
A Review on Phyto-Therapeutic Approaches in Alzheimer’s Disease





Abstract
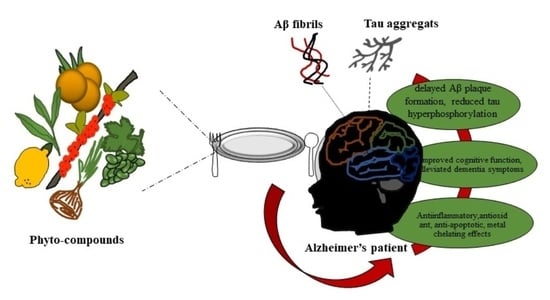
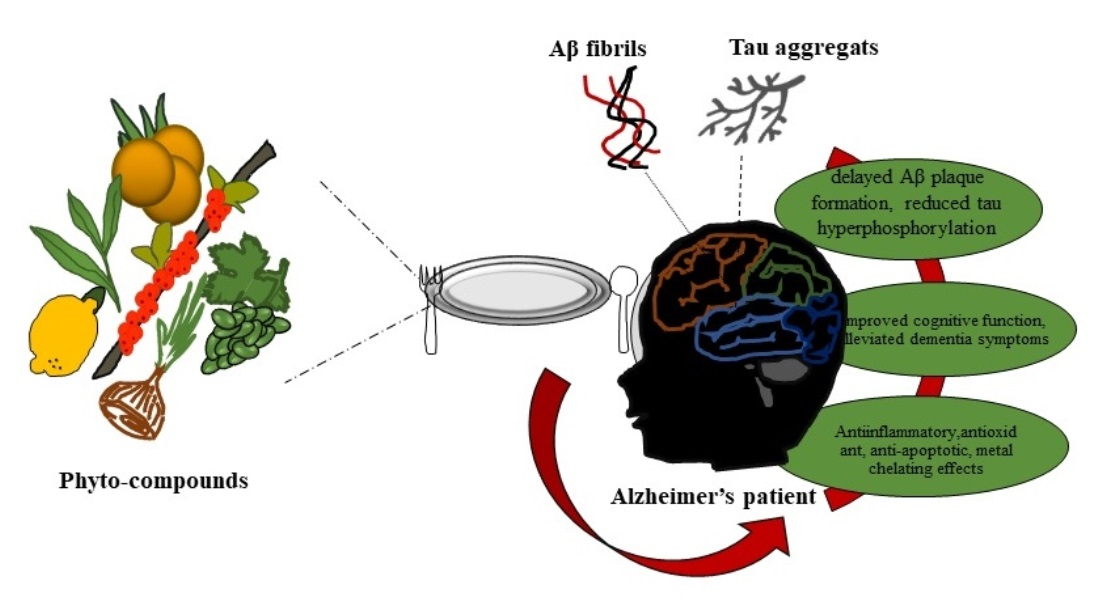
:
1. Introduction
2. Epidemiology of AD
3. Etiology and Pathogenesis of AD
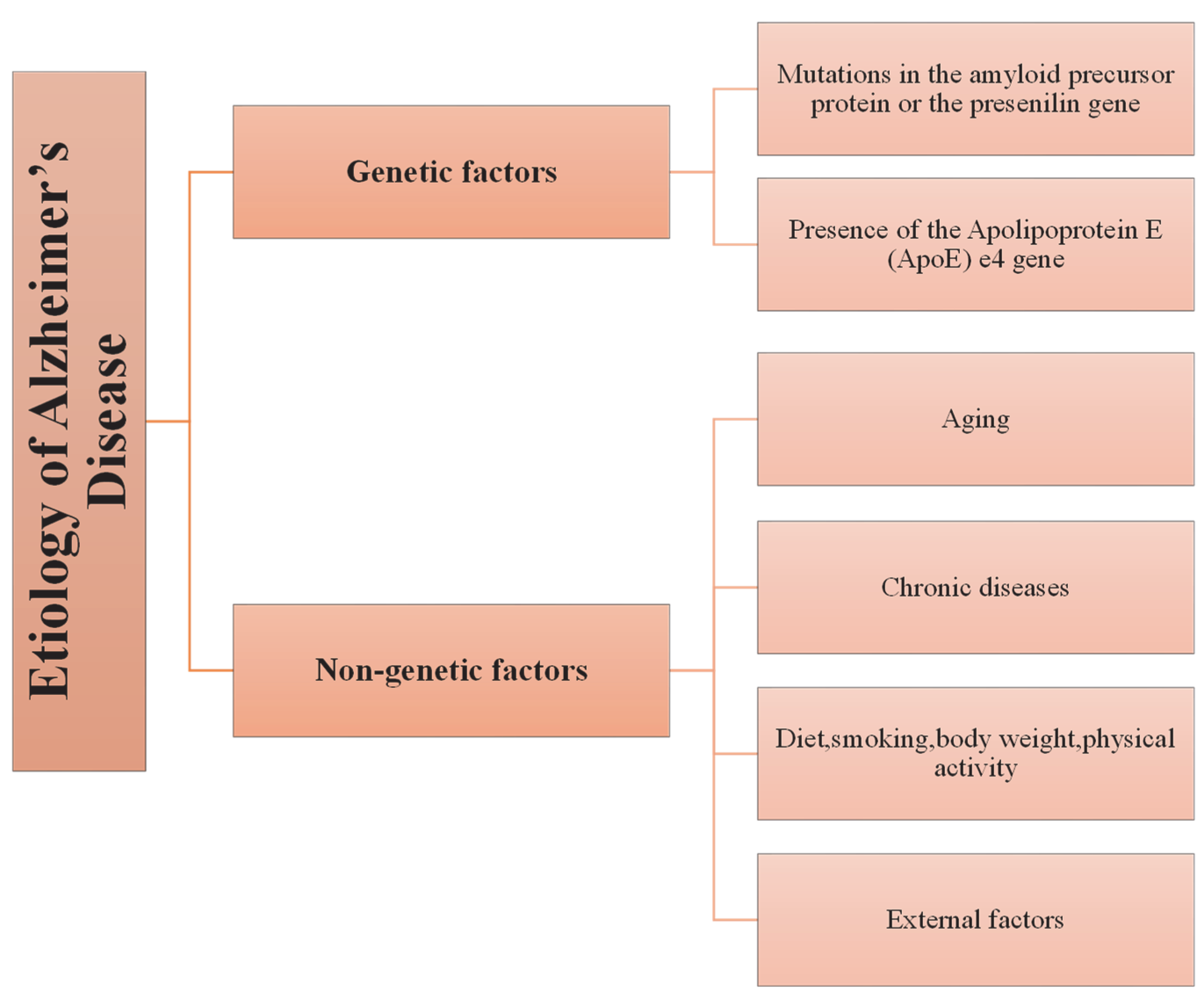
3.1. Etiology of AD
3.1.1. Genetic Predisposition to AD
3.1.2. Non-Genetic Factors in the Pathogenesis of AD
3.1.3. Relationship between Chronic Diseases and AD Progression
- Cerebrovascular Disease–AD relationship.
