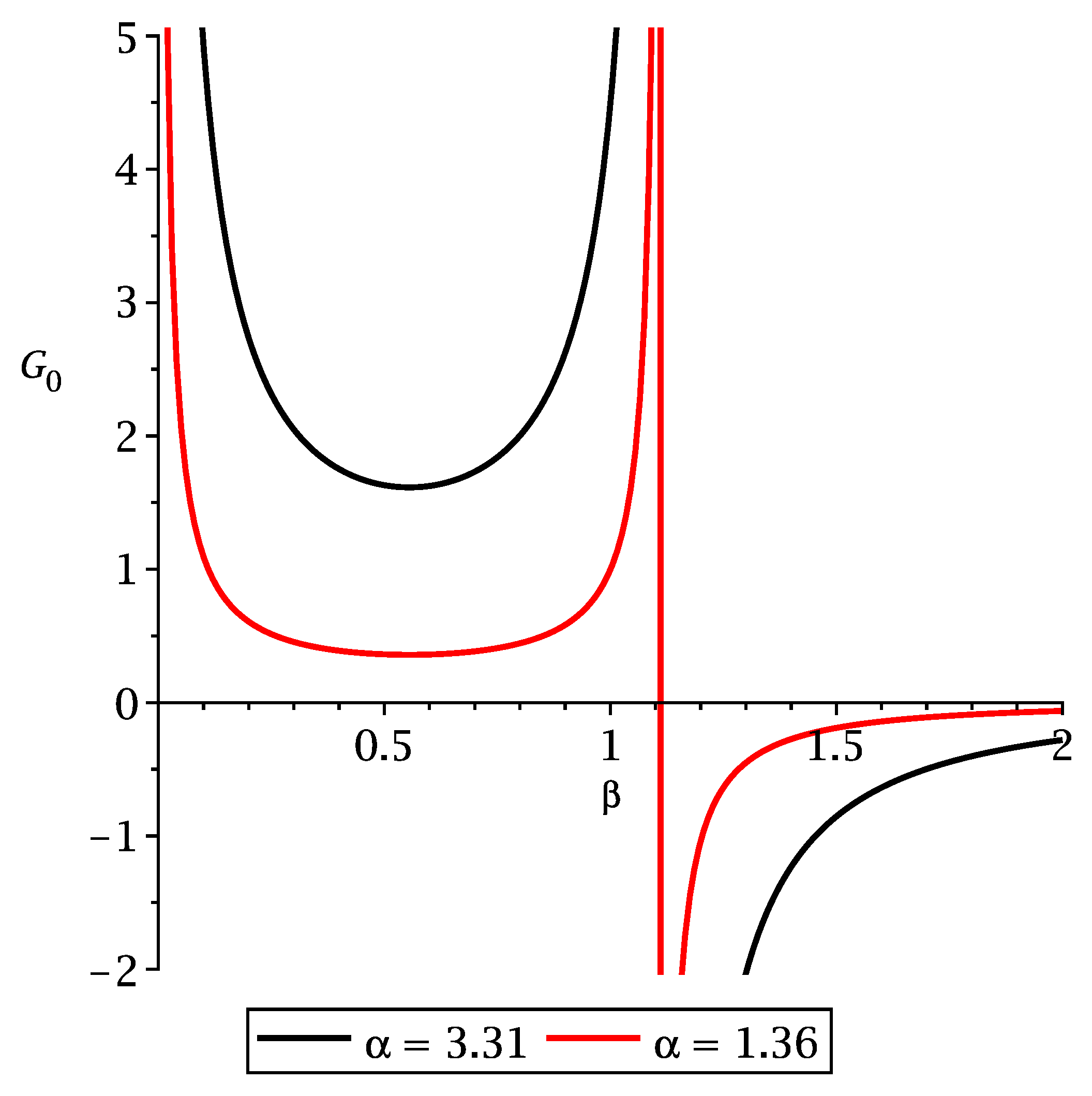
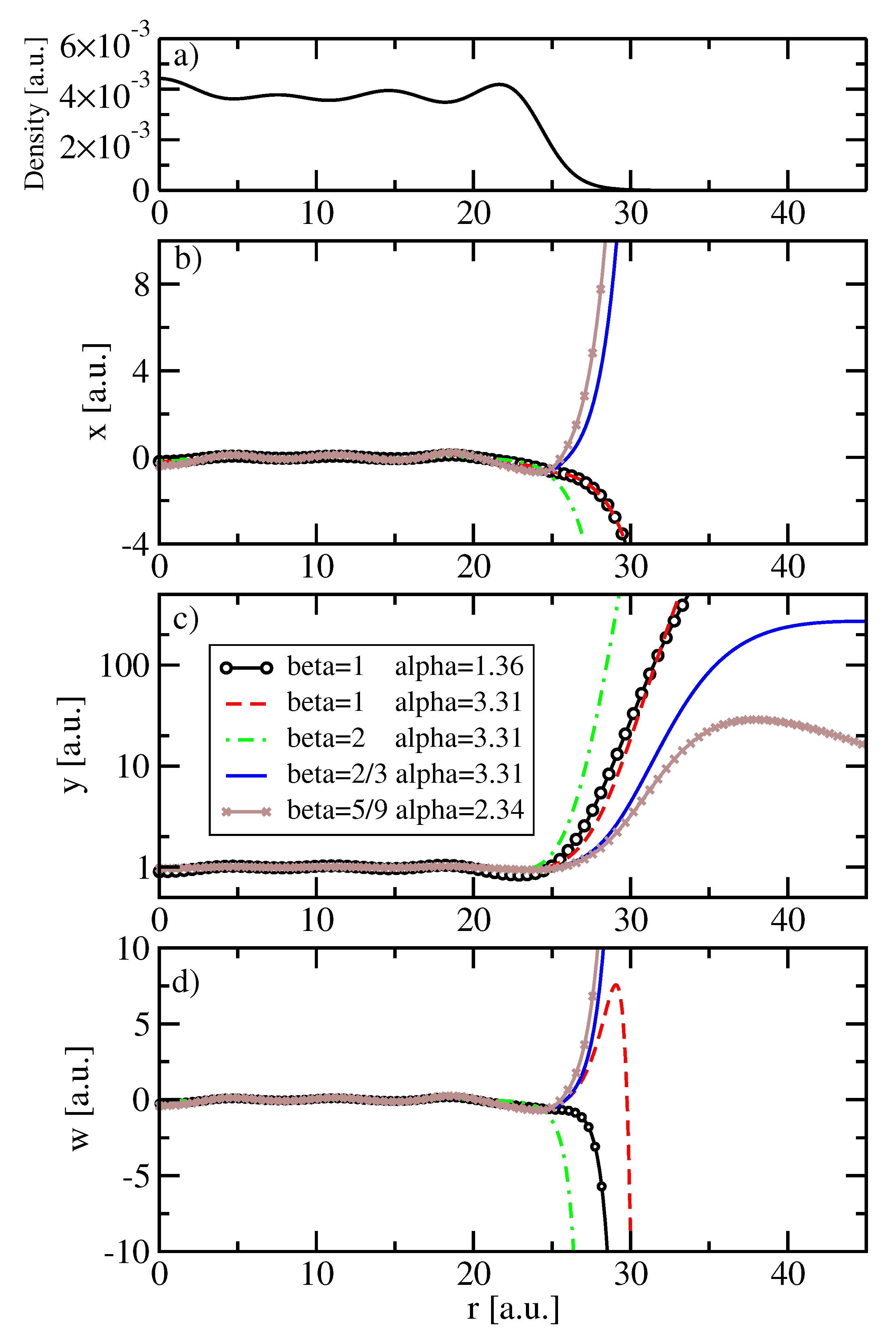
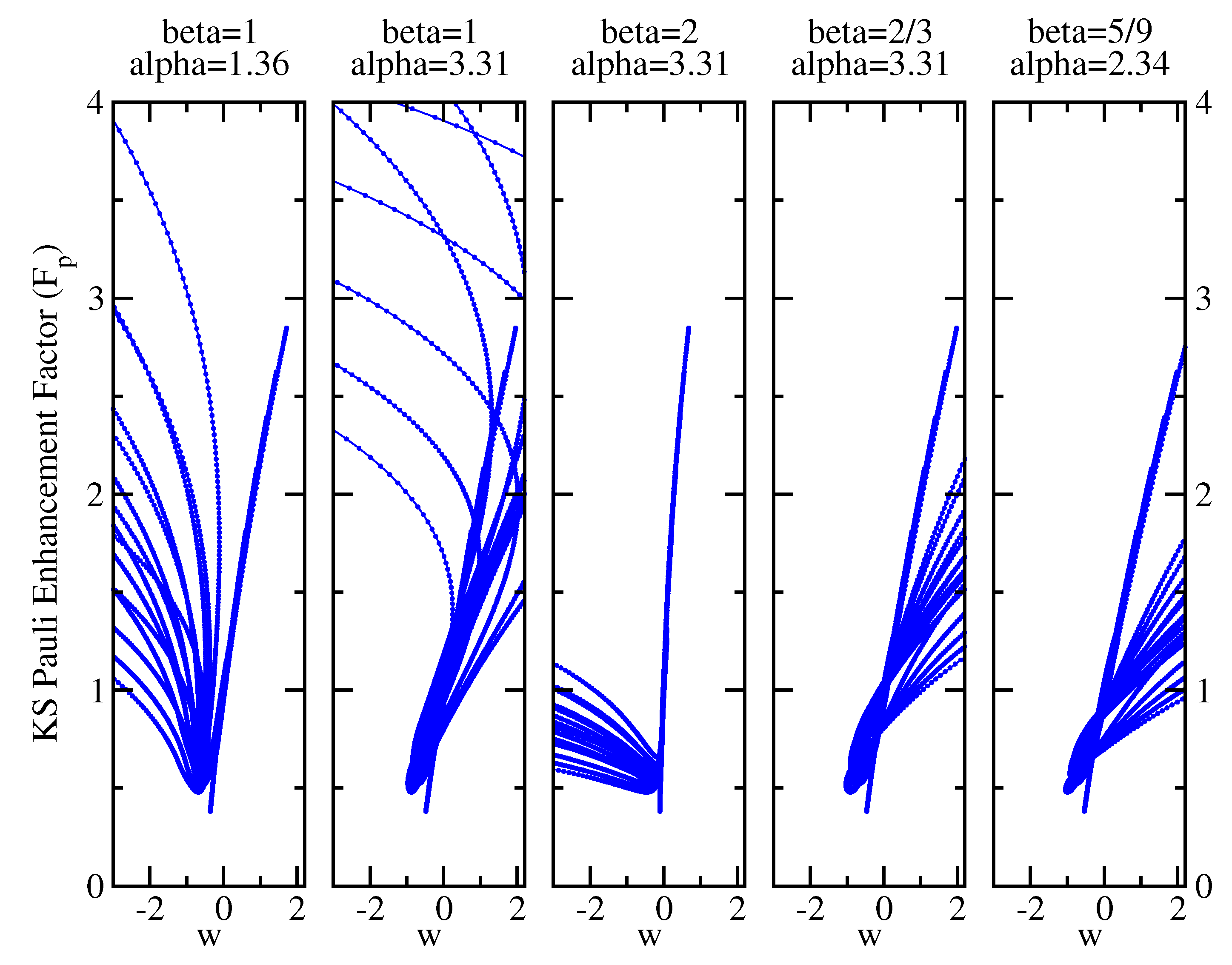
5. Results
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A. Weak Perturbation of y
Appendix B. Integrals for the Generalized Yukawa Expansion
Appendix C. Asymptotics
References
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Dreizler, R.M.; Gross, E.K.U. Density Functional Theory; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Levy, M. Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem. Proc. Nat. Acad. Sci. USA 1979, 76, 6062–6065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.A.; Carter, E.A. Orbital-free kinetic-energy density functional theory. In Theoretical Methods in Condensed Phase Chemistry; Springer: Berlin/Heidelberg, Germany, 2002; pp. 117–184. [Google Scholar]
- Wesolowski, T.A.; Wang, Y.A. (Eds.) Recent Progress in Orbital-Free Density Functional Theory; World Scientific: Singapore, 2013. [Google Scholar] [CrossRef] [Green Version]
- Gavini, V.; Bhattacharya, K.; Ortiz, M. Quasi-continuum orbital-free density-functional theory: A route to multi-million atom non-periodic DFT calculation. J. Mech. Phys. Sol. 2007, 55, 697–718. [Google Scholar] [CrossRef]
- Witt, W.C.; Beatriz, G.; Dieterich, J.M.; Carter, E.A. Orbital-free density functional theory for materials research. J. Mater. Res. 2018, 33, 777–795. [Google Scholar] [CrossRef]
- Götz, A.W.; Beyhan, S.M.; Visscher, L. Performance of Kinetic Energy Functionals for Interaction Energies in a Subsystem Formulation of Density Functional Theory. J. Chem. Theory Comput. 2009, 5, 3161–3174. [Google Scholar] [CrossRef]
- Jacob, C.R.; Neugebauer, J. Subsystem density-functional theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 325–362. [Google Scholar] [CrossRef]
- Wesolowski, T.A.; Shedge, S.; Zhou, X. Frozen-Density Embedding Strategy for Multilevel Simulations of Electronic Structure. Chem. Rev. 2015, 115, 5891–5928. [Google Scholar] [CrossRef] [Green Version]
- Neugebauer, J. Chromophore-specific theoretical spectroscopy: From subsystem density functional theory to mode-specific vibrational spectroscopy. Phys. Rep. 2010, 489, 1–87. [Google Scholar] [CrossRef]
- Krishtal, A.; Sinha, D.; Genova, A.; Pavanello, M. Subsystem density-functional theory as an effective tool for modeling ground and excited states, their dynamics and many-body interactions. J. Phys. Condens. Matter 2015, 27, 183202. [Google Scholar] [CrossRef]
