1. Introduction
Collagen and its denatured derivatives, such as gelatin, are used for many biomedical applications including wound dressings, graft coatings, suture material, drug release, and tissue engineering [
1,
2,
3]. These materials, despite their wide use, possess several disadvantages associated with their origin and method of production. Collagen and its derivatives are sourced from animal tissue, which brings the risk of allergic reactions, as well as the transmission of pathogens like viruses and prions. The age and physiological state of the tissue of origin brings further variability to the final collagen product. Gelatin is produced from denatured collagen by acidic or alkaline extraction [
4]. This commercial process delivers wide variation in batch-to-batch product quality, which can represent a challenge for medical applications [
5].
The production of recombinant collagen-based materials allows the circumvention of the above-mentioned drawbacks associated with animal-derived materials. Therefore, many groups have aimed to produce recombinant collagen-based materials in various organisms. Recombinant collagen I has already been produced in transgenic mice, tobacco plants, silk worms, insect cell lines, and mammalian cell lines [
5]. A production in simple microorganisms like yeast and bacteria would make a recombinant collagen mimetic protein more attractive in terms of achievable yields and production costs [
6]. However, the complexity of helical collagens makes it challenging to integrate them into a microbial recombinant system [
7].
Type I collagen, the most commonly used collagen type in biomedical applications, is a heterotrimer consisting of two alpha 1 and one alpha 2 chains. The central helical domain of this molecule consists of repetitive Gly-Xaa-Yaa sequences, with Xaa and Yaa being proline and hydroxyproline respectively. The thermal stability of collagen is dependent on the hydroxylation of proline with the enzyme prolyl-4-hydroxylase (P4H), which is present in sufficient amounts only in mammalian cell expression systems. In order to produce correctly-folded collagen in most expression systems like plants, insect cells, bacteria, and yeasts, it is therefore necessary to also clone both subunits of the P4H enzyme. The group of Myllyharju accomplished the successful cloning, production and expression of human collagen types I, II and III in the yeast
Pichia pastoris (
P. pastoris) [
8]. To achieve this, the enzyme P4H was also cloned into the yeast strains. The authors could show that the recombinant collagens are triple-helical, but as a result, cannot be exported from the yeast cell [
9]. The recombinant collagen from
P. pastoris has been successfully commercialized [
10].
In this study, we aimed to produce a recombinant collagen fragment which can be secreted from the cell into the media and be produced in simple microorganisms using animal component-free media. Also, we wanted to circumvent the production of P4H and the cloning of its associated subunits, but still obtain a gelling product. The cloning of collagen fragments into
Escherichia coli (
E. coli) or yeast without P4H results in the production of random-coiled collagens, which resemble gelatin in their structure. Therefore, in this work we present the production of a non-hydroxylated gelatin-mimetic protein (GelMP) in
P. pastoris. We selected this yeast, as it is known that yeasts are able to convert repetitive genes better than
E. coli and because we were aiming for extracellular production [
11]. Our goal was to design a cost-efficient, simple and quick process starting from the cloning procedure to the final purification of the material. In order to simplify the process, we cloned a single gene coding for a 400 amino acid segment from the helical region of the human collagen I alpha1 chain and included repetitive prolyl-glycyl-prolyl (PGP)-sequences flanking on both sides of the collagen sequence. Such artificial repeats are inspired by the Gly-Xaa-Yaa structure of collagen and are known to possess thermal trimerization capacity under specific circumstances [
12]. The group of Werten et al. demonstrated that gels from these telechelic triblock (ABA) protein polymers are formed on long incubation times at appropriate concentrations, in accordance with studies performed on synthetic (Pro-Gly-Pro)
9 peptides (PGP) [
13]. In our study, we combined a sequence from the helical region of collagen I to introduce bioactivity and PGP repeats to incorporate gelling behavior without hydroxylation by P4H.
3. Materials and Methods
3.1. Vector Construction
The recombinant GelMP consists of two segments: a 400 amino acid sequence sourced from the alpha chain of type I human collagen (amino acid sequence 541 to 940), and (Pro-Gly-Pro)
9 (PGP) repeats flanking this sequence on each side (
Figure 7). Cleavage sites for SpeI and PacI were inserted between the collagen sequence and the PGP repeats. In addition, an EcoRI and NotI site were inserted at the 5′ and 3′-ends, respectively. The GelMP designed in this manner was codon-optimized and produced by a gene synthesis service (Thermo Fischer, Waltham, MA, USA). The synthetic gene was delivered in a pMK-RQ vector construct. Utilizing the added restrictions sites EcoRI and NotI, the construct was cloned in frame into the cloning/expression region of pPIC9K (InvitrogenTM, Carslbad, CA, USA). The sequence was verified by automated DNA sequencing (Eurofins Genomics, Ebersberg, Germany).