- Cardiovascular disease–AD relationship.
- Cardiovascular disease–AD relationship.
3.1.4. Other Non-Genetic Risk Factors in AD
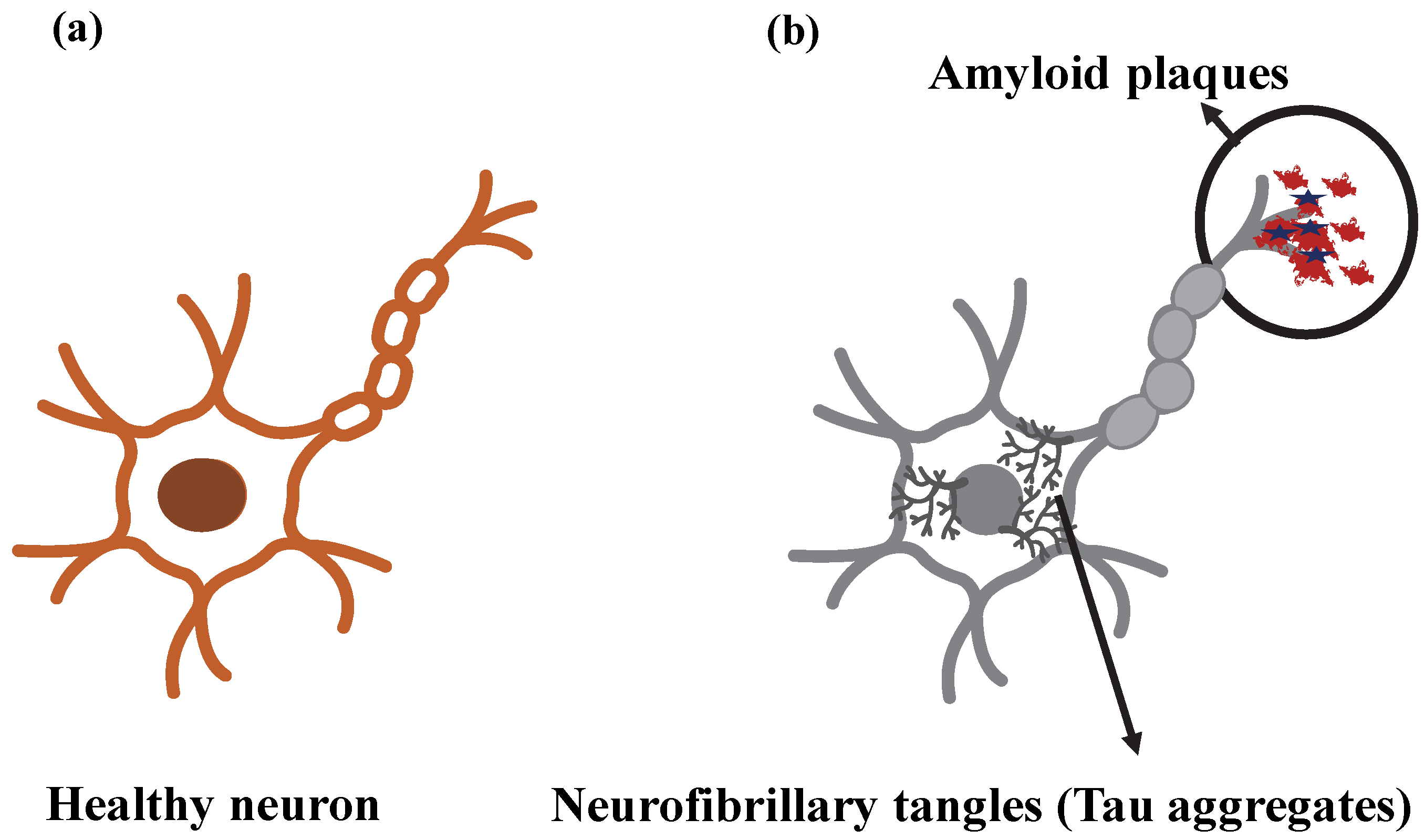
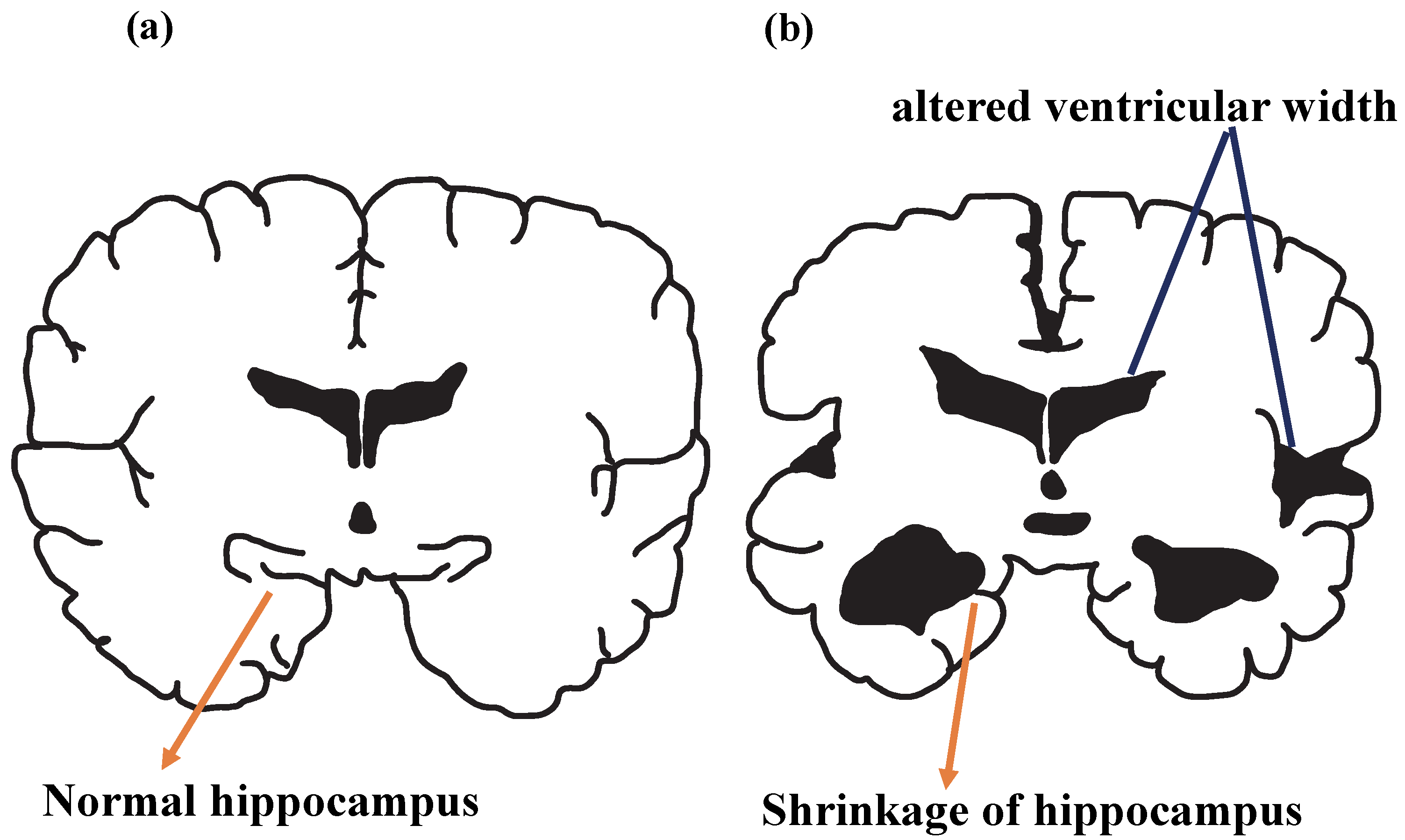
3.2. Pathogenesis of AD
4. Conventional Approaches to AD
4.1. The Role of Acetylcholinesterase Inhibitors
4.2. Memantine
4.3. Other Treatment Approaches in AD
4.3.1. Immunotherapy-Based Approaches to AD
4.3.2. The Use of Antipsychotic and Antidepressant Drugs in AD
4.3.3. Effects of Statins in AD
4.3.4. The Role of Vitamin Supplements in AD Therapy
5. Limiting Factors in Current Approaches to AD
6. Novel Phyto-Therapeutic Approaches in AD
6.1. The Effect of Resveratrol on AD
6.2. The Effect of Tannic Acid on AD
6.3. The Effect of Apigenin on AD
6.4. The Effect of Psoralea corylifolia L. on AD
6.5. The Role of Curcuminı in AD and Preventatıon
6.6. The Effect of Rutin on AD
6.7. The Role of Quercetın in Treatment and Preventatıon of AD
6.8. The Effect of Caffeic Acid on AD
6.9. The Effect of Hesperidin on AD
6.10. The Effect of Limonene on AD
6.11. The Effect of Berberine on AD
6.12. The Effect of Cinnamon on AD
6.13. The Effect of Ginger on AD
6.14. The Role of Luteolin in AD Treatment
6.15. Potential Effects of Ginkgo biloba in AD Treatment and Prevention
7. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Religa, D.; Laudon, H.; Styczynska, M.; Winblad, B.; Näslund, J.; Haroutunian, V. Amyloid β Pathology in Alzheimer’s Disease and Schizophrenia. Am. J. Psychiatry 2003, 160, 867–872. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Kalaria, R.N.; Maestre, G.E.; Arizaga, R.; Friedland, R.P.; Galasko, D.; Hall, K.; Luchsinger, J.A.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Kleij, L.A.; Petersen, E.T.; Siebner, H.R.; Hendrikse, J.; Frederiksen, K.S.; Sobol, N.A.; Hasselbalch, S.G.; Garde, E. The effect of physical exercise on cerebral blood flow in Alzheimer’s disease. NeuroImage Clin. 2018, 20, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R.S.; Ganzer, C.A.; Hristov, H.; Hackett, K.; Caesar, E.; Cohen, R.; Kachko, R.; Meléndez-Cabrero, J.; Rahman, A.; Scheyer, O.; et al. The clinical practice of risk reduction for Alzheimer’s disease: A precision medicine approach. Alzheimer’s Dement. 2018, 14, 1663–1673. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Mantzavinos, V.; Alexiou, A. Biomarkers for Alzheimer’s Disease Diagnosis. Curr. Alzheimer Res. 2017, 14, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Arevalo-Rodriguez, I.; Smailagic, N.; Roqué i Figuls, M.; Ciapponi, A.; Sanchez-Perez, E.; Giannakou, A.; Pedraza, O.L.; Bonfill Cosp, X.; Cullum, S. Mini-Mental State Examination (MMSE) for the detection of Alzheimer’s disease and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2015, 3, 1–62. [Google Scholar] [CrossRef]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in Moderate-to-Severe Alzheimer’s Disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef]
- Thal, D.R.; Attems, J.; Ewers, M. Spreading of Amyloid, Tau, and Microvascular Pathology in Alzheimer’s Disease: Findings from Neuropathological and Neuroimaging Studies. J. Alzheimer’s Dis. 2014, 42, S421–S429. [Google Scholar] [CrossRef]
- DeKosky, S.T. Pathology and Pathways of Alzheimer’s Disease with an Update on New Developments in Treatment. J. Am. Geriatr. Soc. 2003, 51, S314–S320. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.J.; Lipton, R.B.; Hall, C.B.; Zimmerman, M.E.; Sanders, A.E.; Verghese, J.; Dickson, D.W.; Derby, C.A. Age-specific and Sex-specific Prevalence and Incidence of Mild Cognitive Impairment, Dementia, and Alzheimer Dementia in Blacks and Whites. Alzheimer Dis. Assoc. Disord. 2012, 26, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, Y.S.R.; Etman, S.M.; Abdelmonsif, D.A.; Abdallah, O.Y. Intranasal Piperine-Loaded Chitosan Nanoparticles as Brain-Targeted Therapy in Alzheimer’s Disease: Optimization, Biological Efficacy, and Potential Toxicity. J. Pharm. Sci. 2015, 104, 3544–3556. [Google Scholar] [CrossRef]