- Laricchia, S.; Fabiano, E.; Della Sala, F. Frozen density embedding with hybrid functionals. J. Chem. Phys. 2010, 133, 164111. [Google Scholar] [CrossRef] [PubMed]
- Śmiga, S.; Fabiano, E.; Laricchia, S.; Constantin, L.A.; Della Sala, F. Subsystem density functional theory with meta-generalized gradient approximation exchange-correlation functionals. J. Chem. Phys. 2015, 142, 154121. [Google Scholar] [CrossRef] [Green Version]
- Toscano, G.; Straubel, J.; Kwiatkowski, A.; Rockstuhl, C.; Evers, F.; Xu, H.; Asger Mortensen, N.; Wubs, M. Resonance Shifts and Spill-out Effects in Self-Consistent Hydrodynamic Nanoplasmonics. Nat. Commun. 2015, 6, 7132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciracì, C.; Della Sala, F. Quantum hydrodynamic theory for plasmonics: Impact of the electron density tail. Phys. Rev. B 2016, 93, 205405. [Google Scholar] [CrossRef] [Green Version]
- Moldabekov, Z.A.; Bonitz, M.; Ramazanov, T.S. Theoretical Foundations of Quantum Hydrodynamics for Plasmas. Phys. Plasmas 2018, 25, 031903. [Google Scholar] [CrossRef]
- Baghramyan, H.M.; Della Sala, F.; Ciracì, C. Laplacian-Level Quantum Hydrodynamic Theory for Plasmonics. Phys. Rev. X 2021, 11, 011049. [Google Scholar] [CrossRef]
- Mejia-Rodriguez, D.; Trickey, S.B. Deorbitalization strategies for meta-generalized-gradient-approximation exchange-correlation functionals. Phys. Rev. A 2017, 96, 052512. [Google Scholar] [CrossRef] [Green Version]
- Mejia-Rodriguez, D.; Trickey, S.B. Deorbitalized meta-GGA exchange-correlation functionals in solids. Phys. Rev. B 2018, 98, 115161. [Google Scholar] [CrossRef] [Green Version]
- Mejía-Rodríguez, D.; Trickey, S.B. Meta-GGA performance in solids at almost GGA cost. Phys. Rev. B 2020, 102, 121109. [Google Scholar] [CrossRef]
- Tran, F.; Kovács, P.; Kalantari, L.; Madsen, G.K.H.; Blaha, P. Orbital-free approximations to the kinetic-energy density in exchange-correlation MGGA functionals: Tests on solids. J. Chem. Phys. 2018, 149, 144105. [Google Scholar] [CrossRef] [Green Version]
- Tran, F.; Baudesson, G.; Carrete, J.; Madsen, G.K.H.; Blaha, P.; Schwarz, K.; Singh, D.J. Shortcomings of meta-GGA functionals when describing magnetism. Phys. Rev. B 2020, 102, 024407. [Google Scholar] [CrossRef]
- Acharya, P.K.; Bartolotti, L.J.; Sears, S.B.; Parr, R.G. An atomic kinetic energy functional with full Weizsacker correction. Proc. Nat. Acad. Sci. USA 1980, 77, 6978–6982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, A.J. Comparison of kinetic-energy density functionals. Phys. Rev. A 1992, 46, 6920. [Google Scholar] [CrossRef]
