
Simulation Frameworks for Morphogenetic Problems

{kind=link}
{kind=link}
Abstract
:1. Introduction
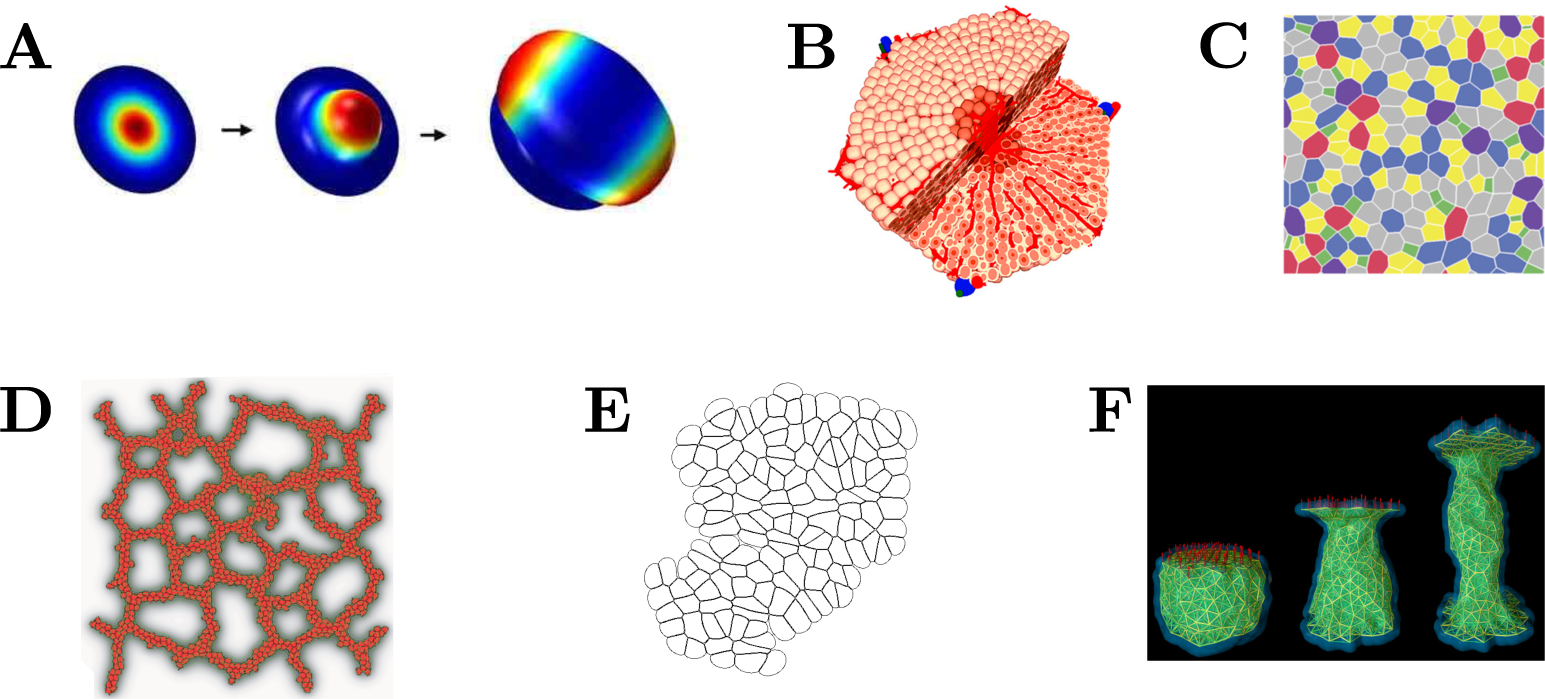
2. Tissue Models
2.1. Continuum Tissue Models
2.2. Spheroid Models
2.3. Vertex Models
2.4. Cellular Potts Model
2.5. Immersed Boundary Cell Model
2.6. Subcellular Element Models
3. Software Frameworks and Standards
3.1. Chaste
3.2. CompuCell3D
3.3. CellSys
3.4. EPISIM
3.5. Other Frameworks
3.6. Mark-Up Languages, Standards and Guidelines
4. Discussion and Future Directions
Acknowledgments
Conflicts of Interest
References
- Craver, C.F. When mechanistic models explain. Synthese 2006, 153, 355–376. [Google Scholar]
- Dillon, R.H.; Hans, G. A mathematical model for outgrowth and spatial patterning of the vertebrate limb bud. J. Theor. Boil. 1999, 197, 295–330. [Google Scholar]
- Bittig, T.; Wartlick, O.; Kicheva, A.; González-Gaitán, M.; Jülicher, F. Dynamics of anisotropic tissue growth. New J. Phys. 2008, 10, 063001. [Google Scholar]
- Osborne, J.M.; Walter, A.; Kershaw, S.K.; Mirams, G.R.; Fletcher, A.G.; Pathmanathan, P.; Gavaghan, D.J.; Jensen, O.E.; Maini, P.K.; Byrne, H.M. A hybrid approach to multi-scale modelling of cancer. Philos. Trans. Ser. A 2010, 368, 5013–5028. [Google Scholar] [Green Version]
- Murray, P.J.; Walter, A.; Fletcher, A.G.; Edwards, C.M.; Tindall, M.J.; Maini, P.K. Comparing a discrete and continuum model of the intestinal crypt. Phys. Boil. 2011, 8, 026011. [Google Scholar]
- Menshykau, D.; Erkan Ünal, P.B.; Sapin, V.; Iber, D. An interplay of geometry and signaling enables robust lung branching morphogenesis. Development 2014, 141, 4526–4536. [Google Scholar]
- Hoehme, S.; Brulport, M.; Bauer, A.; Bedawy, E.; Schormann, W.; Hermes, M.; Puppe, V.; Gebhardt, R.; Zellmer, S.; Schwarz, M.; et al. Prediction and validation of cell alignment along microvessels as order principle to restore tissue architecture in liver regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 10371–10376. [Google Scholar]
- Farhadifar, R.; Röper, J.-C.; Aigouy, B.; Eaton, S.; Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Curr. Boil. 2007, 17, 2095–2104. [Google Scholar]
- Merks, R.M.H.; Perryn, E.D.; Shirinifard, A.; Glazier, J.A. Contact-inhibited chemotaxis in de novo and sprouting blood-vessel growth. PLoS Comput. Boil. 2008, 4, e1000163. [Google Scholar]
- Sandersius, S.A.; Weijer, C.J.; Newman, T.J. Emergent cell and tissue dynamics from subcellular modeling of active biomechanical processes. Phys. Boil. 2011, 8, 045007. [Google Scholar]
- Dillon, R.H.; Othmer, H.G. A mathematical model for outgrowth and spatial patterning of the vertebrate limb bud. J. Theor. Boil. 1999, 197, 295–330. [Google Scholar]
- Rodriguez, E.K.; Hoger, A.; McCulloch, A.D. Stress-dependent finite growth in soft elastic tissues. J. Biomech. 1994, 27, 455–467. [Google Scholar]
- Taber, L.A. Iomechanics of Cardiovascular Development. Annu. Rev. Biomed. Eng. 2001, 3, 1–25. [Google Scholar]
- Taber, L.A. Towards a unified theory for morphomechanics. Philos. Trans. Ser. A 2009, 367, 3555–3583. [Google Scholar]
- Taber, L.A. Theoretical Study of Beloussov’s Hyper-Restoration Hypothesis for Mechanical Regulation of Morphogenesis. Biomech. Model. Mechanobiol. 2009, 7, 427–441. [Google Scholar]