3.2. Organism and Transformation
The expression vector pPIC9K-GelMP was linearized with SalI and used to transform competent GS115 (his4) cells according to the protocol of Lin-Cereghino [
20]. Transformants were spread on minimal dextrose plates and incubated for 72 h at 30 °C. Clones were screened using colony PCR and potential Mut+ transformants were identified. Recombinants were then tested for multiple insertion of the gene-of-interest on geneticin-containing yeast extract–peptone–dextrose plates (range of G418: 0.5 to 4 mg·mL
−1). Selected clones growing on the 4 mg mL
−1 G418 plate were screened for protein expression in shake flasks as described by ref. [
21]. The highest producer was selected for further work and transferred to a bioreactor for scale up of production.
3.3. Fermentative Production of GelMP in P. pastoris
A 30 L stainless steel bench top bioreactor (Biostat® Cplus, Sartorius, Göttingen, Germany) was equipped with a dissolved oxygen electrode (InPro6820, Mettler-Toledo, Columbus, OH, USA), a pH-electrode (EasyFerm Plus K8 200, Hamilton, Martinsried, Germany), a methanol sensor system (Raven Bio-Tech Inc., Canada), and a peristaltic pump (Ismatec Reglo ICC, Wertheim Germany). A Biostat® Cplus control unit (Sartorius, Göttingen, Germany) was connected to the bioreactor for fermentation control. The methanol sensor system was connected to a single-board computer for data processing. Bioreactor operation data was logged using ProfiSignal (Delphin Technology, Germany). A 1 mL aliquot of a cryo culture (OD600 = 50) was thawed and placed in 100 mL medium (per liter: 25 g (NaPO3)6, 0.93 g CaSO4, 18.2 g K2SO4, 14.9 g MgSO4·7H2O, 9 g (NH4)2SO4, 40 g glycerol and 0.435% PTM1 (per liter: 6 g CuSO4·5H2O, 0.08 g NaI, 3 g MnSO4·H2O, 0.2 g Na2MoO4·2H2O, 0.02 g H3BO3, 0.5 g CoCl2, 20 g ZnCl2, 65 g FeSO4·7H2O, 0.2 g Biotin, 5 mL H2SO4). The pre-culture was cultivated until the cells reached OD600 = 10–15. The bioreactor was filled with 10 L fermentation medium (per liter: 25 g (NaPO3)6, 0.93 g CaSO4, 18.2 g K2SO4, 14.9 g MgSO4·7H2O, 9 g (NH4)2SO4, 120 g glycerol and 0.435% PTM1) and was inoculated with cells from the pre-culture to an OD600 = 0.1. The operation parameters were pH 5.0, 30 °C, dissolved oxygen (DO) level of 30% air saturation and a gas flow rate of 2L·min−1. The DO level was controlled using the stirrer cascade system of the BIOSTAT® Cplus and by supplying pure oxygen if necessary. After depletion of glycerol, the methanol feed (100% methanol with 12 mL·L−1 PTM1) was set up to 0.2% v/v methanol. The cultivation ended after 80 h.
3.4. Purification of GelMP from the P. pastoris Culture Supernatant
The supernatant was harvested after 80 h of cultivation by centrifugation (5000× g, 30 min, 4 °C) and stored at −20 °C until further processing. GelMP was purified directly from the fermentation broth using an ÄKTA Pure system (GE Healthcare, Chicago, IL, USA). Then 15 mL of supernatant was loaded on a Sephadex G-25 column (HiPrep™ 26/10 Desalting, GE Healthcare) previously equilibrated with PBS puffer (pH 7.4). GelMP protein was eluted with PBS buffer at a flow rate of 5 mL min−1 for 40 min monitoring UV-absorption and conductivity. The peaks eluted from the column were analyzed by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Fractions of 8 mL were collected and pooled for lyophilization.
A polishing step for the removal of potential endotoxins was also introduced. The target protein was dissolved in Tris-HCl (pH 7.5, 20 mM) to a concentration of 1 mg/mL and was passed through an anion exchange membrane in a flow-through mode (Sartobind® pico, membrane area: 2.9 cm−2, membrane volume: 0.08 mL, Sartorius, Göttingen, Germany). The endotoxin concentration after the polishing step was measured with an Endosafe® nexgen-PTS™ system (Charles River, Wilmington, MA, USA).
3.5. Bicinchoninic Acid Protein Assay
For the determination of protein concentration, a bicinchoninic acid (BCA) assay (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific, Waltham, MA, USA) was performed. Native collagen I from human placenta (Santa Cruz Biotech., Dallas, TX, USA) was used as a reference.
