
A Guide to Phylogenetic Reconstruction Using Heterogeneous Models—A Case Study from the Root of the Placental Mammal Tree

Abstract
:1. Introduction
2. A Practical Guide for Phylogeny Reconstruction

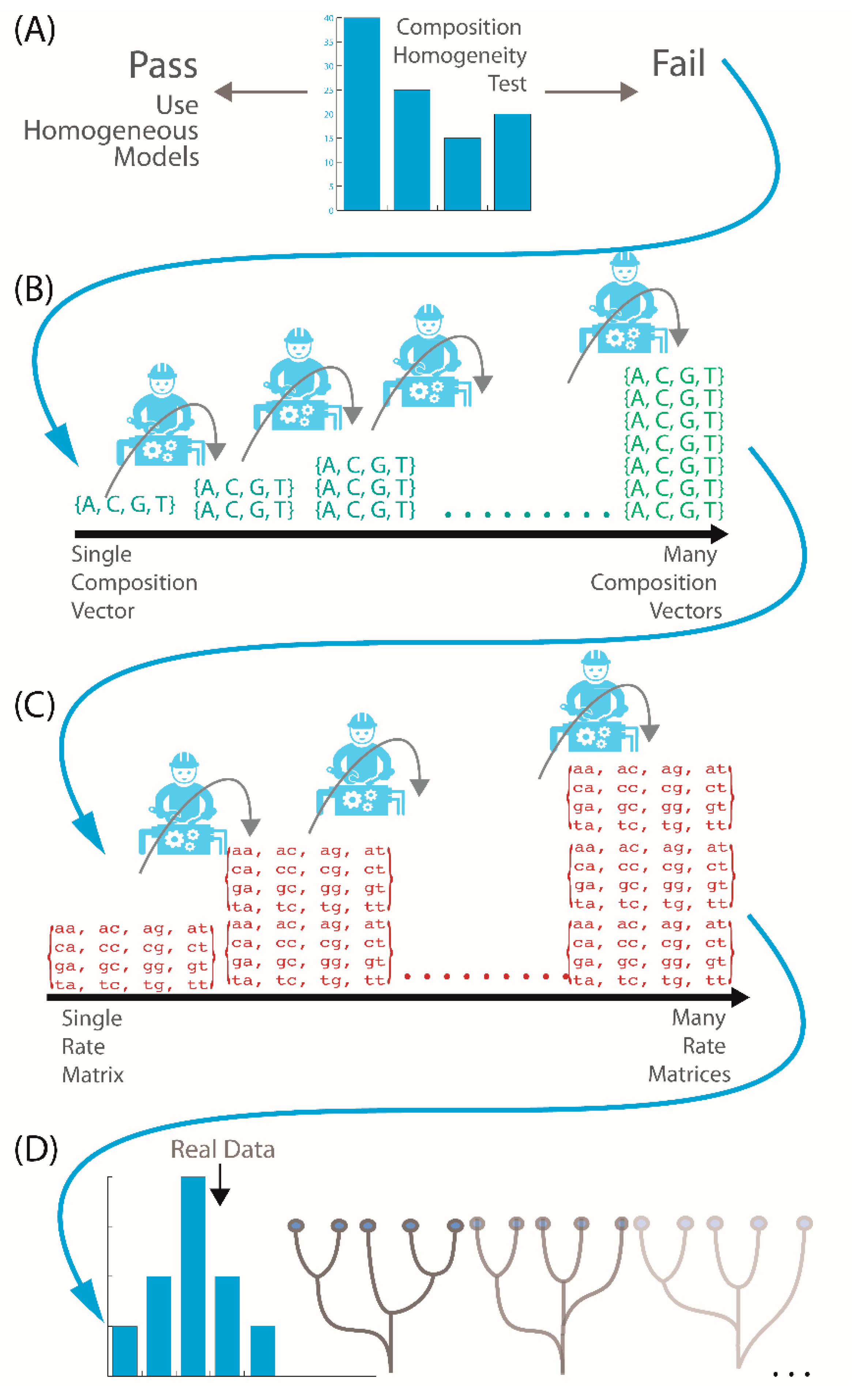{kind=link}
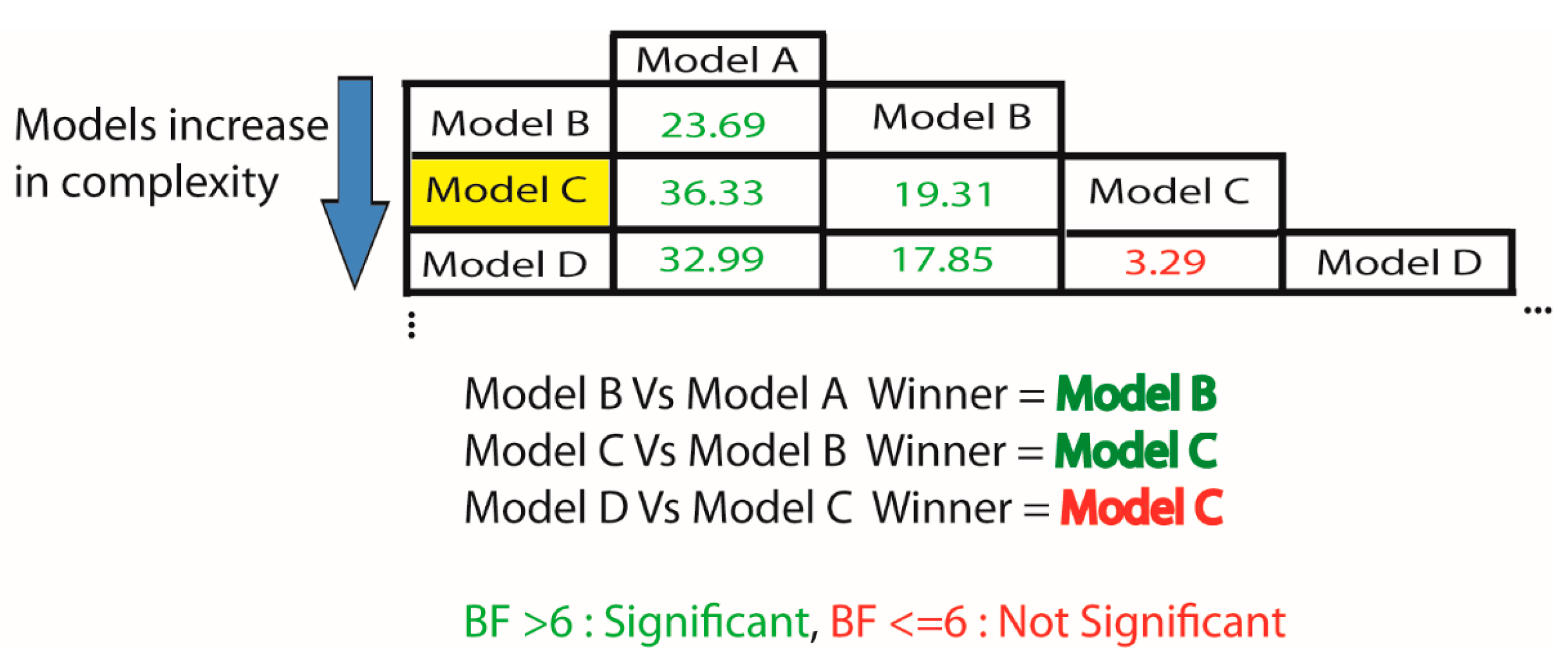{kind=link}
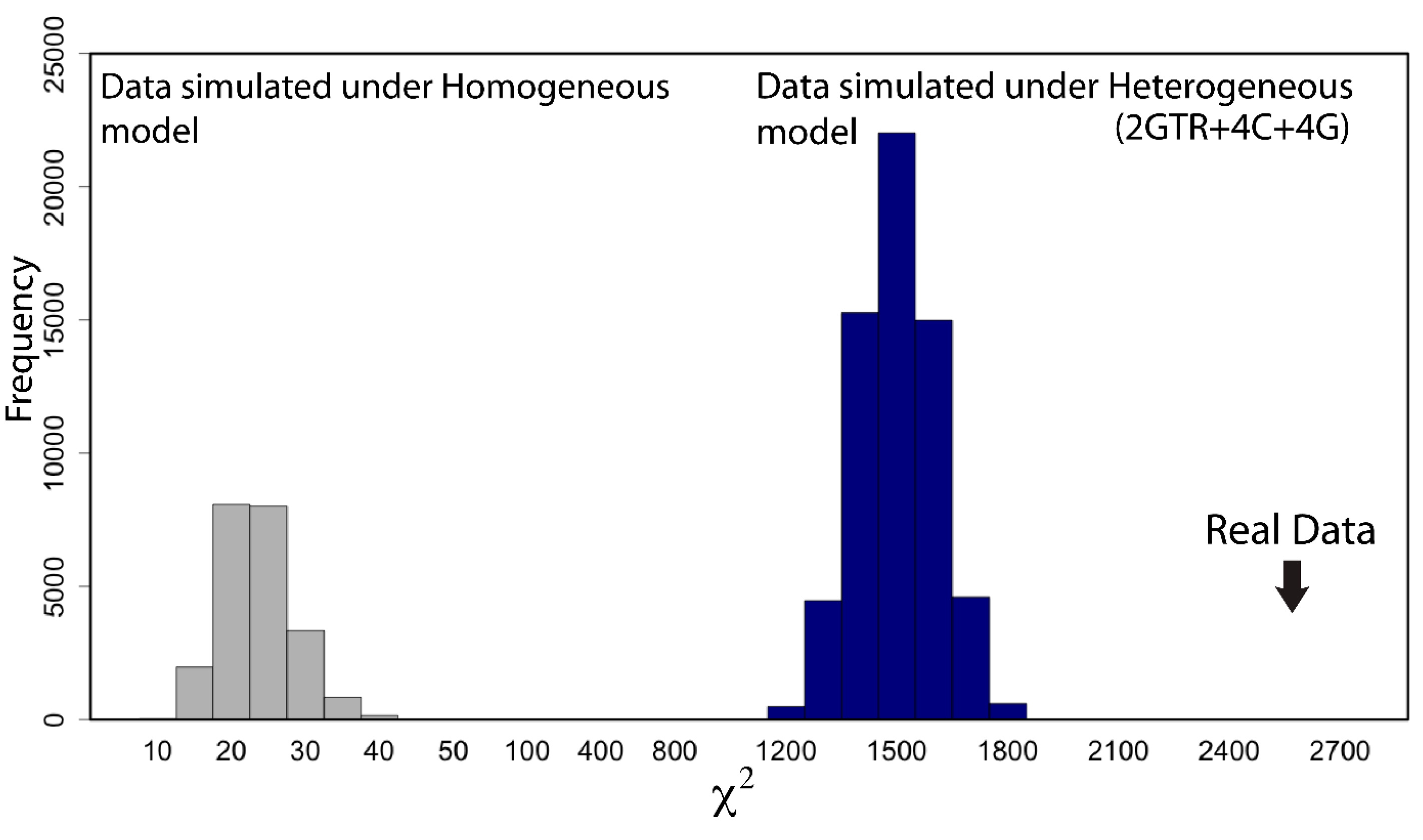{kind=link}