- Wyczalkowski, M.A.; Chen, Z.; Filas, B.A.; Varner, V.D.; Taber, L.A. Computational models for mechanics of morphogenesis. Birth Defects Res. Part C—Embryo Today Rev. 2012, 96, 132–152. [Google Scholar]
- Muñoz, J.J.; Barrett, K.; Miodownik, M. A deformation gradient decomposition method for the analysis of the mechanics of morphogenesis. J. Biomech. 2007, 40, 1372–1380. [Google Scholar]
- Munoz, J.J.; Conte, V.; Miodownik, M. Stress-dependent morphogenesis: Continuum mechanics and truss systems. Biomech. Model. Mechanobiol. 2010, 9, 451–467. [Google Scholar]
- Varner, V.D.; Voronov, D.A.; Taber, L.A. Mechanics of head fold formation: Investigating tissue-level forces during early development. Development 2010, 137, 3801–3811. [Google Scholar]
- Filas, B.A.; Oltean, A.; Beebe, D.C.; Okamoto, R.J.; Bayly, P.V.; Taber, L.A. A potential role for differential contractility in early brain development and evolution. Biomech. Model. Mechanobiol. 2012, 11, 1251–1262. [Google Scholar]
- Kim, H.Y.; Varner, V.D.; Nelson, C.M. Apical constriction initiates new bud formation during monopodial branching of the embryonic chicken lung. Development 2013, 140, 3146–3155. [Google Scholar]
- Shi, Y.; Yao, J.; Xu, G.; Taber, L.A. Bending of the looping heart: differential growth revisited. J. Biomech. Eng. 2014, 136, 1–15. [Google Scholar]
- Murray, J.D.; Oster, G.; Harris, A.K. A mechanical model for mesenchymal morphogenesis. J. Math. Boil. 1983, 17, 125–129. [Google Scholar]
- Foty, R.A.; Forgacs, G.; Pfleger, C.M.; Steinberg, M.S. Liquid properties of embryonic tissues: Measurement of interfacial tensions. Phys. Rev. Lett. 1994, 72, 2298–2301. [Google Scholar]
- Forgacs, G.; Foty, R.A.; Shafrir, Y.; Steinberg, M.S. Viscoelastic properties of living embryonic tissues: A quantitative study. Biophys. J. 1998, 74, 2227–2234. [Google Scholar]
- Ranft, J.; Basan, M.; Elgeti, J.; Joanny, J.-F.; Prost, J.; Jülicher, F. Fluidization of tissues by cell division and apoptosis. Proc. Natl. Acad. Sci. USA 2010, 107, 20863–20868. [Google Scholar]
- Erkan Ünal, ZK.; Menshykau, D.; Iber, D. Simulating Organogenesis in COMSOL: Image-Based Modeling. ArXiv E-Prints 2014. arXiv:1408.1589. [Google Scholar]
- Iber, D.; Tanaka, S.; Fried, P.; Germann, P.; Menshykau, D. Simulating tissue morphogenesis and signaling. Methods Mol. Boil. 2015, 1189, 323–338. [Google Scholar]
- Keller, E.F.; Segel, L.A. Initiation of slime mold aggregation viewed as an instability. J. Theor. Boil. 1970, 26, 399–415. [Google Scholar]
- Chu, Y.-S.; Dufour, S.; Thiery, J.P.; Perez, E.; Pincet, F. Johnson-Kendall-Roberts theory applied to living cells. Phys. Rev. Let. 2005, 94, 028102. [Google Scholar]
- Drasdo, D.; Hoehme, S. A single-cell-based model of tumor growth in vitro: Monolayers and spheroids. Phys. Boil. 2005, 2, 133–147. [Google Scholar]
- Drasdo, D.; Hoehme, S.; Block, M. On the Role of Physics in the Growth and Pattern Formation of Multi-Cellular Systems: What Can We Learn from Individual-Cell Based Models? J. Stat. Phys. 2007, 128, 287–345. [Google Scholar]
- Drasdo, D.; Kree, R.; McCaskill, J.S. Monte Carlo approach to tissue-cell populations. Phys. Rev. E 1995, 52, 6635–6657. [Google Scholar]
- Palsson, E. A three-dimensional model of cell movement in multicellular systems. Future Gener. Comput. Syst. 2001, 17, 835–852. [Google Scholar]