- Larsson, S.; Orsini, N. Coffee Consumption and Risk of Dementia and Alzheimer’s Disease: A Dose-Response Meta-Analysis of Prospective Studies. Nutrients 2018, 10, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.; Albanese, E.; Guerchet, M.; Prina, M. World Alzheimer Report 2014 Dementia and Risk Reduction; Alzheimer’s Disease International: London, UK, 2014. [Google Scholar]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.; Wu, Y.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Gao, S.; Hendrie, H.C.; Hall, K.S.; Hui, S. The Relationships Between Age, Sex, and the Incidence of Dementia and Alzheimer Disease. Arch. Gen. Psychiatry 1998, 55, 809. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-C.; Li, C.-Y.; Sun, Y.; Hu, S.C. Gender and Age Differences and the Trend in the Incidence and Prevalence of Dementia and Alzheimer’s Disease in Taiwan: A 7-Year National Population-Based Study. Biomed Res. Int. 2019, 2019, 5378540. [Google Scholar] [CrossRef] [Green Version]
- Niu, H.; Álvarez-Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalencia e incidencia de la enfermedad de Alzheimer en Europa: Metaanálisis. Neurología 2017, 32, 523–532. [Google Scholar] [CrossRef]
- Fiest, K.M.; Roberts, J.I.; Maxwell, C.J.; Hogan, D.B.; Smith, E.E.; Frolkis, A.; Cohen, A.; Kirk, A.; Pearson, D.; Pringsheim, T.; et al. The Prevalence and Incidence of Dementia Due to Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Can. J. Neurol. Sci./J. Can. Des. Sci. Neurol. 2016, 43, S51–S82. [Google Scholar] [CrossRef]
- Fratiglioni, L.; Launer, L.J.; Andersen, K.; Breteler, M.M.; Copeland, J.R.; Dartigues, J.F.; Lobo, A.; Martinez-Lage, J.; Soininen, H.; Hofman, A. Incidence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000, 54, S10–S15. [Google Scholar] [PubMed]
- Andersen, K.; Launer, L.J.; Dewey, M.E.; Letenneur, L.; Ott, A.; Copeland, J.R.M.; Dartigues, J.-F.; Kragh-Sorensen, P.; Baldereschi, M.; Brayne, C.; et al. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. Neurology 1999, 53, 1992. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Tom, S.E.; Hubbard, R.A.; Crane, P.K.; Haneuse, S.J.; Bowen, J.; McCormick, W.C.; McCurry, S.; Larson, E.B. Characterization of Dementia and Alzheimer’s Disease in an Older Population: Updated Incidence and Life Expectancy with and without Dementia. Am. J. Public Health 2015, 105, 408–413. [Google Scholar] [CrossRef]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef] [PubMed]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef]
- Rocca, W.A.; Petersen, R.C.; Knopman, D.S.; Hebert, L.E.; Evans, D.A.; Hall, K.S.; Gao, S.; Unverzagt, F.W.; Langa, K.M.; Larson, E.B.; et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimer’s Dement. 2011, 7, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Giaccone, G. The existence of primary age-related tauopathy suggests that not all the cases with early braak stages of neurofibrillary pathology are Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 48, 919–921. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Katzman, R.; Kawas, C. The epidemiology of dementia and Alzheimer disease. In Alzheimer Disease; UC Irvine: Irvine, CA, USA, 1994; pp. 105–122. [Google Scholar]
- Nizzari, M.; Thellung, S.; Corsaro, A.; Villa, V.; Pagano, A.; Porcile, C.; Russo, C.; Florio, T. Neurodegeneration in Alzheimer Disease: Role of Amyloid Precursor Protein and Presenilin 1 Intracellular Signaling. J. Toxicol. 2012, 2012, 187297. [Google Scholar] [CrossRef] [PubMed]