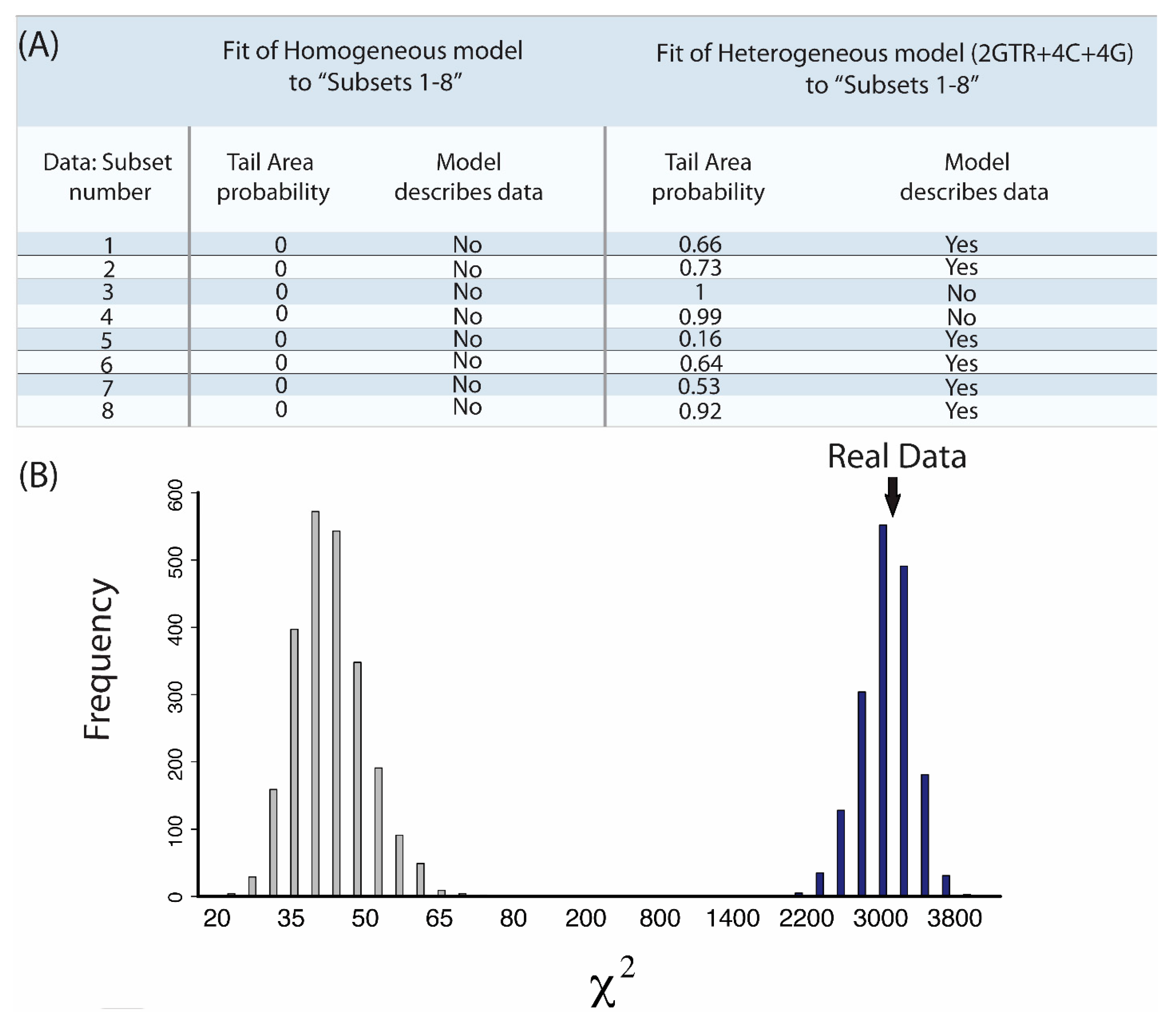{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages |
|---|---|---|
| RAxML [35] (Maximum likelihood) | Very Fast Easy to use Recommended for homogeneous modeling | No heterogeneous modeling Often too simplistic |
| P4 [10] (Bayesian) | Models heterogeneity across lineages Extensible Strong statistical framework | Python knowledge helpful Time/computationally consuming Convergence diagnosis difficult |
