Enhanced Osteogenic Differentiation of Human Fetal Cartilage Rudiment Cells on Graphene Oxide-PLGA Hybrid Microparticles



Abstract
:
1. Introduction
2. Results and Discussion
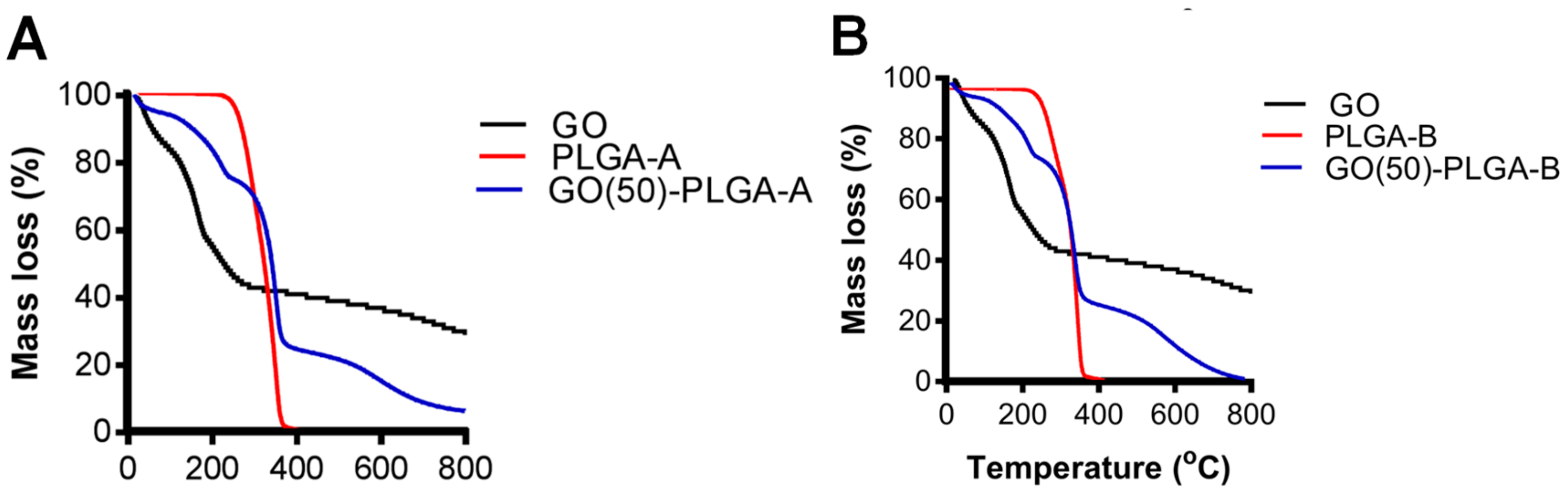
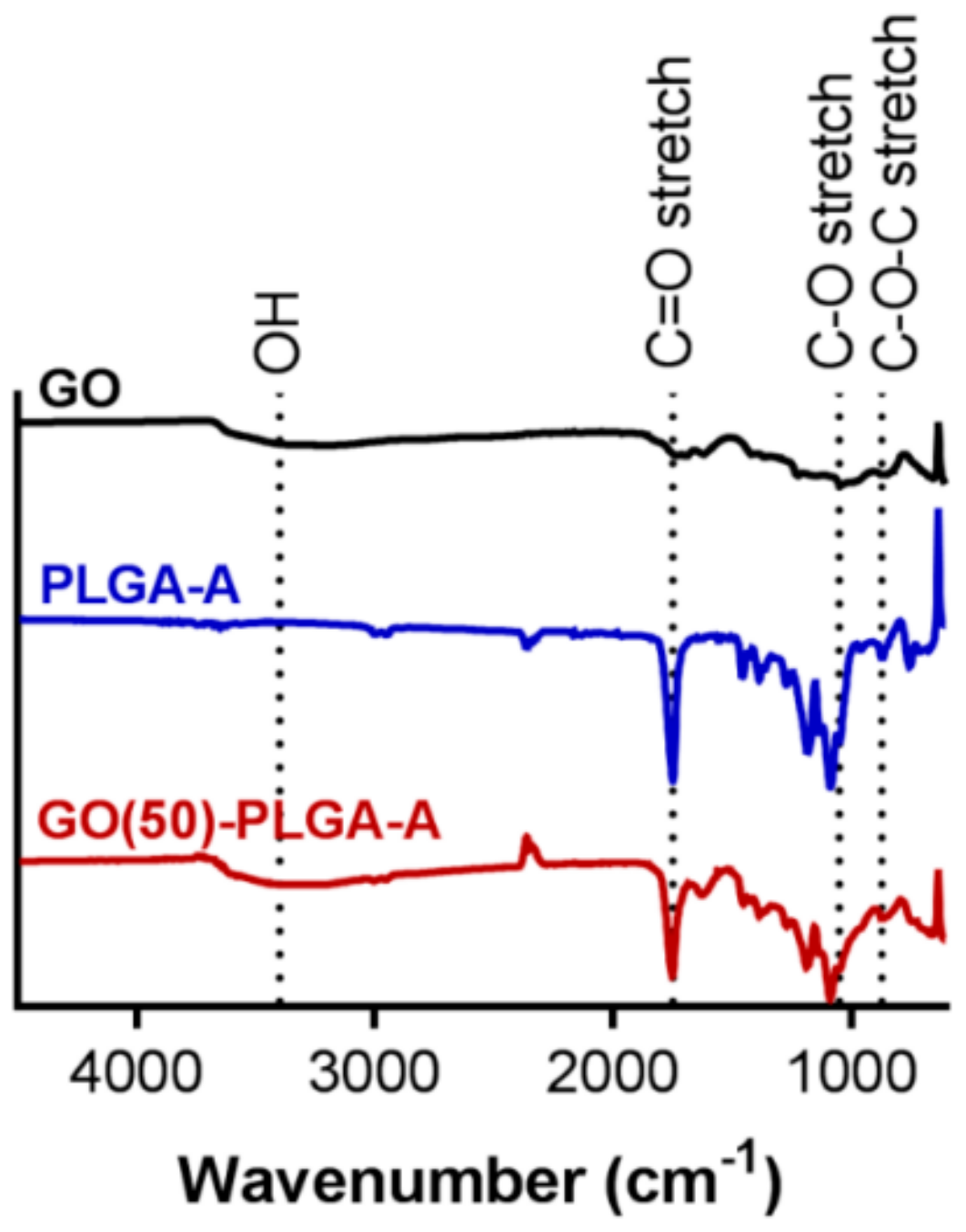
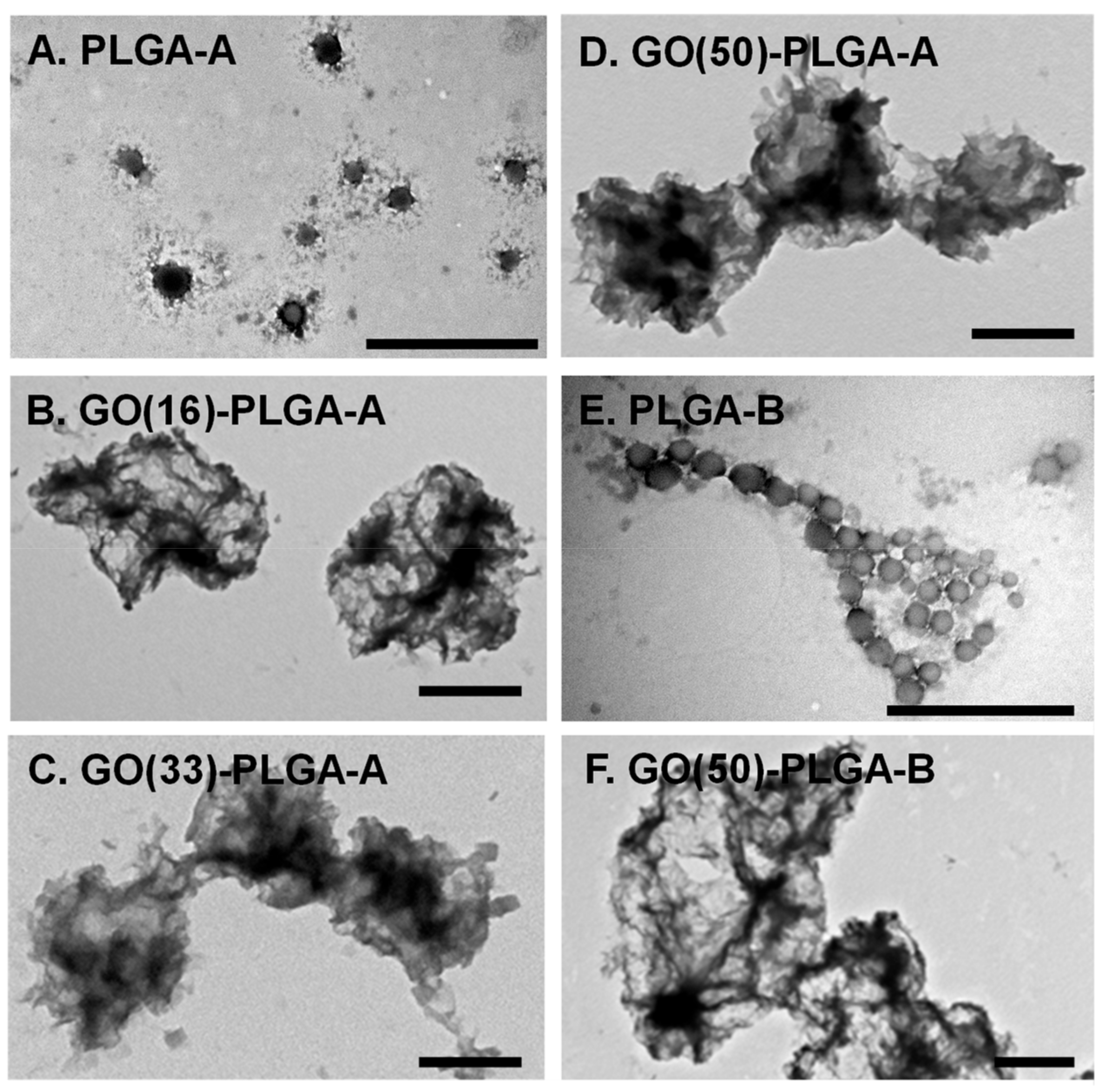
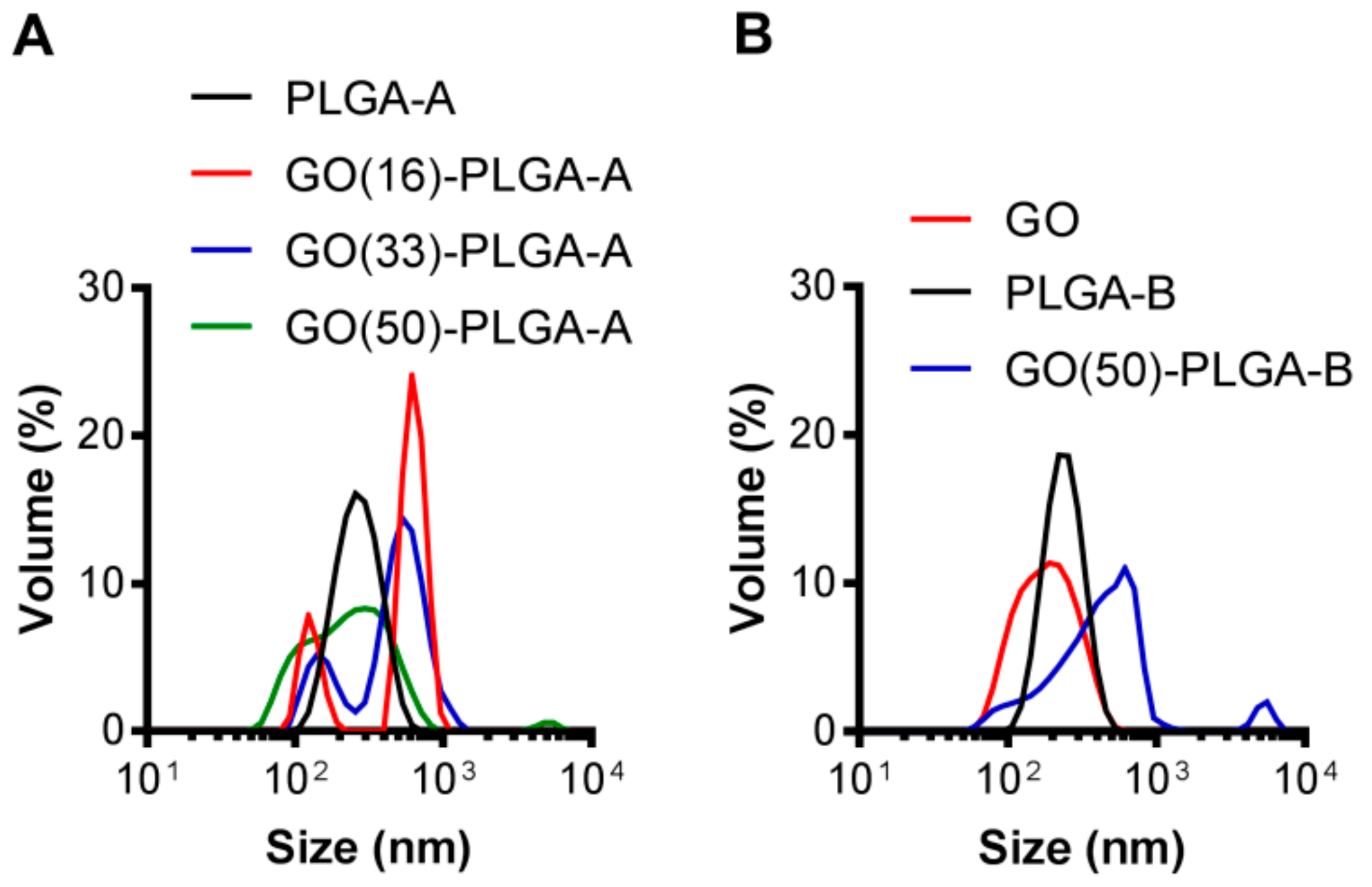
2.1. Characterization of GO-PLGA Microparticles
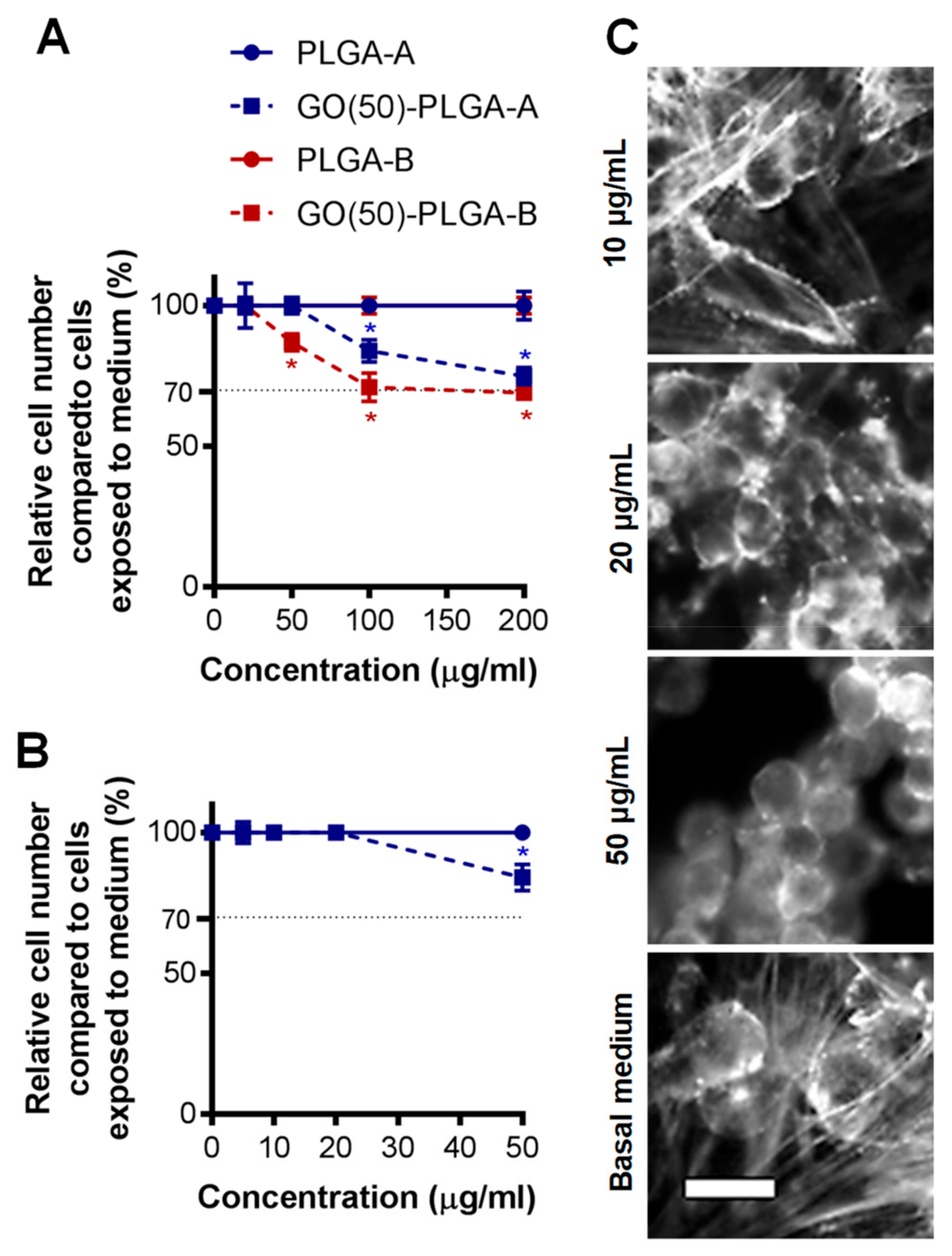
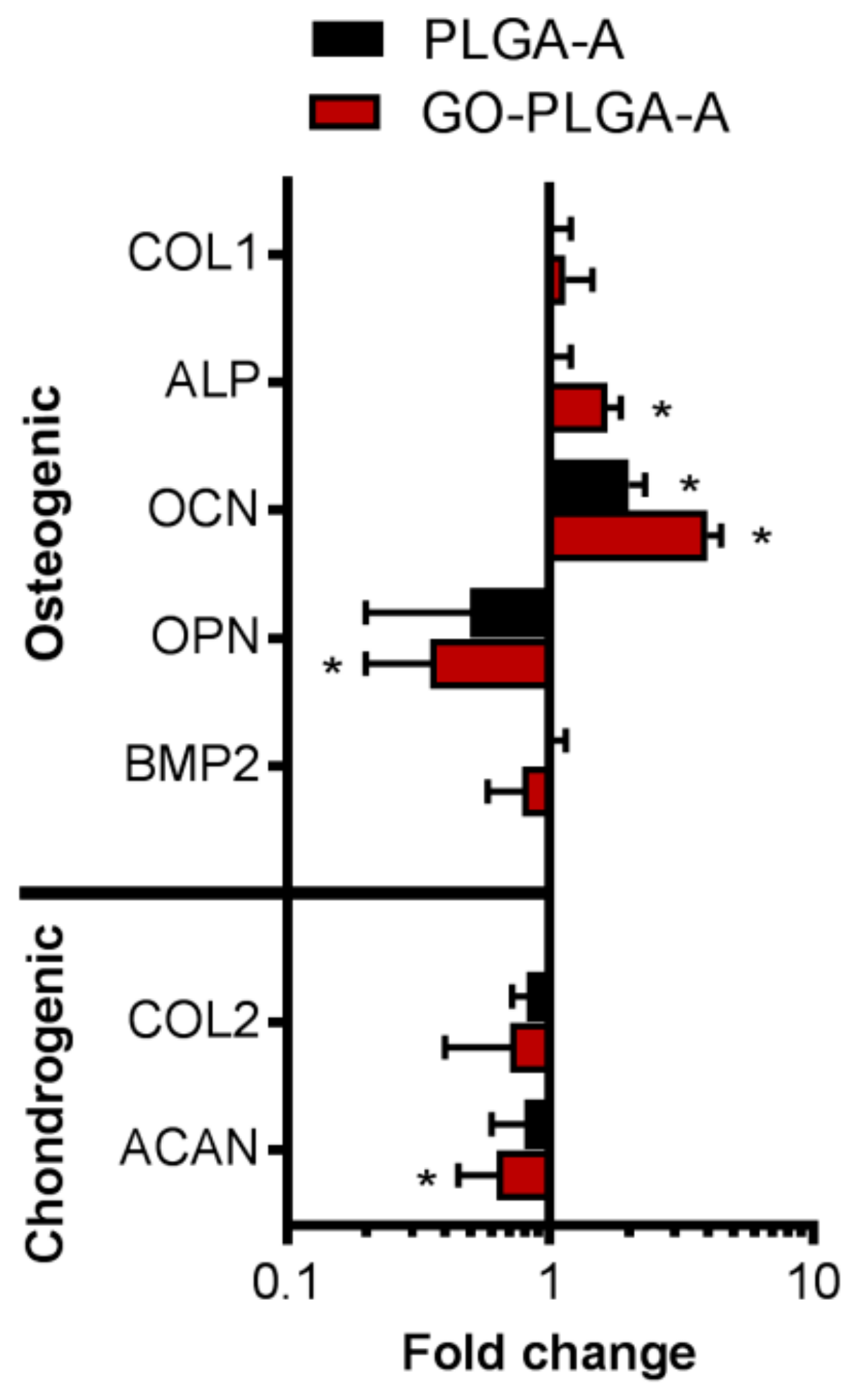
2.2. Cellular Interactions with GO-PLGA Microparticles
3. Materials and Methods
3.1. Materials
3.2. Preparation of Graphite Oxide
3.3. Preparation of GO-PLGA Hybrid Particles
3.4. Characterization of Particles
3.5. Culture of Cells
3.6. Cell Viability
3.7. qPCR
3.8. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Champa Jayasuriya, A.; Ebraheim, N. Evaluation of bone matrix and demineralized bone matrix incorporated PLGA matrices for bone repair. J. Mater. Sci. Mater. Med. 2009, 20, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Hu, Y.; Zhang, C.; Li, X.; Lv, R.; Qin, L.; Zhu, R. Cartilage regeneration using mesenchymal stem cells and a PLGA–gelatin/chondroitin/hyaluronate hybrid scaffold. Biomaterials 2006, 27, 4573–4580. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.H.; Han, D.-W.; Matsumura, K.; Tsutsumi, S.; Hyon, S.