- Lembarki, A.; Chermette, H. Obtaining a gradient–corrected kinetic–energy functional from the Perdew–Wang exchange functional. Phys. Rev. A 1994, 50, 5328. [Google Scholar] [CrossRef]
- Tran, F.; Wesołowski, T.A. Link between the kinetic-and exchange-energy functionals in the generalized gradient approximation. Int. J. Quant. Chem. 2002, 89, 441–446. [Google Scholar] [CrossRef]
- Constantin, L.A.; Fabiano, E.; Laricchia, S.; Della Sala, F. Semiclassical neutral atom as a reference system in density functional theory. Phys. Rev. Lett. 2011, 106, 186406. [Google Scholar] [CrossRef]
- Laricchia, S.; Fabiano, E.; Constantin, L.A.; Della Sala, F. Generalized Gradient Approximations of the Noninteracting Kinetic Energy from the Semiclassical Atom Theory: Rationalization of the Accuracy of the Frozen Density Embedding Theory for Nonbonded Interactions. J. Chem. Theory Comput. 2011, 7, 2439–2451. [Google Scholar] [CrossRef]
- Karasiev, V.V.; Chakraborty, D.; Shukruto, O.A.; Trickey, S.B. Nonempirical generalized gradient approximation free-energy functional for orbital-free simulations. Phys. Rev. B 2013, 88, 161108(R). [Google Scholar] [CrossRef] [Green Version]
- Borgoo, A.; Tozer, D.J. Density scaling of noninteracting kinetic energy functionals. J. Chem. Theory Comput. 2013, 9, 2250–2255. [Google Scholar] [CrossRef]
- Trickey, S.B.; Karasiev, V.V.; Chakraborty, D. Comment on “Single-point kinetic energy density functionals: A pointwise kinetic energy density analysis and numerical convergence investigation”. Phys. Rev. B 2015, 92, 117101. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Carter, E.A. Reply to “Comment on ‘Single-point kinetic energy density functionals: A pointwise kinetic energy density analysis and numerical convergence investigation’”. Phys. Rev. B 2015, 92, 117102. [Google Scholar] [CrossRef] [Green Version]
- Constantin, L.A.; Fabiano, E.; Śmiga, S.; Della Sala, F. Jellium-with-gap model applied to semilocal kinetic functionals. Phys. Rev. B 2017, 95, 115153. [Google Scholar] [CrossRef] [Green Version]
- Luo, K.; Karasiev, V.V.; Trickey, S. A simple generalized gradient approximation for the noninteracting kinetic energy density functional. Phys. Rev. B 2018, 98, 041111(R). [Google Scholar] [CrossRef] [Green Version]
- Lehtomäki, J.; Lopez-Acevedo, O. Semilocal kinetic energy functionals with parameters from neutral atoms. Phys. Rev. B 2019, 100, 165111. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Constantin, L.A. Laplacian-level density functionals for the kinetic energy density and exchange-correlation energy. Phys. Rev. B 2007, 75, 155109. [Google Scholar] [CrossRef] [Green Version]