| PhyloBayes [36] (Bayesian) | Employs CAT model of heterogeneity Very easy to use Multi-processor (MPI) version available Convergence is easily assessed and can be done automatically | Computationally intensive and time consuming, even with MPI. (However, newer computer clusters are more readily prepared for this.) |
2.1. What Type of Molecular Sequence Data Should I Use?
2.2. Is My Data Compositionally Homogeneous?

2.3. What If My Sequences Vary in Rates of Change across Their Length?
2.4. What If My Sequences Have Both Compositional Heterogeneity and Rate Heterogeneity?
2.5. What If My Sequences Have Both Compositional and Rate Heterogeneity across Sites?
2.6. How Do I Select the Optimum Model for My Data?
2.7. Does My Model Fit My Data?



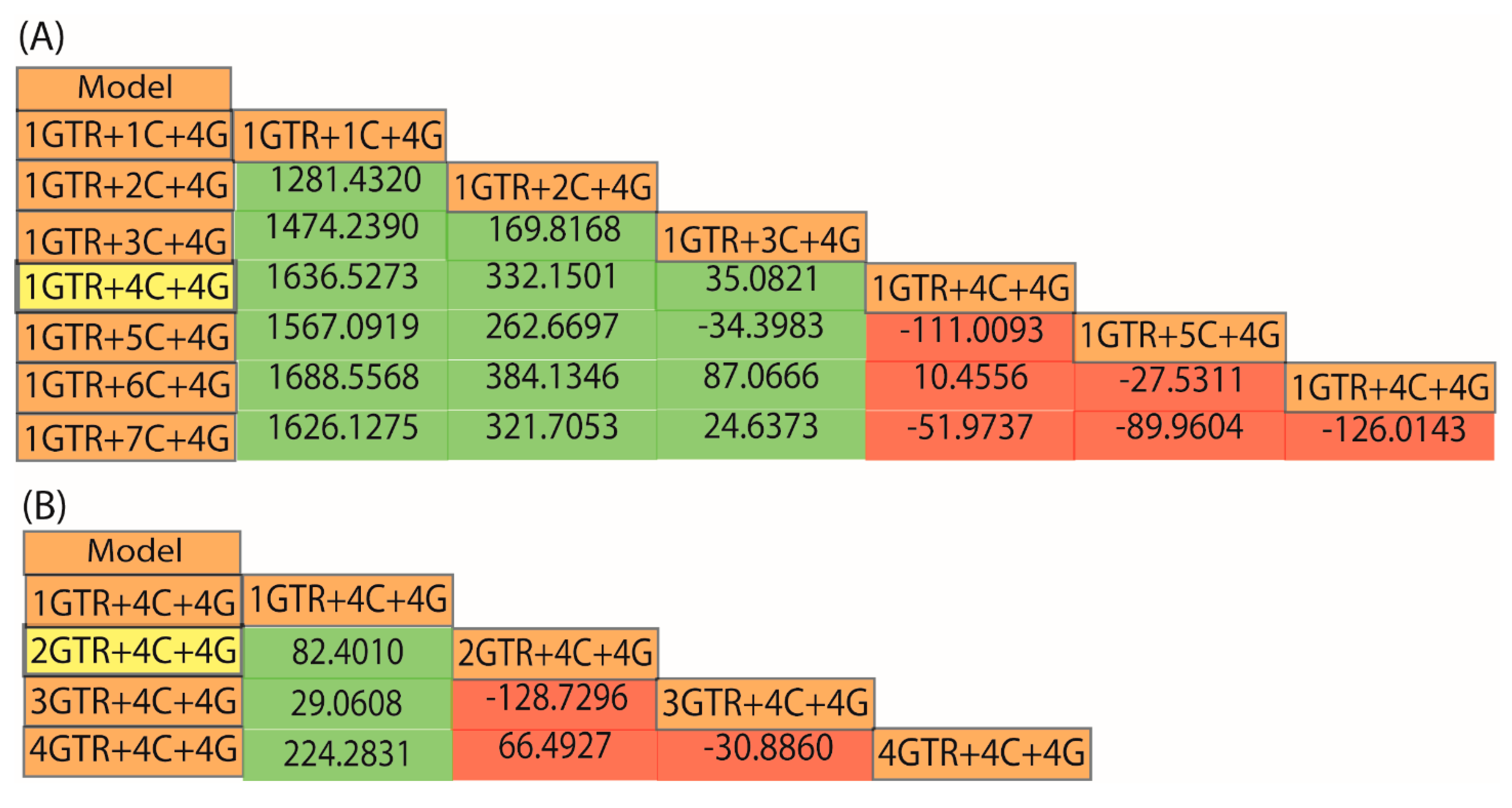
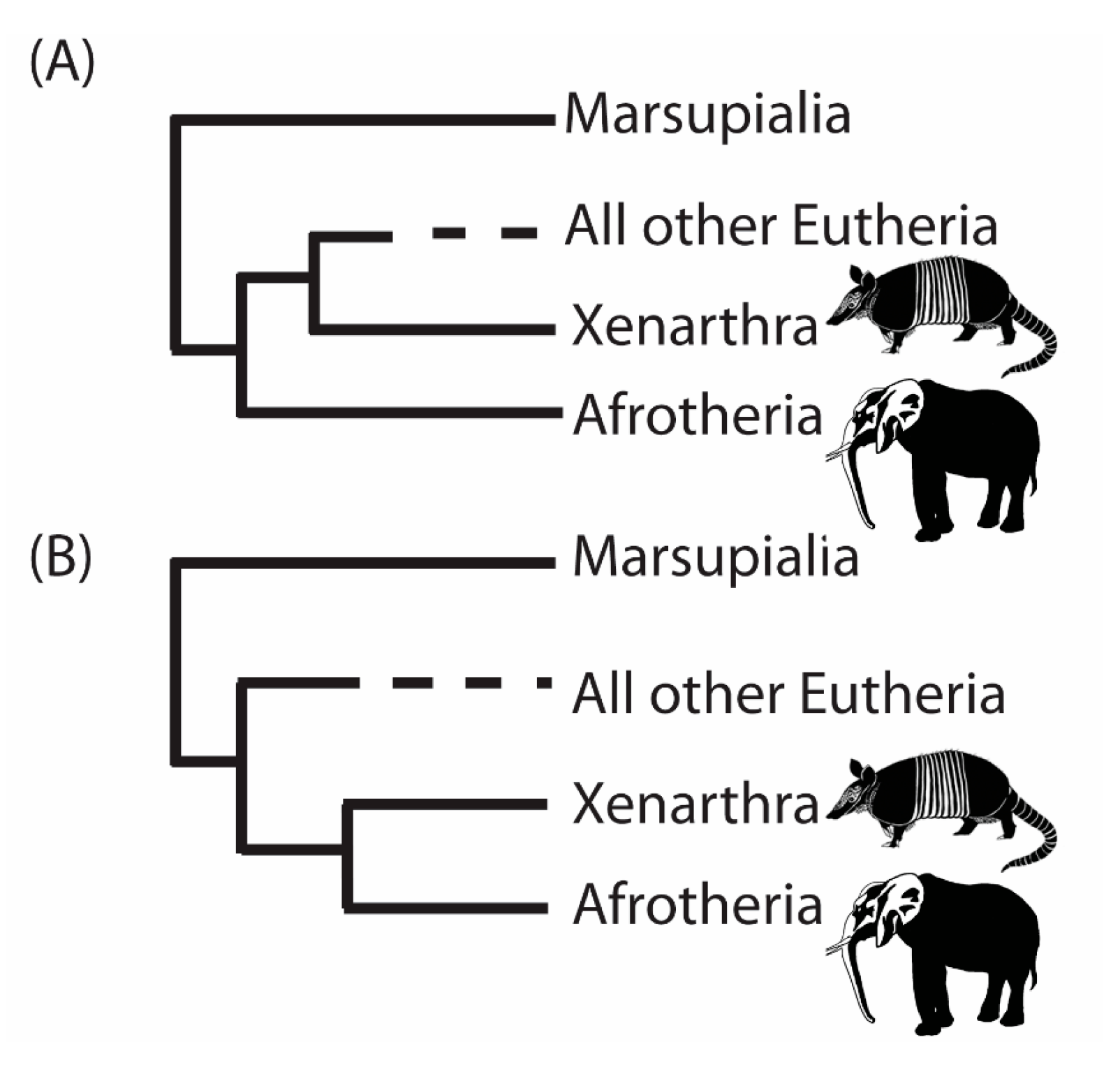
3. A Case Study from the Root of the Placental Mammal Tree
- (1)
- “RomiguierTopAT”: This is a concatenated alignment using 100 of the most AT-rich genes. We wished to assess the impact of heterogeneous modeling on these data and on the support for the proposed Afrotheria hypothesis.