-H. The behavior of vascular smooth muscle cells and platelets onto epigallocatechin gallate-releasing poly(l-lactide-co-ε-caprolactone) as stent-coating materials. Biomaterials 2008, 29, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An Overview of Poly(lactic-co-glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering. Int J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Herrera, C.E.; Singh, B.; Singh, S.; Singh, R.K.; Kumar, R. Graphene oxide: An efficient material and recent approach for biotechnological and biomedical applications. Mat. Sci. Eng. C 2018, 86, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Pang, D.W.P.; Hwang, S.M.; Tuan, H.Y.; Hu, Y.C. A graphene-based platform for induced pluripotent stem cells culture and differentiation. Biomaterials 2012, 33, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Yoon, O.; Sohn, I.; Kim, D.; Lee, N.-E. Enhancement of thermomechanical properties of poly(D,L-lactic-co-glycolic acid) and graphene oxide composite films for scaffolds. Macromol. Res. 2012, 20, 789–794. [Google Scholar] [CrossRef]
- Luo, Y.; Shen, H.; Fang, Y.; Cao, Y.; Huang, J.; Zhang, M.; Dai, J.; Shi, X.; Zhang, Z. Enhanced proliferation and osteogenic differentiation of mesenchymal stem cells on graphene oxide-incorporated electrospun poly(lactic-co-glycolic acid) nanofibrous mats. ACS Appl. Mater. Interfaces 2015, 7, 6331–6339. [Google Scholar] [CrossRef]