- Drasdo, D. Center-Based Single-Cell Models: An Approach to Multi-Cellular Organization Based on a Conceptual Analogy to Colloidal Particles. In Single Cell Based Models in Biology and Medicine; Birkhäuser Basel: Reinach, Switzerland, 2007; pp. 171–196. [Google Scholar]
- Palsson, E.; Othmer, H.G. A model for individual and collective cell movement in Dictyostelium discoideum. Proc. Natl. Acad. Sci. USA 2000, 97, 10448–10453. [Google Scholar]
- Drasdo, D.; Hoehme, S. Individual-based approaches to birth and death in avascu1ar tumors. Math. Comput. Model. 2003, 37, 1163–1175. [Google Scholar]
- Galle, J.; Loeffler, M.; Drasdo, D. Modeling the effect of deregulated proliferation and apoptosis on the growth dynamics of epithelial cell populations in vitro. Biophys. J. 2005, 88, 62–75. [Google Scholar]
- Macklin, P.; Edgerton, M.E.; Thompson, A.M.; Cristini, V. Patient-calibrated agent-based modelling of ductal carcinoma in situ (DCIS): From microscopic measurements to macroscopic predictions of clinical progression. J. Theor. Boil. 2012, 301, 122–140. [Google Scholar]
- Dallon, J.C.; Othmer, H.G. How cellular movement determines the collective force generated by the Dictyostelium discoideum slug. J. Theor. Boil. 2004, 231, 203–222. [Google Scholar]
- Honda, H. Description of cellular patterns by Dirichlet domains: The two-dimensional case. J. Theor. Boil. 1978, 72, 523–543. [Google Scholar]
- Honda, H.; Tanemura, M.; Yoshida, A. Differentiation of wing epidermal scale cells in a butterfly under the lateral inhibition model—Appearance of large cells in a polygonal pattern. Acta Biotheor. 2000, 48, 121–136. [Google Scholar]
- Meineke, F.A.; Potten, C.S.; Loeffler, M. Cell migration and organization in the intestinal crypt using a lattice-free model. Cell Prolif. 2001, 34, 253–266. [Google Scholar]
- Gevertz, J.L.; Torquato, S. Modeling the effects of vasculature evolution on early brain tumor growth. J. Theor. Boil. 2006, 243, 517–531. [Google Scholar]
- Van Leeuwen, I.M.M.; Mirams, G.R.; Walter, A.; Fletcher, A.G.; Murray, P.J.; Osborne, J.M.; Varma, S.; Young, S.J.; Cooper, J.; Doyle, B.; et al. An integrative computational model for intestinal tissue renewal. Cell Prolif. 2009, 42, 617–636. [Google Scholar]
- Kim, Y.; Stolarska, M.A.; Othmer, H.G. A Hybrid Model for Tumor Spheroid Growth In Vitro I: Theoretical Development and Early Results. Math. Models Methods Appl. Sci. 2007, 17, 1773–1798. [Google Scholar]
- Kim, Y.; Othmer, H.G. A hybrid model of tumor-stromal interactions in breast cancer. Bull. Math. Boil. 2013, 75, 1304–1350. [Google Scholar]
- Pathmanathan, P.; Cooper, J.; Fletcher, A.G.; Mirams, G.R.; Murray, P.J.; Osborne, J.M.; Pitt-Francis, J.; Walter, A.; Chapman, J.S. A computational study of discrete mechanical tissue models. Phys. Boil. 2009, 6, 036001. [Google Scholar]
- Honda, H.; Eguchi, G. How much does the cell boundary contract in a monolayered cell sheet? J. Theor. Biol. 1980, 84, 575–588. [Google Scholar]
- Fletcher, A.G.; Osborne, J.M.; Maini, P.K.; Gavaghan, D.J. Implementing vertex dynamics models of cell populations in biology within a consistent computational framework. Progr. Biophys. Mol. Biol. 2013, 113, 299–326. [Google Scholar]
- Fletcher, A.G.; Osterfield, M.; Baker, R.E.; Shvartsman, S.Y. Vertex models of epithelial morphogenesis. Biophys. J. 2014, 106, 2291–2304. [Google Scholar]