3.6. SDS PAGE and Western Blot Analysis
Protein samples were analyzed with SDS-PAGE using 10% Tris-HCl gels and a Mini Protean Tetra Cell System (BioRad, USA). The culture supernatant was mixed 1:1 with Laemmli buffer (2×) and 10 µL were loaded onto the gel. A Precision Plus Protein Standard (BioRad, Hercules, CA, USA) was used for the determination of molecular weight. Proteins were visualized with Coomassie Brilliant Blue G 250 staining. SDS-PAGE protein bands were blotted onto a PVDF membrane [
22], which was then subjected to Western blot analysis using the Western Breeze Immunodetection Kit (Invitrogen, USA) according to the manufacturer’s instructions and using a collagen-I-antibody (Acris Antibodies, Nr. R1038, Herford, Germany). N-terminal sequencing was performed in the Helmholtz Zentrum (HZI) Braunschweig.
3.7. Circular Dichroism Spectroscopy
Circular dichroism (CD)-spectra were recorded using a spectropolarimeter J-815 (Jasco, Pfungstadt, Germany). The purified GelMP and gelatin A (Sigma Aldrich, St. Louis, MO, USA) were dissolved in PBS buffer at a concentration of 1 mg/mL. The human collagen I (Biotechnology Inc., USA) was dissolved in acetic acid (1 mg/mL) and was also treated at 90 °C for 10 min for thermal denaturation. Test solutions were placed in a cuvette with a 1 mm path length and spectra were recorded in the 190–240 nm region. The spectra were obtained with a scanning speed of 50 nm/min at a resolution of 0.5 nm. Melting curves from 10 to 60 °C were derived by increasing the temperature in steps of 10 °C. Measurements were performed 5 times and the averaged scan values were plotted.
3.8. Label-Free Differential Scanning Fluorimetry
Thermostability was measured by nano differential scanning fluorimetry (DSF) on a Tycho NT 6 instrument (NanoTemper Technologies, Munich, Germany) with a back-reflection aggregation detection at a range from 20–95 °C and with a heating rate of 30 °C/min. Protein unfolding was followed by tryptophan/tyrosine fluorescence intensity at 330 and 350 nm in various buffers covering pH 5–8.5. The apparent melting temperature (Tm) was determined by detecting the maximum of the first derivative of the fluorescence ratios (F350/F330) after fitting experimental data with a polynomial function.
3.9. Rheological Characterization
The gel formation of a 2.5 mM GelMP solution in PBS (pH = 7.4) was investigated at 15 °C using a MCR 302 Modular Rheometer, Anton Paar, Austria) equipped with a plate-plate geometry (20 mm diameter, 1 mm gap size). The trimerization was recorded with a time sweep oscillatory test under constant strain amplitude of 0.1% and at a constant frequency of 1 Hz for a period of 20 h. A solvent trap was used to prevent sample loss by evaporation.
3.10. Cell Culture and Proliferation
Cell proliferation on GelMP coatings was assessed by using a culture of human adipose-derived mesenchymal stem cells (hAD-MSC). hAD-MSCs were expanded in alpha-MEM medium (Thermo Fisher Scientific, Braunschweig, Germany) containing 1 g L−1 glucose, 2 mM L-glutamine, 10% human serum (CC-pro, Oberdorla, Germany) and 50 µg·mL−1 gentamicin (Biochrom, Berlin, Germany), harvested by accutase treatment (Sigma Aldrich, St. Louis, MO, USA). Experiments were performed with hAD-MSCs of passages two to twelve.