- Shao, W.; He, J.; Wang, Q.; Cui, S.; Ding, B. Biomineralized Poly(l-lactic-co-glycolic acid)/Graphene Oxide/Tussah Silk Fibroin Nanofiber Scaffolds with Multiple Orthogonal Layers Enhance Osteoblastic Differentiation of Mesenchymal Stem Cells. ACS Biomater. Sci. Eng. 2017, 3, 1370–1380. [Google Scholar] [CrossRef]
- Fu, C.; Bai, H.; Hu, Q.; Gao, T.; Bai, Y. Enhanced proliferation and osteogenic differentiation of MC3T3-E1 pre-osteoblasts on graphene oxide-impregnated PLGA-gelatin nanocomposite fibrous membranes. RSC Advances 2017, 7, 8886–8897. [Google Scholar] [CrossRef]
- Fu, C.; Yang, X.; Tan, S.; Song, L. Enhancing cell proliferation and osteogenic differentiation of MC3T3-E1 pre-osteoblasts by BMP-2 delivery in graphene oxide-incorporated PLGA/HA biodegradable microcarriers. Sci. Rep. 2017, 7, 12549. [Google Scholar] [CrossRef] [PubMed]
- Astete, C.E.; Sabliov, C.M. Synthesis of Poly(D,L-Lactide-Co-Glycolide) Nanoparticles with entrapped magnetite by emulsion evaporation method. Particul. Sci. Technol. 2006, 24, 321–328. [Google Scholar] [CrossRef]
- Astete, C.E.; Sabliov, C.M. Synthesis and characterization of PLGA nanoparticles. J. Biomater. Sci. Polym. Ed. 2006, 17, 247–289. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, R.; Seperhi, N.; Manoochehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Bouchemal, K. Methods for the preparation and manufacture of polymeric nanoparticles. Pharm. Res. 2009, 26, 1025–1058. [Google Scholar] [CrossRef]