- Karasiev, V.V.; Jones, R.S.; Trickey, S.B.; Harris, F.E. Properties of constraint-based single-point approximate kinetic energy functionals. Phys. Rev. B 2009, 80, 245120. [Google Scholar] [CrossRef] [Green Version]
- Laricchia, S.; Constantin, L.A.; Fabiano, E.; Della Sala, F. Laplacian-Level kinetic energy approximations based on the fourth-order gradient expansion: Global assessment and application to the subsystem formulation of density functional theory. J. Chem. Theory Comput. 2013, 10, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Cancio, A.C.; Stewart, D.; Kuna, A. Visualization and analysis of the Kohn-Sham kinetic energy density and its orbital-free description in molecules. J. Chem. Phys. 2016, 144, 084107. [Google Scholar] [CrossRef] [Green Version]
- Cancio, A.C.; Redd, J.J. Visualisation and orbital-free parametrisation of the large-Z scaling of the kinetic energy density of atoms. Mol. Phys. 2017, 115, 618–635. [Google Scholar] [CrossRef] [Green Version]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Semilocal Pauli–Gaussian Kinetic Functionals for Orbital-Free Density Functional Theory Calculations of Solids. J. Phys. Chem. Lett. 2018, 9, 4385–4390. [Google Scholar] [CrossRef] [Green Version]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Performance of Semilocal Kinetic Energy Functionals for Orbital-Free Density Functional Theory. J. Chem. Theory Comput. 2019, 15, 3044–3055. [Google Scholar] [CrossRef] [PubMed]
- Śmiga, S.; Constantin, L.A.; Della Sala, F.; Fabiano, E. The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development. Computation 2019, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Carter, E.A. Single-point kinetic energy density functionals: A pointwise kinetic energy density analysis and numerical convergence investigation. Phys. Rev. B 2015, 91, 045124. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.W.; Teter, M.P. Kinetic-energy functional of the electron density. Phys. Rev. B 1992, 45, 13196. [Google Scholar] [CrossRef]
- Perrot, F. Hydrogen-hydrogen interaction in an electron gas. J. Phys. Condens. Matter 1994, 6, 431. [Google Scholar] [CrossRef]
- Smargiassi, E.; Madden, P.A. Orbital-free kinetic-energy functionals for first-principles molecular dynamics. Phys. Rev. B 1994, 49, 5220. [Google Scholar] [CrossRef]
- García-González, P.; Alvarellos, J.E.; Chacón, E. Nonlocal kinetic-energy-density functionals. Phys. Rev. B 1996, 53, 9509. [Google Scholar] [CrossRef]