- (2)
- “Subsets 1–8”: Each of the eight subsets are a concatenated alignment using 25 genes chosen at random with varying GC-richness. We wished to assess whether heterogeneous models can adequately model the GC variation.


4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Posada, D. Selecting Models of Evolution. In The Phylogenetic Handbook: A Practical Approach to DNA and Protein Phylogeny; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Yang, Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol. Evol. 1996, 11, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Li, W.-H. Phylogenetic analysis based on rRNA sequences supports the archaebacterial rather than the eocyte tree. Nature 1989, 339, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Tourasse, N.J.; Gouy, M. Accounting for evolutionary rate variation among sequence sites consistently changes universal phylogenies deduced from rRNA and protein-coding genes. Mol. Phylogenet. Evol. 1999, 13, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-H.; Tanimura, M.; Sharp, P.M. An evaluation of the molecular clock hypothesis using mammalian DNA sequences. J. Mol. Evol. 1987, 25, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Romiguier, J.; Ranwez, V.; Douzery, E.J.P.; Galtier, N. Contrasting GC-content dynamics across 33 mammalian genomes: Relationship with life-history traits and chromosome sizes. Genome Res. 2010, 20, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.C.; Foster, P.G.; Webb, A.E.; Pisani, D.; McInerney, J.O.; O’Connell, M.J. Heterogeneous models place the root of the placental mammal phylogeny. Mol. Biol. Evol. 2013, 30, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Duret, L. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 2007, 23, 273–277. [Google Scholar]
- Galtier, N.; Piganeau, G.; Mouchiroud, D.; Duret, L. GC-content evolution in mammalian genomes: The biased gene conversion hypothesis. Genetics 2001, 159, 907–911. [Google Scholar] [PubMed]
- Foster, P.G. Modeling compositional heterogeneity. Syst. Biol. 2004, 53, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Olmstead, R.G. Molecular Systematics, 2nd ed.; Hillis, D.M., Moritz, C., Mable, B.K., Eds.; Sinauer Associates: Sunderland, MA, USA, 1996. [Google Scholar]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J.A.A. MrModeltest v2. Program Distributed by the Author; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004. [Google Scholar]
- Keane, T.M.; Naughton, T.J.; McInerney, J.O. ModelGenerator: Amino Acid and Nucleotide Substitution Model Selection; National University of Ireland: Maynooth, Ireland, 2004. [Google Scholar]
- Dayhoff, M.O.; Schwartz, R.M. A Model of Evolutionary Change in Proteins. In Atlas of Protein Sequence and Structure; National Biomedical Research Foundation: Washington, DC, USA, 1978. [Google Scholar]
- Liò, P.; Goldman, N. Models of molecular evolution and phylogeny. Genome Res. 1998, 8, 1233–1244. [Google Scholar] [PubMed]
- Phillips, M.J.; Penny, D. The root of the mammalian tree inferred from whole mitochondrial genomes. Mol. Phylogenet. Evol. 2003, 28, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.A.; Inagaki, Y.; Hashimoto, T. RY-coding and non-homogeneous models can ameliorate the maximum-likelihood inferences from nucleotide sequence data with parallel compositional heterogeneity. Evolut. Bioinform. Online 2012, 8, 357. [Google Scholar] [CrossRef]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mamm. Protein Metab. 1969, 3, 121–132. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J.; Churchill, G.A. A Hidden Markov Model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 1996, 13, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. A mutation data matrix for transmembrane proteins. FEBS Lett. 1994, 339, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Posada, D.; Zardoya, R. MtArt: A new model of amino acid replacement for Arthropoda. Mol. Biol. Evol. 2007, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Waddell, P.J.; Martin, W.; Hasegawa, M. Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA. J. Mol. Evol. 2000, 50, 348–358. [Google Scholar] [PubMed]
- Cao, Y.; Adachi, J.; Janke, A.; Pääbo, S.; Hasegawa, M. Phylogenetic relationships among eutherian orders estimated from inferred sequences of mitochondrial proteins: Instability of a tree based on a single gene. J. Mol. Evol. 1994, 39, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Nickle, D.C.; Heath, L.; Jensen, M.A.; Gilbert, P.B.; Mullins, J.I.; Kosakovsky Pond, S.L. HIV-specific probabilistic models of protein evolution. PLoS ONE 2007, 2, e503. [Google Scholar] [CrossRef] [PubMed]
- Dimmic, M.W.; Rest, J.S.; Mindell, D.P.; Goldstein, R.A. rtREV: An amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J. Mol. Evol. 2002, 55, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Vingron, M. Modeling amino acid replacement. J. Comput. Biol. 2000, 7, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Phylogenetic models of rate heterogeneity: A high performance computing perspective. In Proceedings of the 20th International Parallel and Distributed Processing Symposium (IPDPS 2006), Rhodes Island, Greece, 25–29 April 2006.