- Pickering, S.U. CXCVI-Emulsions. J. Chem. Soc. Faraday Trans. 1907, 91, 2001–2021. [Google Scholar]
- Binks, B.P. Particles as surfactants—Similarities and differences. Curr. Opin. Colloid. Interface Sci. 2002, 7, 21–41. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces, 2nd ed.; Academic Press: London, UK, 1992. [Google Scholar]
- Hu, Y.; Zou, S.; Yang, Y.; Tong, Z.; Wang, C. Facile fabrication of macroporous PLGA microspheres via double-Pickering emulsion templates. Macromol. Chem. Phys. 2015, 216, 714–720. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, C.; Liu, H.; Zou, S.; Tong, Z. Facile fabrication of biocompatible PLGA drug-carrying microspheres by O/W pickering emulsions. Colloids Surf. B Biointerfaces 2012, 91, 97–105. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, C.; Zou, S.; Liu, H.; Tong, Z. Fe2O3 nanoparticles as particulate emulsifier: Preparation of magnetic and biocompatible PLGA microcapsules. Colloids Surf. A 2011, 392, 116–123. [Google Scholar] [CrossRef]
- Iwamoto, T.; Terada, T.; Kogai, Y.; Okada, M.; Fujii, S.; Furuzono, T. Development of microspheres covered with hydroyapatite nanocrystals as cell scaffold for angiogenesis. Funct Mater. Lett 2012, 5, 1260010. [Google Scholar] [CrossRef]