- García-González, P.; Alvarellos, J.E.; Chacón, E. Kinetic-energy density functional: Atoms and shell structure. Phys. Rev. A 1996, 54, 1897. [Google Scholar] [CrossRef]
- García-González, P.; Alvarellos, J.E.; Chacón, E. Nonlocal symmetrized kinetic-energy density functional: Application to simple surfaces. Phys. Rev. B 1998, 57, 4857. [Google Scholar] [CrossRef]
- Wang, Y.A.; Govind, N.; Carter, E.A. Orbital-free kinetic-energy functionals for the nearly free electron gas. Phys. Rev. B 1998, 58, 13465. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.A.; Govind, N.; Carter, E.A. Orbital-free kinetic-energy density functionals with a density-dependent kernel. Phys. Rev. B 1999, 60, 16350. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Ligneres, V.L.; Carter, E.A. Improving the orbital-free density functional theory description of covalent materials. J. Chem. Phys. 2005, 122, 044103. [Google Scholar] [CrossRef]
- Garcia-Aldea, D.; Alvarellos, J. Fully nonlocal kinetic energy density functionals: A proposal and a general assessment for atomic systems. J. Chem. Phys. 2008, 129, 074103. [Google Scholar] [CrossRef]
- Garcia-Aldea, D.; Alvarellos, J.E. Approach to kinetic energy density functionals: Nonlocal terms with the structure of the von Weizsäcker functional. Phys. Rev. A 2008, 77, 022502. [Google Scholar] [CrossRef]
- Huang, C.; Carter, E.A. Nonlocal orbital-free kinetic energy density functional for semiconductors. Phys. Rev. B 2010, 81, 045206. [Google Scholar] [CrossRef] [Green Version]
- Shin, I.; Carter, E.A. Enhanced von Weizsäcker Wang-Govind-Carter kinetic energy density functional for semiconductors. J. Chem. Phys. 2014, 140, 18A531. [Google Scholar] [CrossRef]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Nonlocal kinetic energy functional from the jellium-with-gap model: Applications to orbital-free density functional theory. Phys. Rev. B 2018, 97, 205137. [Google Scholar] [CrossRef] [Green Version]
- Mi, W.; Genova, A.; Pavanello, M. Nonlocal kinetic energy functionals by functional integration. J. Chem. Phys. 2018, 148, 184107. [Google Scholar] [CrossRef] [Green Version]
- Mi, W.; Pavanello, M. Orbital-free density functional theory correctly models quantum dots when asymptotics, nonlocality, and nonhomogeneity are accounted for. Phys. Rev. B 2019, 100, 041105. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Lv, J.; Wang, Y.; Ma, Y. Nonlocal kinetic energy density functionals for isolated systems obtained via local density approximation kernels. Phys. Rev. B 2020, 101, 045110. [Google Scholar] [CrossRef] [Green Version]