- Weliky, M.; Oster, G. The mechanical basis of cell rearrangement. I. Epithelial morphogenesis during Fundulus epiboly. Development 1990, 109, 373–386. [Google Scholar]
- Weliky, M.; Minsuk, S.; Keller, R.; Oster, G. Notochord morphogenesis in Xenopus laevis: Simulation of cell behavior underlying tissue convergence and extension. Development 1991, 113, 1231–1244. [Google Scholar]
- Nagai, T.; Honda, H. A dynamic cell model for the formation of epithelial tissues. Philos. Mag. Part B. 2001, 81, 699–719. [Google Scholar]
- Chen, H.H.; Brodland, W.G. Cell-level finite element studies of viscous cells in planar aggregates. J. Biomech. Eng. 2000, 122, 394–401. [Google Scholar]
- Brodland, W.G.; Chen, H.H. The Mechanics of Heterotypic Cell Aggregates: Insights from Computer Simulations. J. Biomech. Eng. 2000, 122, 402–407. [Google Scholar]
- Rudge, T.; Haseloff, J. A Computational Model of Cellular Morphogenesis in Plants. In Advances in Artificial Life, Lecture Notes in Computer Science; Capcarrère, M.S., Freitas, A.A., Bentley, P.J., Johnson, C.G., Timmis, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3630, pp. 78–87. [Google Scholar]
- Merks, R.M.H.; Dirk Inzé, M.G.; Beemster, G.T.S. VirtualLeaf: An open-source framework for cell-based modeling of plant tissue growth and development. Plant Physiol. 2011, 155, 656–666. [Google Scholar]
- Graner, F.; Glazier, J.A. Simulation of biological cell sorting using a two-dimensional extended Potts model. Phys. Rev. Lett. 1992, 69, 2013–2016. [Google Scholar]
- Schilling, S.; Willecke, M.; Aegerter-Wilmsen, T.; Cirpka, O.A.; Basler, K.; von Mering, C. Cell-sorting at the A/P boundary in the Drosophila wing primordium: A computational model to consolidate observed non-local effects of Hh signaling. PLoS Comput. Boil. 2011, 7, e1002025. [Google Scholar]
- Smith, A.M.; Baker, R.E.; Kay, D.; Maini, P.K. Incorporating chemical signalling factors into cell-based models of growing epithelial tissues. J. Math. Boil. 2011, 65, 441–463. [Google Scholar] [Green Version]
- Osterfield, M.; Du, X.; Schüpbach, T.; Wieschaus, E.F.; Shvartsman, S.Y. Three-dimensional epithelial morphogenesis in the developing Drosophila egg. Dev. Cell. 2013, 24, 400–410. [Google Scholar]
- Honda, H.; Tanemura, M.; Nagai, T. A three-dimensional vertex dynamics cell model of space-filling polyhedra simulating cell behavior in a cell aggregate. J. Theor. Boil. 2004, 226, 439–453. [Google Scholar]
- Honda, H.; Motosugi, N.; Nagai, T.; Tanemura, M.; Hiiragi, T. Computer simulation of emerging asymmetry in the mouse blastocyst. Development 2008, 135, 1407–1414. [Google Scholar]
- Okuda, S.; Inoue, Y.; Eiraku, M.; Adachi, T.; Sasai, Y. Vertex dynamics simulations of viscosity-dependent deformation during tissue morphogenesis. Biomech. Model. Mechanobiol. 2014, 14, 413–425. [Google Scholar]
- Hufnagel, L.; Teleman, A.A.; Rouault, H.; Cohen, S.M.; Shraiman, B.I. On the mechanism of wing size determination in fly development. Proc. Natl. Acad. Sci. USA 2007, 104, 3835–3840. [Google Scholar]
- Brodland, W.G.; Viens, D.; Veldhuis, J.H. A new cell-based FE model for the mechanics of embryonic epithelia. Comput. Methods Biomech. Biomed. Eng. 2007, 10, 121–128. [Google Scholar]
- Glazier, J.A.; Graner, F. Simulation of the differential adhesion driven rearrangement of biological cells. Phys. Rev. E 1993, 47, 2128–2154. [Google Scholar]