3.11. Cell Adhesion Experiments
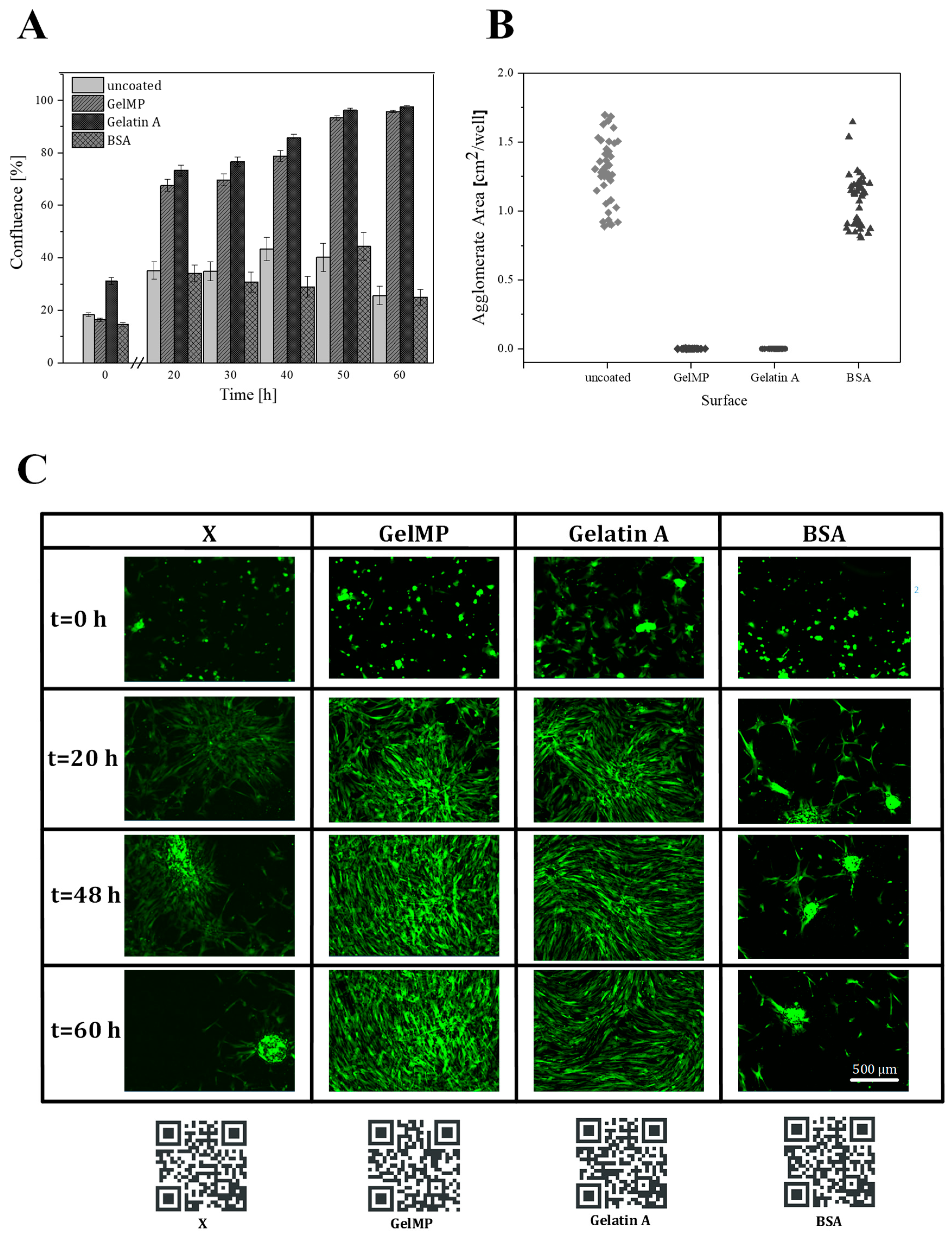
The lyophilized GelMP was dissolved in PBS-buffer (pH 7.4) at a concentration of 2 mg mL−1. Then 400 µL of this GelMP solution was added in each well and incubated at 4 °C for 12 h to allow protein adsorption to the well surfaces. Next, wells were rinsed with several milliliters of PBS buffer to remove excess protein. As control for the adhesion experiment, a set of wells were coated with bovine serum albumin (BSA) and gelatin (Sigma Aldrich, gelatin type A, St. Louis, MO, USA) with the same concentration and using the same procedure as for GelMP.
For adhesion experiments, hAD-MSCs were seeded onto a coated 48-well-plate (Eppendorf, Germany) at a density of 15,000 cells/cm2 with 300 µL medium per well. After seeding, cells were cultivated for 60 h in alpha-MEM containing 10% fetal bovine serum (FBS) and 50 µg mL−1 gentamicin in a humidified atmosphere containing 5% CO2 and 21% O2 at 37 °C. During the cultivation, microscopic pictures were taken every 2 h (four fields imaged per well) using an IncuCyte® Live-Cell Imaging System (Sartorius, Göttingen, Germany). Data were analyzed using the IncuCyte® Confluence software (IncuCyte® S3 2018 A), which quantified cell surface area coverage as confluence values.
3.12. Biocompatibility of GelMP by a Quantitative Assay of Caspase-3/7 Kinetic Activation
The hAD-MSCs were seeded in 48 well-plates at a density of 15,000 cells/well in 300 µL culture medium (alpha-MEM containing 10% FBS, 50 µg mL−1 gentamicin, and 0.5 µM IncuCyte® Caspase-3/7 reagent). The medium was previously mixed with GelMP, gelatin and BSA, resulting in a final concentration of 5 mg·mL−1. A quantitative analysis of apoptosis of hAD-MSCs over time was performed during the cultivation in treated medium (GelMP, BSA, gelatin) and with regular cell culture medium serving as a control. Once treated, the cells were immediately placed inside the IncuCyte® Live-Cell Analysis System (Sartorius Stedim Biotech GmbH, Göttingen, Germany) with a 10× objective in a standard cell culture incubator and both phase-contrast and fluorescent images were collected every 2 h.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