- Finzel, K. Shell-structure-based functionals for the kinetic energy. Theor. Chem. Acc. 2015, 134, 106. [Google Scholar] [CrossRef]
- Finzel, K. Local conditions for the Pauli potential in order to yield self-consistent electron densities exhibiting proper atomic shell structure. J. Chem. Phys. 2016, 144, 034108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludeña, E.V.; Salazar, E.X.; Cornejo, M.H.; Arroyo, D.E.; Karasiev, V.V. The Liu-Parr power series expansion of the Pauli kinetic energy functional with the incorporation of shell-inducing traits: Atoms. Int. J. Quantum Chem. 2018, 118, e25601. [Google Scholar] [CrossRef]
- Salazar, E.X.; Guarderas, P.F.; Ludena, E.V.; Cornejo, M.H.; Karasiev, V.V. Study of some simple approximations to the non-interacting kinetic energy functional. Int. J. Quantum Chem. 2016, 116, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.; Parkhill, J. Kinetic Energy of Hydrocarbons as a Function of Electron Density and Convolutional Neural Networks. J. Chem. Theory Comput. 2016, 12, 1139–1147. [Google Scholar] [CrossRef]
- Alonso, J.A.; Girifalco, L.A. Nonlocal approximation to the exchange potential and kinetic energy of an inhomogeneous electron gas. Phys. Rev. B 1978, 17, 3735. [Google Scholar] [CrossRef]
- Chacón, E.; Alvarellos, J.E.; Tarazona, P. Nonlocal kinetic energy functional for nonhomogeneous electron systems. Phys. Rev. B 1985, 32, 7868. [Google Scholar] [CrossRef]
- Sarcinella, F.; Fabiano, E.; Constantin, L.A.; Della Sala, F. Nonlocal kinetic energy functionals in real space using a Yukawa-potential kernel: Properties, linear response, and model functionals. Phys. Rev. B 2021, 103, 155127. [Google Scholar] [CrossRef]
- Kumar, S.; Borda, E.L.; Sadigh, B.; Zhu, S.; Hamel, S.; Gallagher, B.; Bulatov, V.; Klepeis, J.; Samanta, A. Accurate parameterization of the kinetic energy functional. J. Chem. Phys. 2022, 156, 024110. [Google Scholar] [CrossRef]
- Karasiev, V.V.; Chakraborty, D.; Trickey, S.B. Progress on New Approaches to Old Ideas: Orbital-Free Density Functionals. In Many-Electron Approaches in Physics, Chemistry and Mathematics; Bach, V., Delle Site, L., Eds.; Springer: Cham, Switzerland, 2014; pp. 113–134. [Google Scholar] [CrossRef]
- March, N.; Santamaria, R. Non-local relation between kinetic and exchange energy densities in Hartree–Fock theory. Int. J. Quantum Chem. 1991, 39, 585–592. [Google Scholar] [CrossRef]