- Dries, D.; Yu, G. Assembly, Maturation, and Trafficking of the γ-Secretase Complex in Alzheimers Disease. Curr. Alzheimer Res. 2008, 5, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Koike, M.; Spusta, S.C.; Niemi, E.C.; Yenari, M.; Nakamura, M.C.; Seaman, W.E. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 2009, 109, 1144–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, C.R. The Molecular Pathology of Alzheimer’s Disease. Neuroimaging Clin. N. Am. 2012, 22, 11–22. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef]
- Fein, G.; Di Sclafani, V.; Tanabe, J.; Cardenas, V.; Weiner, M.W.; Jagust, W.J.; Reed, B.R.; Norman, D.; Schuff, N.; Kusdra, L.; et al. Hippocampal and cortical atrophy predict dementia in subcortical ischemic vascular disease. Neurology 2000, 55, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Sparks, D.L. Coronary Artery Disease, Hypertension, ApoE, and Cholesterol: A Link to Alzheimer’s Disease? Ann. N. Y. Acad. Sci. 1997, 826, 128–146. [Google Scholar] [CrossRef]
- Launer, L.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.; Foley, D.; White, L.R.; Havlik, R.J. Midlife blood pressure and dementia: The Honolulu–Asia aging study☆. Neurobiol. Aging 2000, 21, 49–55. [Google Scholar] [CrossRef]
- Petrovitch, H.; White, L.; Izmirilian, G.; Ross, G.; Havlik, R.; Markesbery, W.; Nelson, J.; Davis, D.; Hardman, J.; Foley, D.; et al. Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: The HAAS☆. Neurobiol. Aging 2000, 21, 57–62. [Google Scholar] [CrossRef]
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in the etiology of Alzheimer’s disease. J. Alzheimer’s Dis. 2002, 4, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Bales, K.R.; Wu, S.; Bhat, P.; Parsadanian, M.; Fagan, A.M.; Chang, L.K.; Sun, Y.; Paul, S.M. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer’s disease. J. Clin. Investig. 1999, 103, R15–R21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Jaspan, J.B.; Huang, W.; Kastin, A.J. Transport of Insulin Across the Blood-Brain Barrier: Saturability at Euglycemic Doses of Insulin. Peptides 1997, 18, 1423–1429. [Google Scholar] [CrossRef]
- Selkoe, D.J. The Origins of Alzheimer Disease. JAMA 2000, 283, 1615. [Google Scholar] [CrossRef]
- Martinez, A.; Gil, C.; Perez, D.I. Glycogen Synthase Kinase 3 Inhibitors in the Next Horizon for Alzheimer’s Disease Treatment. Int. J. Alzheimers. Dis. 2011, 2011, 280502. [Google Scholar] [CrossRef] [Green Version]
- Koponen, S.; Taiminen, T.; Kairisto, V.; Portin, R.; Isoniemi, H.; Hinkka, S.; Tenovuo, O. APOE-ε4 predicts dementia but not other psychiatric disorders after traumatic brain injury. Neurology 2004, 63, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Tsitsopoulos, P.P.; Marklund, N. Amyloid-β Peptides and Tau Protein as Biomarkers in Cerebrospinal and Interstitial Fluid Following Traumatic Brain Injury: A Review of Experimental and Clinical Studies. Front. Neurol. 2013, 4, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breunig, J.J.; Guillot-Sestier, M.-V.; Town, T. Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-Analysis of Alzheimer’s Disease Risk with Obesity, Diabetes, and Related Disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer Disease and Oxidative Stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Collin, L.; Bohrmann, B.; Göpfert, U.; Oroszlan-Szovik, K.; Ozmen, L.; Grüninger, F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer‘s disease. Brain 2014, 137, 2834–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Landau, S.M.; Snitz, B.E.; Klunk, W.E.; Blennow, K.; Zetterberg, H. Fluid and PET biomarkers for amyloid pathology in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 97, 3–17. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Haroutunian, V. Regional Distribution of Neuritic Plaques in the Nondemented Elderly and Subjects with Very Mild Alzheimer Disease. Arch. Neurol. 1998, 55, 1185–1191. [Google Scholar] [CrossRef]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The Neurotrophins and Their Role in Alzheimers Disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Benveniste, E.N.; Nguyen, V.T.; O’Keefe, G.M. Immunological aspects of microglia: Relevance to Alzheimer’s disease. Neurochem. Int. 2001, 39, 381–391. [Google Scholar] [CrossRef]
- Dickson, D.W. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: Cause or effect? J. Clin. Investig. 2004, 114, 23–27. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.A.; Schneider, J.A.; Wilson, R.S.; Bienias, J.L.; Arnold, S.E. Neurofibrillary Tangles Mediate the Association of Amyloid Load with Clinical Alzheimer Disease and Level of Cognitive Function. Arch. Neurol. 2004, 61, 378. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleisher, A.S.; Chen, K.; Quiroz, Y.T.; Jakimovich, L.J.; Gutierrez Gomez, M.; Langois, C.M.; Langbaum, J.B.S.; Roontiva, A.; Thiyyagura, P.; Lee, W.; et al. Associations Between Biomarkers and Age in the Presenilin 1 E280A Autosomal Dominant Alzheimer Disease Kindred. JAMA Neurol. 2015, 72, 316. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Cai, J.; Song, X.; Zhu, H. Bayesian hidden Markov models for delineating the pathology of Alzheimer’s disease. Stat. Methods Med. Res. 2019, 28, 2112–2124. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.S. Using Positron Emission Tomography and Florbetapir F 18 to Image Cortical Amyloid in Patients with Mild Cognitive Impairment or Dementia Due to Alzheimer Disease. Arch. Neurol. 2011, 68, 1404. [Google Scholar] [CrossRef] [Green Version]
- Ashford, J.W.; Salehi, A.; Furst, A.; Bayley, P.; Frisoni, G.B.; Jack, C.R., Jr.; Sabri, O.; Adamson, M.M.; Coburn, K.L.; Olichney, J.; et al. Imaging the Alzheimer Brain. J. Alzheimer’s Dis. 2011, 26, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Manning, F.C. Tacrine therapy for the dementia of Alzheimer’s disease. Am. Fam. Phys. 1994, 50, 819–826. [Google Scholar]
- Suner, S.S.; Sahiner, M.; Ayyala, R.S.; Bhethanabotla, V.R.; Sahiner, N. Versatile Fluorescent Carbon Dots from Citric Acid and Cysteine with Antimicrobial, Anti-biofilm, Antioxidant, and AChE Enzyme Inhibition Capabilities. J. Fluoresc. 2021, 31, 1705–1717. [Google Scholar] [CrossRef]
- Grossberg, G.T. Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease: Getting on and staying on. Curr. Ther. Res. 2003, 64, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Raskind, M.A.; Peskind, E.R.; Wessel, T.; Yuan, W. Galantamine in AD. Neurology 2000, 54, 2261–2268. [Google Scholar] [CrossRef]
- Birks, J.S. Cholinesterase inhibitors for Alzheimer’s disease. In Cochrane Database of Systematic Reviews; Birks, J.S., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Seo, J.-H.; Kim, S.-H.; Kim, H.-S.; Park, C.H.; Jeong, S.-J.; Lee, J.-H.; Choi, S.H.; Chang, K.-A.; Rah, J.-C.; Koo, J.; et al. Effects of nicotine on APP secretion and Aβ- or CT105-induced toxicity. Biol. Psychiatry 2001, 49, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.; Schneider, L.; Davis, K.; Talwalker, S.; Smith, F.; Hoover, T.; Gracon, S. Long-term tacrine (Cognex) treatment. Neurology 1996, 47, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Suner, S.S.; Sahiner, M.; Ayyala, R.S.; Bhethanabotla, V.R.; Sahiner, N. Nitrogen-Doped Arginine Carbon Dots and Its Metal Nanoparticle Composites as Antibacterial Agent. C—J. Carbon Res. 2020, 6, 58. [Google Scholar] [CrossRef]