- Kim, J.; Cote, L.J.; Kim, F.; Yuan, W.; Shull, K.R.; Huang, J. Graphene oxide sheets at interfaces. J. Am. Chem. Soc. 2010, 132, 8180–8186. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Cote, L.J.; Tung, V.C.; Tan, A.T.L.; Goins, P.E.; Wu, J.; Huang, J. Graphene oxide nanocolloids. J. Am. Chem. Soc. 2010, 132, 17667–17669. [Google Scholar] [CrossRef] [PubMed]
- Thickett, S.C.; Zetterlund, P.B. Functionalization of graphene oxide for the production of novel graphene-based polymeric and colloidal materials. Curr. Org. Chem. 2013, 17, 956–974. [Google Scholar] [CrossRef]
- Creighton, M.A.; Ohata, Y.; Miyawaki, J.; Bose, A.; Hurt, R.H. Two-dimensional materials as emulsion stabilizers: Interfacial thermodynamics and molecular barrier properties. Langmuir 2014, 30, 3687–3696. [Google Scholar] [CrossRef] [PubMed]
- Che Man, S.H.; Mohd Yusof, N.Y.; Whittaker, M.R.; Thickett, S.C.; Zetterlund, P.B. Influence of monomer type on miniemulsion polymerization systems stabilized by graphene oxide as sole surfactant. J. Polym. Sci. A Polym. Chem. 2013, 51, 5153–5162. [Google Scholar] [CrossRef]
- Che Man, S.H.; Thickett, S.C.; Whittaker, M.R.; Zetterlund, P.B. Synthesis of polystyrene nanoparticles “armoured” with nanodimensional graphene oxide sheets by miniemulsion polymerization. J. Polym. Sci. A Polym. Chem. 2013, 51, 47–58. [Google Scholar] [CrossRef]
- Thickett, S.C.; Wood, N.; Ng, Y.H.; Zetterlund, P.B. Hollow hybrid polymer-graphene oxide nanoparticles via Pickering miniemulsion polymerization. Nanoscale 2014, 6, 8590–8594. [Google Scholar] [CrossRef]