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Douady, C.J.; Delsuc, F.; Boucher, Y.; Doolittle, W.F.; Douzery, E.J.P. Comparison of Bayesian and maximum likelihood bootstrap measures of phylogenetic reliability. Mol. Biol. Evol. 2003, 20, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Crandall, K.A. Phylogeny estimation and hypothesis testing using maximum likelihood. Annu. Rev. Ecol. Syst. 1997, 28, 437–466. [Google Scholar] [CrossRef]
- Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Biol. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Brown, J.K. Bootstrap hypothesis tests for evolutionary trees and other dendrograms. Proc. Natl. Acad. Sci. USA 1994, 91, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Shafer, G. A Mathematical Theory of Evidence; Princeton University Press: Princeton, NJ, USA, 1976; Volume 1. [Google Scholar]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Hastings, W.K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Altekar, G.; Dwarkadas, S.; Huelsenbeck, J.P.; Ronquist, F. Parallel metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 2004, 20, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Gatesy, J.; Springer, M.S. Phylogenetic analysis at deep timescales: Unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol. Phylogenet. Evol. 2014, 80, 231–266. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Liu, L.; Edwards, S.V.; Wu, S. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc. Natl. Acad. Sci. USA 2012, 109, 14942–14947. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Cummins, C.A.; McInerney, J.O. A method for inferring the rate of evolution of homologous characters that can potentially improve phylogenetic inference, resolve deep divergence and correct systematic biases. Syst. Biol. 2011, 60, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Frandsen, P.B.; Calcott, B.; Mayer, C.; Lanfear, R. Automatic selection of partitioning schemes for phylogenetic analyses using iterative k-means clustering of site rates. BMC Evolut. Biol. 2015, 15, 13. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Kainer, D.; Mayer, C.; Stamatakis, A. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evolut. Biol. 2014, 14, 82. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldn, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.A.; Ellis, J.T. Effects of nucleotide sequence alignment on phylogeny estimation: A case study of 18S rDNAs of Apicomplexa. Mol. Biol. Evol. 1997, 14, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Creevey, C.J.; Thompson, J.D.; Arendt, D.; Bork, P. AQUA: Automated quality improvement for multiple sequence alignments. Bioinformatics 2010, 26, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C. Recent evolutions of multiple sequence alignment algorithms. PLoS Comput. Biol. 2007, 3, e123. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.; Janies, D.; Wheeler, W. Multiple sequence alignment in phylogenetic analysis. Mol. Phylogenet. Evol. 2000, 16, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Thierry, J.C.; Poch, O. RASCAL: Rapid scanning and correction of multiple sequence alignments. Bioinformatics 2003, 19, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Blackburne, B.P.; Whelan, S. Measuring the distance between multiple sequence alignments. Bioinformatics 2012, 28, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.; Gowri-Shankar, V.; Higgs, P.G.; Rattray, M. A comprehensive analysis of mammalian mitochondrial genome base composition and improved phylogenetic methods. Mol. Biol. Evol. 2005, 22, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Kjer, K.M.; Honeycutt, R.L. Site specific rates of mitochondrial genomes and the phylogeny of eutheria. BMC Evolut. Biol. 2007, 7, 8. [Google Scholar] [CrossRef]