- Schmidt, R.; Hofer, E.; Bouwman, F.H.; Buerger, K.; Cordonnier, C.; Fladby, T.; Galimberti, D.; Georges, J.; Heneka, M.T.; Hort, J.; et al. EFNS-ENS/EAN Guideline on concomitant use of cholinesterase inhibitors and memantine in moderate to severe Alzheimer’s disease. Eur. J. Neurol. 2015, 22, 889–898. [Google Scholar] [CrossRef]
- Atri, A.; Molinuevo, J.L.; Lemming, O.; Wirth, Y.; Pulte, I.; Wilkinson, D. Memantine in patients with Alzheimer’s disease receiving donepezil: New analyses of efficacy and safety for combination therapy. Alzheimer's Res. Ther. 2013, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, M. Cerebral Hemorrhage after Passive Anti-Abeta Immunotherapy. Science 2002, 298, 1379. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.; Birks, J.; Dening, T. Antidepressants for treating depression in dementia. Cochrane Database Syst. Rev. 2002, 4, 1–31. [Google Scholar] [CrossRef]
- Jick, H.; Zornberg, G.; Jick, S.; Seshadri, S.; Drachman, D. Statins and the risk of dementia. Lancet 2000, 356, 1627–1631. [Google Scholar] [CrossRef]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease -amyloid peptides A 42 and A 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Bookheimer, S.Y.; Strojwas, M.H.; Cohen, M.S.; Saunders, A.M.; Pericak-Vance, M.A.; Mazziotta, J.C.; Small, G.W. Patterns of Brain Activation in People at Risk for Alzheimer’s Disease. N. Engl. J. Med. 2000, 343, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.C. Dietary Intake of Antioxidant Nutrients and the Risk of Incident Alzheimer Disease in a Biracial Community Study. JAMA 2002, 287, 3230. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Haughey, N.; Lee, J.; Evans, M.; Mattson, M.P. Folic Acid Deficiency and Homocysteine Impair DNA Repair in Hippocampal Neurons and Sensitize Them to Amyloid Toxicity in Experimental Models of Alzheimer’s Disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Torres, C.J.; Reynolds, J.O.; Pautler, R.G. Use of Magnetization Transfer Contrast MRI to Detect Early Molecular Pathology in Alzheimer’s Disease. Magn. Reson. Med. 2014, 71, 333–338. [Google Scholar] [CrossRef]
- Kasper, S. Phytopharmaceutical treatment of anxiety, depression, and dementia in the elderly: Evidence from randomized, controlled clinical trials. Wien. Med. Wochenschr. 2015, 165, 217–228. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Hong, K.H.; Ning, Y.; Yu, P.; Ren, J.; Ji, M.; Cai, J. Design, synthesis and evaluation of a novel metal chelator as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 87, 720–727. [Google Scholar] [CrossRef]
- El Gaamouch, F.; Liu, K.; Lin, H.; Wu, C.; Wang, J. Development of grape polyphenols as multi-targeting strategies for Alzheimer’s disease. Neurochem. Int. 2021, 147, 105046. [Google Scholar] [CrossRef]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S.; Toyoda, T.; Abe, K.; Hamaguchi, T.; Ono, K.; et al. Rosmarinic acid suppresses Alzheimer’s disease development by reducing amyloid β aggregation by increasing monoamine secretion. Sci. Rep. 2019, 9, 8711. [Google Scholar] [CrossRef] [Green Version]
- Sahiner, M.; Blake, D.A.; Fullerton, M.L.; Suner, S.S.; Sunol, A.K.; Sahiner, N. Enhancement of biocompatibility and carbohydrate absorption control potential of rosmarinic acid through crosslinking into microparticles. Int. J. Biol. Macromol. 2019, 137, 836–843. [Google Scholar] [CrossRef]