- Wu, J.; Ma, G.H. Recent studies of pickering emulsions: Particles make the difference. Small 2016, 12, 4633–4648. [Google Scholar] [CrossRef]
- Liang, C.; Luo, Y.; Yang, G.; Xia, D.; Liu, L.; Zhang, X.; Wang, H. Graphene oxide hybridized nHAC/PLGA scaffolds facilitate the proliferation of MC3T3-E1 cells. Nanoscale Res. Lett. 2018, 13, 15. [Google Scholar] [CrossRef]
- Thickett, S.C.; Zetterlund, P.B. Graphene oxide (GO) nanosheets as oil-in-water emulsion stabilizers: Influence of oil phase polarity. J. Colloid Interface Sci. 2015, 442, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Tsoi, B.M.; Farrugia, B.L.; Simon Ting, S.R.; Baker, S.; Wiesmann, W.P.; Whitelock, J.M. Synthesis and characterization of water soluble biomimetic chitosans for bone and cartilage tissue regeneration. J. Mater. Chem. B 2014, 2, 6517–6526. [Google Scholar] [CrossRef]
- Huang, W.; Carlsen, B.; Rudkin, G.; Berry, M.; Ishida, K.; Yamaguchi, D.T.; Miller, T.A. Osteopontin is a negative regulator of proliferation and differentiation in MC3T3-E1 pre-osteoblastic cells. Bone 2004, 34, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Long, M.W. Osteogenesis and bone-marrow-derived cells. Blood Cells Mol. Dis. 2001, 27, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, J.; Li, C.; Yu, Y. Role of bone