- Ising, E. Beitrag zur Theorie des Ferromagnetismus. Zeitschrift für Physik 1925, 31, 253–258, In German. [Google Scholar]
- Potts, R.B. Some generalized order-disorder transformations. Math. Proc. Camb. Philos. Soc. 1952, 48, 106–109. [Google Scholar]
- Glazier, J.A.; Balter, A.; Poplawski, N.J. Magnetization to Morphogenesis: A Brief History of the Glazier-Graner-Hogeweg Model. In Single Cell Based Models in Biology and Medicine; Birkhäuser Basel: Reinach, Switzerland, 2007; pp. 79–106. [Google Scholar]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar]
- Marée, A.F.M.; Grieneisen, V.A.; Hogeweg, P. The Cellular Potts Model and Biophysical Properties of Cells, Tissues and Morphogenesis. In Single Cell Based Models in Biology and Medicine; Birkhäuser Basel: Reinach, Switzerland, 2007; pp. 107–136. [Google Scholar]
- Swat, M.H.; Thomas, G.L.; Belmonte, J.M.; Shirinifard, A.; Hmeljak, D.; Glazier, J.A. Multi-Scale Modeling of Tissues Using CompuCell3D. Methods Cell Biol. 2012, 110, 325–366. [Google Scholar]
- Hogeweg, P. Evolving mechanisms of morphogenesis: On the interplay between differential adhesion and cell differentiation. J. Theor. Boil. 2000, 203, 317–333. [Google Scholar]
- Hogeweg, P. Computing an organism: On the interface between informatic and dynamic processes. Biosystems 2002, 64, 97–109. [Google Scholar]
- Hogeweg, P.; Takeuchi, N. Multilevel selection in models of prebiotic evolution: Compartments and spatial self-organization. Orig. Life Evol. Biosph. 2003, 33, 375–403. [Google Scholar]
- Merks, R.M.H.; Brodsky, S.V.; Goligorksy, M.S.; Newman, S.A.; Glazier, J.A. Cell elongation is key to in silico replication of in vitro vasculogenesis and subsequent remodeling. Dev. Biol. 2006, 289, 44–54. [Google Scholar]
- Szabó, A.; Czirók, A. The Role of Cell-Cell Adhesion in the Formation of Multicellular Sprouts. Math. Model. Nat. Phenom. 2010, 5, 106–122. [Google Scholar]
- Bauer, A.L.; Jackson, T.L.; Jiang, Y. A cell-based model exhibiting branching and anastomosis during tumor-induced angiogenesis. Biophys. J. 2007, 92, 3105–3121. [Google Scholar]
- Hirashima, T.; Iwasa, Y.; Morishita, Y. Dynamic modeling of branching morphogenesis of ureteric bud in early kidney development. J. Theor. Boil. 2009, 259, 58–66. [Google Scholar]
- Hester, S.D.; Belmonte, J.M.; Gens, S.J.; Clendenon, S.G.; Glazier, J.A. A multi-cell, multi-scale model of vertebrate segmentation and somite formation. PLoS Comput. Boil. 2011, 7, e1002155. [Google Scholar]
- Poplawski, N.J.; Swat, M.H.; Gens, S.J.; Glazier, J.A. Adhesion between cells, diffusion of growth factors, and elasticity of the AER produce the paddle shape of the chick limb. Phys. A 2007, 373, 521–532. [Google Scholar]
- Rejniak, K.A.; Kliman, H.J.; Fauci, L.J. A computational model of the mechanics of growth of the villous trophoblast bilayer. Bull. Math. Boil. 2004, 66, 199–232. [Google Scholar]
- Rejniak, K.A. An immersed boundary framework for modelling the growth of individual cells: An application to the early tumour development. J. Theor. Boil. 2007, 247, 186–204. [Google Scholar]
- Dillon, R.H.; Owen, M.R.; Painter, K. A single-cell-based model of multicellular growth using the immersed boundary method. Contemp. Math. 2008, 466, 1–15. [Google Scholar]
- Peskin, C.S. The immersed boundary method. Acta Numer. 2003, 11, 479–511. [Google Scholar]