- Della Sala, F.; Fabiano, E.; Constantin, L.A. Kohn-Sham kinetic energy density in the nuclear and asymptotic regions: Deviations from the von Weizsäcker behavior and applications to density functionals. Phys. Rev. B 2015, 91, 035126. [Google Scholar] [CrossRef] [Green Version]
- Howard, I.A.; March, N.H.; Van Doren, V.E. r- and p-space electron densities and related kinetic and exchange energies in terms of s states alone for the leading term in the 1/Z expansion for nonrelativistic closed-shell atomic ions. Phys. Rev. A 2001, 63, 062501. [Google Scholar] [CrossRef]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Kinetic and Exchange Energy Densities near the Nucleus. Computation 2016, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Śmiga, S.; Siecińska, S.; Fabiano, E. Methods to generate reference total and Pauli kinetic potentials. Phys. Rev. B 2020, 101, 165144. [Google Scholar] [CrossRef]
- Snyder, J.C.; Rupp, M.; Hansen, K.; Müller, K.R.; Burke, K. Finding density functionals with machine learning. Phys. Rev. Lett. 2012, 108, 253002. [Google Scholar] [CrossRef]
- Li, L.; Snyder, J.C.; Pelaschier, I.M.; Huang, J.; Niranjan, U.N.; Duncan, P.; Rupp, M.; Müller, K.R.; Burke, K. Understanding machine-learned density functionals. Int. J. Quant. Chem. 2016, 116, 819–833. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, F.H.; Kais, S. Kinetic energy density for orbital-free density functional calculations by axiomatic approach. Int. J. Quantum Chem. 2017, 117, e25373. [Google Scholar] [CrossRef]
- Seino, J.; Kageyama, R.; Fujinami, M.; Ikabata, Y.; Nakai, H. Semi-local machine-learned kinetic energy density functional with third-order gradients of electron density. J. Chem. Phys. 2018, 148, 241705. [Google Scholar] [CrossRef]
- Golub, P.; Manzhos, S. Kinetic energy densities based on the fourth order gradient expansion: Performance in different classes of materials and improvement via machine learning. Phys. Chem. Chem. Phys. 2019, 21, 378–395. [Google Scholar] [CrossRef] [Green Version]
- Manzhos, S.; Golub, P. Data-driven kinetic energy density fitting for orbital-free DFT: Linear vs Gaussian process regression. J. Chem. Phys. 2020, 153, 074104. [Google Scholar] [CrossRef]
- Meyer, R.; Weichselbaum, M.; Hauser, A.W. Machine Learning Approaches toward Orbital-free Density Functional Theory: Simultaneous Training on the Kinetic Energy Density Functional and Its Functional Derivative. J. Chem. Theory Comput. 2020, 16, 5685–5694. [Google Scholar] [CrossRef] [PubMed]