morphogenetic protein-2 in osteogenic differentiation of mesenchymal stem cells. Mol. Med. Rep. 2015, 12, 4230–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.W.; LaChaud, G.; Shen, J.; Asatrian, G.; Nguyen, V.; Zhang, X.; Ting, K.; Soo, C. A review of the clinical side effects of bone morphogenetic protein-2. Tissue Eng. Part B Rev. 2016, 22, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.Y.; Shahin, K.; Lord, M.S.; Melrose, J.; Doran, P.M.; Whitelock, J.M. The cartilage matrix molecule components produced by human foetal cartilage rudiment cells within scaffolds and the role of exogenous growth factors. Biomaterials 2012, 33, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Czekanska, E.M.; Stoddart, M.J.; Richards, R.G.; Hayes, J.S. In search of an osteoblast cell model for in vitro research. Eur. Cell. Mater. 2012, 24, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, F.; Handschel, J. Effects of dexamethasone, ascorbic acid and beta-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res. Ther. 2013, 4, 117. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLGA | PLGA Lactide: Glycolide Ratio | GO wt % | TEM Diameter (nm) | DLS Diameter/PDI (nm) | Zeta Potential (mV) |
|---|---|---|---|---|---|
| A | 75:25 | 0 | 170 ± 50 | 236/0.12 | −32.4 |
| 16 | 1100 ± 280 | 734/0.64 | −42.2 | ||
| 33 | 1220 ± 340 | 506/0.50 | −48.2 | ||
| 50 | 1860 ± 350 | 270/0.27 | −46.6 | ||