- Mittal, R.; Iaccarino, G. Immersed Boundary Methods. Annu. Rev. Fluid Mech. 2005, 37, 239–261. [Google Scholar]
- Rejniak, K.A.; Dillon, R.H. A Single Cell-Based Model of the Ductal Tumour Microarchitecture. Comput. Math. Methods Med. 2007, 8, 51–69. [Google Scholar]
- Le, D.-V.; White, J.; Peraire, J.; Lim, K.M.; Khoo, B.C. An implicit immersed boundary method for three-dimensional fluid-membrane interactions. J. Comput. Phys. 2009, 228, 8427–8445. [Google Scholar]
- Newman, T.J.; Modeling, Multicellular; Systems, Using. Subcellular Elements. Math. Biosci. Eng. 2005, 2, 613–624. [Google Scholar]
- Christley, S.; Lee, B.; Dai, X.; Nie, Q. Integrative multicellular biological modeling: A case study of 3D epidermal development using GPU algorithms. BMC Syst. Biol. 2010, 4, 107. [Google Scholar]
- Milde, F.; Tauriello, G.; Haberkern, H.; Koumoutsakos, P. SEM++: A particle model of cellular growth, signaling and migration. Comput. Part. Mech. 2014, 1, 211–227. [Google Scholar]
- Sandersius, S.A.; Newman, T.J. Modeling cell rheology with the Subcellular Element Model. Phys. Boil. 2008, 5, 015002. [Google Scholar]
- Pitt-Francis, J.; Pathmanathan, P.; Bernabeu, M.Q.; Bordas, R.; Cooper, J.; Fletcher, A.G.; Mirams, G.R.; Murray, P.J.; Osborne, J.M.; Walter, A. Chaste: A test-driven approach to software development for biological modelling. Comput. Phys. Commun. 2009, 180, 2452–2471. [Google Scholar]
- Mirams, G.R.; Arthurs, C.J.; Bernabeu, M.O.; Bordas, R.; Cooper, J.; Corrias, A.; Davit, Y.; Dunn, S.J.; Fletcher, A.G.; Harvey, D.G.; et al. Chaste: An open source C++ library for computational physiology and biology. PLoS Comput. Boil. 2013, 9, e1002970. [Google Scholar]
- Moreira, J.; Deutsch, A. Cellular Automation Models of Tumor Development: A Critical Review. Adv. Complex Syst. 2002, 5, 247–267. [Google Scholar]
- Cuellar, A.A.; Lloyd, C.M.; Nielsen, P.M.F.; Bullivant, D.P.; Nickerson, D.P.; Hunter, P.J. An Overview of CellML 1.1, a Biological Model Description Language. Simulation 2003, 79, 740–747. [Google Scholar]
- Fletcher, A.G.; Breward, C.J.W.; Chapman, J.S. Mathematical modeling of monoclonal conversion in the colonic crypt. J. Theor. Boil. 2012, 300, 118–133. [Google Scholar] [Green Version]
- Mirams, G.R.; Fletcher, A.G.; Maini, P.K.; Byrne, H.M. A theoretical investigation of the effect of proliferation and adhesion on monoclonal conversion in the colonic crypt. J. Theor. Boil. 2012, 312, 143–156. [Google Scholar]
- Dunn, S.J.; Näthke, I.S.; Osborne, J.M. Computational models reveal a passive mechanism for cell migration in the crypt. PLoS ONE 2013, 8, e80516. [Google Scholar]
- Dunn, S.-J.; Fletcher, A.G.; Chapman, J.S.; Gavaghan, D.J.; Osborne, J.M. Modelling the role of the basement membrane beneath a growing epithelial monolayer. J. Theor. Boil. 2012, 298, 82–91. [Google Scholar]
- Dunn, S.-J.; Appleton, P.L.; Nelson, S.A.; Näthke, I.S.; Gavaghan, D.J.; Osborne, J.M. A two-dimensional model of the colonic crypt accounting for the role of the basement membrane and pericryptal fibroblast sheath. PLoS Comput. Boil. 2012, 8, e1002515. [Google Scholar]
- Figueredo, G.P.; Joshi, T.V.; Osborne, J.M.; Byrne, H.M.; Owen, M.R. On-lattice agent-based simulation of populations of cells within the open-source Chaste framework. Interface Focus 2013, 3, 20120081. [Google Scholar]