- Reyes, A.; Gissi, C.; Catzeflis, F.; Nevo, E.; Pesole, G.; Saccone, C. Congruent mammalian trees from mitochondrial and nuclear genes using Bayesian methods. Mol. Biol. Evol. 2004, 21, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Arnason, U.; Janke, A. Mitogenomic analyses of eutherian relationships. Cytogenet. Genome Res. 2002, 96, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Stanhope, M.J.; Madsen, O.; de Jong, W.W. Molecules consolidate the placental mammal tree. Trends Ecol. Evol. 2004, 19, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.C.; Creevey, C.J.; O’Connell, M.J. Mitochondrial data are not suitable for resolving placental mammal phylogeny. Mamm. Genome 2014, 25, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A. Molecular Phylogenetics. In Genomes, 2nd ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Hasegawa, M. Phylogeny and molecular evolution in primates. Jpn. J. Genet. 1990, 65, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-H.; Gouy, M.; Sharp, P.M.; O’Huigin, C.; Yang, Y.-W. Molecular phylogeny of Rodentia, Lagomorpha, Primates, Artiodactyla, and Carnivora and molecular clocks. Proc. Natl. Acad. Sci. USA 1990, 87, 6703–6707. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.H. Heterogeneity in the substitution process of amino acid sites of proteins coded for by mitochondrial DNA. J. Mol. Evol. 1992, 35, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Computational Molecular Evolution; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Mayrose, I.; Friedman, N.; Pupko, T. A Gamma mixture model better accounts for among site rate heterogeneity. Bioinformatics 2005, 21, ii151–ii158. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Gouy, M. Inferring pattern and process: Maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Mol. Biol. Evol. 1998, 15, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Tourasse, N.; Gouy, M. A nonhyperthermophilic common ancestor to extant life forms. Science 1999, 283, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Roberts, D. On the use of nucleic acid sequences to infer early branchings in the tree of life. Mol. Biol. Evol. 1995, 12, 451–458. [Google Scholar] [PubMed]
- Rannala, B. Identifiability of parameters in MCMC Bayesian inference of phylogeny. Syst. Biol. 2002, 51, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI. Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar]
- Newton, M.A.; Raftery, A.E. Approximate Bayesian inference with the weighted likelihood bootstrap. J. R. Statist. Soc. Ser. B Methodol. 1994, 58, 3–48. [Google Scholar]
- Kass, R.E.; Raftery, A.E. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Lopes, H.F.; West, M. Bayesian model assessment in factor analysis. Stat. Sin. 2004, 14, 41–68. [Google Scholar]
- Gelman, A.; Carlin, J.B.; Stern, H.S.; Dunson, D.B.; Vehtari, A.; Rubin, D.B. Bayesian Data Analysis; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Teeling, E.C.; Hedges, S.B. Making the impossible possible: Rooting the tree of placental mammals. Mol. Biol. Evol. 2013, 30, 1999–2000. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Pringle, T.H.; Crider, T.A.; Springer, M.S.; Miller, W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007, 17, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.B.; Allard, M.W.; Green, E.D. Confirming the phylogeny of mammals by use of large comparative sequence data sets. Mol. Biol. Evol. 2008, 25, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Romiguier, J.; Ranwez, V.; Delsuc, F.; Galtier, N.; Douzery, E.J.P. Less is more in mammalian phylogenomics: AT-rich genes minimize tree conflicts and unravel the root of placental mammals. Mol. Biol. Evol. 2013, 30, 2134–2144. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J.A.A.; Wilgenbusch, J.C.; Warren, D.L.; Swofford, D.L. AWTY (are we there yet?): A system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 2008, 24, 581–583. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moran, R.J.; Morgan, C.C.; O'Connell, M.J. A Guide to Phylogenetic Reconstruction Using Heterogeneous Models—A Case Study from the Root of the Placental Mammal Tree. Computation 2015, 3, 177-196. https://doi.org/10.3390/computation3020177
Moran RJ, Morgan CC, O'Connell MJ. A Guide to Phylogenetic Reconstruction Using Heterogeneous Models—A Case Study from the Root of the Placental Mammal Tree. Computation. 2015; 3(2):177-196. https://doi.org/10.3390/computation3020177
Chicago/Turabian StyleMoran, Raymond J., Claire C. Morgan, and Mary J. O'Connell. 2015. "A Guide to Phylogenetic Reconstruction Using Heterogeneous Models—A Case Study from the Root of the Placental Mammal Tree" Computation 3, no. 2: 177-196. https://doi.org/10.3390/computation3020177
APA StyleMoran, R. J., Morgan, C. C., & O'Connell, M. J. (2015). A Guide to Phylogenetic Reconstruction Using Heterogeneous Models—A Case Study from the Root of the Placental Mammal Tree. Computation, 3(2), 177-196. https://doi.org/10.3390/computation3020177