| B | 65:35 | 0 | 125 ± 20 | 219/0.06 | −25.3 |
| 50 | 2350 ± 620 | 456/0.52 | −40.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thickett, S.C.; Hamilton, E.; Yogeswaran, G.; Zetterlund, P.B.; Farrugia, B.L.; Lord, M.S. Enhanced Osteogenic Differentiation of Human Fetal Cartilage Rudiment Cells on Graphene Oxide-PLGA Hybrid Microparticles. J. Funct. Biomater. 2019, 10, 33. https://doi.org/10.3390/jfb10030033
Thickett SC, Hamilton E, Yogeswaran G, Zetterlund PB, Farrugia BL, Lord MS. Enhanced Osteogenic Differentiation of Human Fetal Cartilage Rudiment Cells on Graphene Oxide-PLGA Hybrid Microparticles. Journal of Functional Biomaterials. 2019; 10(3):33. https://doi.org/10.3390/jfb10030033
Chicago/Turabian StyleThickett, Stuart C., Ella Hamilton, Gokulan Yogeswaran, Per B. Zetterlund, Brooke L. Farrugia, and Megan S. Lord. 2019. "Enhanced Osteogenic Differentiation of Human Fetal Cartilage Rudiment Cells on Graphene Oxide-PLGA Hybrid Microparticles" Journal of Functional Biomaterials 10, no. 3: 33. https://doi.org/10.3390/jfb10030033
APA StyleThickett, S. C., Hamilton, E., Yogeswaran, G., Zetterlund, P. B., Farrugia, B. L., & Lord, M. S. (2019). Enhanced Osteogenic Differentiation of Human Fetal Cartilage Rudiment Cells on Graphene Oxide-PLGA Hybrid Microparticles. Journal of Functional Biomaterials, 10(3), 33. https://doi.org/10.3390/jfb10030033