- Glazier, J.A.; Zhang, Y.; Swat, M.H.; Zaitlen, B.L.; Schnell, S. Coordinated action of N-CAM, N-cadherin, EphA4, and ephrinB2 translates genetic prepatterns into structure during somitogenesis in chick. Curr. Top. Dev. Biol. 2008, 81, 205–247. [Google Scholar]
- Dias, A.S.; de Almeida, I.; Belmonte, J.M.; Glazier, J.A.; Stern, C.D. Somites without a clock. Science 2014, 343, 791–795. [Google Scholar]
- Guidolin, D.; Albertin, G.; Sorato, E.; Oselladore, B.; Mascarin, A.; Ribatti, D. Mathematical modeling of the capillary-like pattern generated by adrenomedullin-treated human vascular endothelial cells in vitro. Dev. Dyn. 2009, 238, 1951–1963. [Google Scholar]
- Kleinstreuer, N.; Dix, D.; Rountree, M.; Baker, N.; Sipes, N.; Reif, D.; Spencer, R.; Knudsen, T. A computational model predicting disruption of blood vessel development. PLoS Comput. Boil. 2013, 9, e1002996. [Google Scholar]
- Boas, S.E.M.; Merks, R.M.H. Synergy of cell-cell repulsion and vacuolation in a computational model of lumen formation. J. R. Soc. Interface/R. Soc. 2014, 11, 20131049. [Google Scholar]
- Shirinifard, A.; Gens, S.J.; Zaitlen, B.L.; Poplawski, N.J.; Swat, M.H.; Glazier, J.A. 3D multi-cell simulation of tumor growth and angiogenesis. PLoS ONE 2009, 4, e7190. [Google Scholar]
- Poplawski, N.J.; Shirinifard, A.; Agero, U.; Gens, S.J.; Swat, M.H.; Glazier, J.A. Front instabilities and invasiveness of simulated 3D avascular tumors. PLoS ONE 2010, 5, e10641. [Google Scholar]
- Li, J.F.; Lowengrub, J. The effects of cell compressibility, motility and contact inhibition on the growth of tumor cell clusters using the Cellular Potts Model. J. Theor. Boil. 2014, 343, 79–91. [Google Scholar]
- Vasiev, B.; Balter, A.; Chaplain, M.A.J.; Glazier, J.A.; Weijer, C.J. Modeling gastrulation in the chick embryo: Formation of the primitive streak. PLoS ONE 2010, 5, e10571. [Google Scholar]
- Ray, S.; Yuan, D.; Dhulekar, N.; Oztan, B.; Yener, B.; Larsen, M. Cell-based multi-parametric model of cleft progression during submandibular salivary gland branching morphogenesis. PLoS Comput. Boil. 2013, 9, e1003319. [Google Scholar]
- Hoehme, S.; Drasdo, D. A cell-based simulation software for multi-cellular systems. Bioinformatics 2010, 26, 2641–2642. [Google Scholar]
- Hoehme, S.; Hengstler, J.G.; Brulport, M.; Schäfer, M.; Bauer, A.; Gebhardt, R.; Drasdo, D. Mathematical modelling of liver regeneration after intoxication with CCl4. Chemico-Biol. Interact. 2007, 168, 74–93. [Google Scholar]
- Drasdo, D.; Hoehme, S.; Hengstler, J.G. How predictive quantitative modelling of tissue organisation can inform liver disease pathogenesis. J. Hepatol. 2014, 61, 951–956. [Google Scholar]
- Schliess, F.; Hoehme, S.; Henkel, S.G.; Ghallab, A.; Driesch, D.; Böttger, J.; Guthke, R.; Pfaff, M.; Hengstler, J.G.; Gebhardt, R.; et al. Integrated metabolic spatial-temporal model for the prediction of ammonia detoxification during liver damage and regeneration. Hepatology 2014, 60, 2040–2051. [Google Scholar]
- Sütterlin, T.; Kolb, C.; Dickhaus, H.; Jäger, D.; Grabe, N. Bridging the scales: Semantic integration of quantitative SBML in graphical multi-cellular models and simulations with EPISIM and COPASI. Bioinformatics 2013, 29, 223–239. [Google Scholar]
- EPISIM Multi-Scale Modeling and Simulation Platform. Avaiable online: http://tigacenter.bioquant.uni-heidelberg.de/episim.html accessed on 20 April 2015.