- Imoto, F.; Imada, M.; Oshiyama, A. Order-N orbital-free density-functional calculations with machine learning of functional derivatives for semiconductors and metals. Phys. Rev. Res. 2021, 3, 033198. [Google Scholar] [CrossRef]
- Ryczko, K.; Wetzel, S.J.; Melko, R.G.; Tamblyn, I. Toward Orbital-Free Density Functional Theory with Small Data Sets and Deep Learning. J. Chem. Theory Comput. 2022, 18, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Prodan, E.; Kohn, W. Nearsightedness of electronic matter. Proc. Nat. Acad. Sci. USA 2005, 102, 11635–11638. [Google Scholar] [CrossRef] [Green Version]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Modified Fourth-Order Kinetic Energy Gradient Expansion with Hartree Potential-Dependent Coefficients. J. Chem. Theory Comput. 2017, 13, 4228–4239. [Google Scholar] [CrossRef]
- Lindhard, J. On the properties of a gas of charged particles. Dan. Vid. Selsk Mat.-Fys. Medd. 1954, 28, 8. [Google Scholar]
- Tao, J.; Perdew, J.P.; Almeida, L.M.; Fiolhais, C.; Kümmel, S. Nonempirical density functionals investigated for jellium: Spin-polarized surfaces, spherical clusters, and bulk linear response. Phys. Rev. B 2008, 77, 245107. [Google Scholar] [CrossRef]
- Levy, M.; Ou-Yang, H. Exact properties of the Pauli potential for the square root of the electron density and the kinetic energy functional. Phys. Rev. A 1988, 38, 625–629. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabiano, E.; Sarcinella, F.; Constantin, L.A.; Della Sala, F. Kinetic Energy Density Functionals Based on a Generalized Screened Coulomb Potential: Linear Response and Future Perspectives. Computation 2022, 10, 30. https://doi.org/10.3390/computation10020030
Fabiano E, Sarcinella F, Constantin LA, Della Sala F. Kinetic Energy Density Functionals Based on a Generalized Screened Coulomb Potential: Linear Response and Future Perspectives. Computation. 2022; 10(2):30. https://doi.org/10.3390/computation10020030
Chicago/Turabian StyleFabiano, Eduardo, Fulvio Sarcinella, Lucian A. Constantin, and Fabio Della Sala. 2022. "Kinetic Energy Density Functionals Based on a Generalized Screened Coulomb Potential: Linear Response and Future Perspectives" Computation 10, no. 2: 30. https://doi.org/10.3390/computation10020030
APA StyleFabiano, E., Sarcinella, F., Constantin, L. A., & Della Sala, F. (2022). Kinetic Energy Density Functionals Based on a Generalized Screened Coulomb Potential: Linear Response and Future Perspectives. Computation, 10(2), 30. https://doi.org/10.3390/computation10020030