- Sütterlin, T.; Grabe, N. Graphical Multi-Scale Modeling of Epidermal Homeostasis with EPISIM. In Computational Biophysics of the Skin; Pan Stanford Publishing: Singapore, 2014; Volume 3, pp. 421–460. [Google Scholar]
- Safferling, K.; Sütterlin, T.; Westphal, K.; Ernst, C.; Breuhahn, K.; James, M.; Jäger, D.; Halama, N.; Grabe, N. Wound healing revised: A novel reepithelialization mechanism revealed by in vitro and in silico models. J. Cell Biol. 2013, 203, 691–709. [Google Scholar]
- Kang, S.; Kahan, S.; McDermott, J.; Flann, N.; Shmulevich, I. Biocellion: Accelerating computer simulation of multicellular biological system models. Bioinformatics 2014, 30, 3101–3108. [Google Scholar]
- Starruß, J.; de Back, W.; Brusch, L.; Deutsch, A. Morpheus: A user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30, 1331–1332. [Google Scholar]
- De Back, W.; Zhou, J.X.; Brusch, L. On the role of lateral stabilization during early patterning in the pancreas. J. R. Soc. Interface/R. Soc. 2013, 10, 20120766. [Google Scholar]
- De Back, W.; Zimm, R.; Brusch, L. Transdifferentiation of pancreatic cells by loss of contact-mediated signaling. BMC Syst. Biol. 2013, 7, 77. [Google Scholar]
- Köhn-Luque, A.; de Back, W.; Mattiotti, A.; Deutsch, A.; Pérez-Pomares, J.M.; Herrero, M.A. Early embryonic vascular patterning by matrix-mediated paracrine signalling: A mathematical model study. PLoS ONE 2011, 6, e24175. [Google Scholar]
- Köhn-Luque, A.; de Back, W.; Yamaguchi, Y.; Yoshimura, K.; Herrero, M.; Miura, T. Dynamics of VEGF matrix-retention in vascular network patterning. Phys. Boil. 2013, 10, 066007. [Google Scholar]
- Bradley, C.P.; Bowery, A.; Britten, R.; Budelmann, V.; Camara, O.; Christie, R.G.; Cookson, A.; Frangi, A.F.; Gamage, T.B.; Heidlauf, T.; et al. OpenCMISS: A multi-physics & multi-scale computational infrastructure for the VPH/Physiome project. Progr. Biophys. Mol. Biol. 2011, 107, 32–47. [Google Scholar]
- Tanaka, S.; Sichau, D.; Iber, D. LBIBCell: A Cell-Based Simulation Environment for Morphogenetic Problems. Bioinformatics 2015. [Google Scholar] [CrossRef]
- Li, C.; Donizelli, M.; Rodriguez, N.; Dharuri, H.; Endler, L.; Chelliah, V.; Li, L.; He, E.; Henry, A.; Stefan, M.I.; et al. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst. Biol. 2010, 4, 92. [Google Scholar]
- Le Novère, N.; Finney, A.; Hucka, M.; Bhalla, U.S.; Campagne, F.; Collado-Vides, J.; Crampin, E.J.; Halstead, M.; Klipp, E.; Mendes, P.; et al. Minimum information requested in the annotation of biochemical models (MIRIAM). Nat. Biotechnol. 2005, 23, 1509–1515. [Google Scholar]
- Waltemath, D.; Adams, R.; Beard, D.A.; Bergmann, F.T.; Bhalla, U.S.; Britten, R.; Chelliah, V.; Cooling, M.T.; Cooper, J.; Crampin, E.J.; et al. Minimum Information About a Simulation Experiment (MIASE). PLoS Comput. Boil. 2011, 7, e1001122. [Google Scholar] [Green Version]
- Waltemath, D.; Adams, R.; Bergmann, F.T.; Hucka, M.; Kolpakov, F.; Miller, A.K.; Moraru, I.I.; Nickerson, D.P.; Sahle, S.; Snoep, J.L.; et al. Reproducible computational biology experiments with SED-ML—The Simulation Experiment Description Markup Language. BMC Syst. Biol. 2011, 5, 198. [Google Scholar]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; et al. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar]
- Sluka, J.P.; Shirinifard, A.; Swat, M.H.; Cosmanescu, A.; Heiland, R.W.; Glazier, J.A. The cell behavior ontology: Describing the intrinsic biological behaviors of real and model cells seen as active agents. Bioinformatics 2014, 30, 2367–2374. [Google Scholar]
- Demir, E.; Cary, M.P.; Paley, S.; Fukuda, K.; Lemer, C.; Vastrik, I.; Wu, G.; D’Eustachio, P.; Schaefer, C.; Luciano, J.; et al. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010, 28, 935–942. [Google Scholar]
- Büchel, F.; Wrzodek, C.; Mittag, F.; Dräger, A.; Eichner, J.; Rodriguez, N.; Le Novère, N.; Zell, A. Qualitative translation of relations from BioPAX to SBML qual. Bioinformatics 2012, 28, 2648–2653. [Google Scholar]
- Christie, R.G.; Nielsen, P.M.F.; Blackett, S.A.; Bradley, C.P.; Hunter, P.J. FieldML: Concepts and implementation. Philos. Trans. Ser. A 2009, 367, 1869–1884. [Google Scholar]
- Rejniak, K.A. An immersed boundary framework for modelling the growth of individual cells: An application to the early tumour development. J. Theor. Biol. 2007, 247, 186–204. [Google Scholar]


© 2015 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, S. Simulation Frameworks for Morphogenetic Problems. Computation 2015, 3, 197-221. https://doi.org/10.3390/computation3020197
Tanaka S. Simulation Frameworks for Morphogenetic Problems. Computation. 2015; 3(2):197-221. https://doi.org/10.3390/computation3020197
Chicago/Turabian StyleTanaka, Simon. 2015. "Simulation Frameworks for Morphogenetic Problems" Computation 3, no. 2: 197-221. https://doi.org/10.3390/computation3020197
APA StyleTanaka, S. (2015). Simulation Frameworks for Morphogenetic Problems. Computation, 3(2), 197-221. https://doi.org/10.3390/computation3020197