- Swaraz, A.M.; Sultana, F.; Bari, M.W.; Ahmed, K.S.; Hasan, M.; Islam, M.M.; Islam, M.A.; Satter, M.A.; Hossain, M.H.; Islam, M.S.; et al. Phytochemical profiling of Blumea laciniata (Roxb.) DC. and its phytopharmaceutical potential against diabetic, obesity, and Alzheimer’s. Biomed. Pharmacother. 2021, 141, 111859. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahiner, N. Self-Crosslinked Ellipsoidal Poly(Tannic Acid) Particles for Bio-Medical Applications. Molecules 2021, 26, 2429. [Google Scholar] [CrossRef]
- Suner, S.S.; Sahiner, M.; Mohapatra, S.; Ayyala, R.S.; Bhethanabotla, V.R.; Sahiner, N. Degradable poly(catechin) nanoparticles as a versatile therapeutic agent. Int. J. Polym. Mater. Polym. Biomater. 2022, 71, 1104–1115. [Google Scholar] [CrossRef]
- Benchikha, N.; Messaoudi, M.; Larkem, I.; Ouakouak, H.; Rebiai, A.; Boubekeur, S.; Ferhat, M.A.; Benarfa, A.; Begaa, S.; Benmohamed, M.; et al. Evaluation of Possible Antioxidant, Anti-Hyperglycaemic, Anti-Alzheimer and Anti-Inflammatory Effects of Teucrium polium Aerial Parts (Lamiaceae). Life 2022, 12, 1579. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, M.; Rebiai, A.; Sawicka, B.; Atanassova, M.; Ouakouak, H.; Larkem, I.; Egbuna, C.; Awuchi, C.G.; Boubekeur, S.; Ferhat, M.A.; et al. Effect of Extraction Methods on Polyphenols, Flavonoids, Mineral Elements, and Biological Activities of Essential Oil and Extracts of Mentha pulegium L. Molecules 2021, 27, 11. [Google Scholar] [CrossRef]
- Deshpande, P.; Gogia, N.; Singh, A. Exploring the efficacy of natural products in alleviating Alzheimer’s disease. Neural. Regen. Res. 2019, 14, 1321–1329. [Google Scholar] [CrossRef]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-β peptides. J. Biol. Chem. 2005, 280, 37377–37382. [Google Scholar] [CrossRef] [Green Version]
- Savaskan, E.; Olivieri, G.; Meier, F.; Seifritz, E.; Wirz-Justice, A.; Müller-Spahn, F. Red wine ingredient resveratrol protects from β-amyloid neurotoxicity. Gerontology 2003, 49, 380–383. [Google Scholar] [CrossRef] [Green Version]
- Jayasena, T.; Poljak, A.; Smythe, G.; Braidy, N.; Münch, G.; Sachdev, P. The role of polyphenols in the modulation of sirtuins and other pathways involved in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 867–883. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Oxidative stress modulates Sir2α in rat hippocampus and cerebral cortex. Eur. J. Neurosci. 2006, 23, 2573–2580. [Google Scholar] [CrossRef] [PubMed]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [Green Version]
- Al-Bishri, W.M.; Hamza, A.H.; Farran, S.K. Resveratrol Treatment Attenuates Amyloid Beta, Tau Protein and Markers of Oxidative Stress, and Inflammation in Alzheimer’s disease Rat Model. Int. J. Pharm. Res. Sci. 2017, 6, 71–78. [Google Scholar]
- Lin, Y.T.; Wu, Y.C.; Sun, G.C.; Ho, C.Y.; Wong, T.Y.; Lin, C.H.; Chen, H.H.; Yeh, T.C.; Li, C.J.; Tseng, C.J.; et al. Effect of resveratrol on reactive oxygen species-induced cognitive impairment in rats with angiotensin II-induced early Alzheimer’s disease. J. Clin. Med. 2018, 7, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Q.H.; Li, F.; Jin, F.; Shia, J.S. Resveratrol attenuates neuroinflammation-mediated cognitive deficits in rats. J. Health Sci. 2010, 56, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.S.; Thomas, R.G.; Craft, S.; Van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Juan, M.E.; Maijó, M.; Planas, J.M. Quantification of trans-resveratrol and its metabolites in rat plasma and tissues by HPLC. J. Pharm. Biomed. Anal. 2010, 51, 391–398. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.W.; Grossman, H.; Neugroschl, J.; Parker, S.; Burden, A.; Luo, X.; Sano, M. A randomized, double-blind, placebo-controlled trial of resveratrol with glucose and malate (RGM) to slow the progression of Alzheimer’s disease: A pilot study. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Sahiner, M.; Kurt, S.B.; Sahiner, N. Biodiverse Properties of Tannic Acid-Based Fibers. Fibers Polym. 2021, 22, 2986–2994. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Yoshiike, Y.; Takashima, A.; Yamada, M.; Naiki, H. Nordihydroguaiaretic acid potently breaks down pre-formed Alzheimer’s β-amyloid fibrils in vitro. J. Neurochem. 2002, 81, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer’s β-amyloid fibrils in vitro. Biochim. Biophys. Acta—Mol. Basis Dis. 2004, 1690, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Rezai-Zadeh, K.; Koyama, N.; Arendash, G.W.; Yamaguchi, H.; Kakuda, N.; Horikoshi-Sakuraba, Y.; Tan, J.; Town, T. Tannic acid is a natural β-secretase inhibitor that prevents cognitive impairment and mitigates Alzheimer-like pathology in transgenic mice. J. Biol. Chem. 2012, 287, 6912–6927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Gao, X.; Sun, W.; Yao, T.; Shi, S.; Ji, L. Molecular hairpin: A possible model for inhibition of tau aggregation by tannic acid. Biochemistry 2013, 52, 1893–1902. [Google Scholar] [CrossRef]
- Dourado, N.S.; Souza, C.D.S.; de Almeida, M.M.A.; Bispo da Silva, A.; dos Santos, B.L.; Silva, V.D.A.; De Assis, A.M.; da Silva, J.S.; Souza, D.O.; De Fátima Dias Costa, M.; et al. Neuroimmunomodulatory and Neuroprotective Effects of the Flavonoid Apigenin in in vitro Models of Neuroinflammation Associated with Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 119. [Google Scholar] [CrossRef]
- Balez, R.; Steiner, N.; Engel, M.; Muñoz, S.S.; Lum, J.S.; Wu, Y.; Wang, D.; Vallotton, P.; Sachdev, P.; O’Connor, M.; et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci. Rep. 2016, 6, 31450. [Google Scholar] [CrossRef] [Green Version]
- Sang, Z.; Wang, K.; Shi, J.; Cheng, X.; Zhu, G.; Wei, R.; Ma, Q.; Yu, L.; Zhao, Y.; Tan, Z.; et al. Apigenin-rivastigmine hybrids as multi-target-directed liagnds for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2020, 187, 111958. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.L.; Liu, R.; Li, X.X.; Li, J.F.; Zhang, L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer’s disease mouse model. Molecules 2013, 18, 9949–9965. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.F.; Wu, B.; Yang, J.; Hu, L.M.; Su, Y.F.; Gao, X.M. A rapid method for the analysis of ten compounds in Psoralea corylifolia L. by UPLC. Chromatographia 2009, 70, 199–204. [Google Scholar] [CrossRef]
- Xu, Q.X.; Hu, Y.; Li, G.Y.; Xu, W.; Zhang, Y.T.; Yang, X.W. Multi-target anti-Alzheimer activities of four prenylated compounds from Psoralea Fructus. Molecules 2018, 23, 614. [Google Scholar] [CrossRef] [PubMed]
- Radwan, A.; Alanazi, F. Combined Modeling Study of the Binding Characteristics of Natural Compounds, Derived from Psoralea Fruits, to β-Amyloid Peptide Monomer. Int. J. Mol. Sci. 2022, 23, 3546. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.R.; Zhou, W.X.; Zhang, Y.X. The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res. Rev. 2014, 13, 13–37. [Google Scholar] [CrossRef]
- Chen, Z.J.; Yang, Y.F.; Zhang, Y.T.; Yang, D.H. Dietary total prenylflavonoids from the fruits of Psoralea corylifolia L. Prevents age-related cognitive deficits and down-regulates Alzheimer’s markers in SAMP8 mice. Molecules 2018, 23, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, S.; Wu, Q.; Yao, X.; Zhang, J.; Zhong, W.; Zhao, J.; Liu, Q.; Zhang, M. Inhibitory Effects of Isobavachalcone on Tau Protein Aggregation, Tau Phosphorylation, and Oligomeric Tau-Induced Apoptosis. ACS Chem. Neurosci. 2021, 12, 123–132. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, Q.; Zhao, R.; Yao, X.; Du, X.; Liu, Q.; Lv, G.; Xiao, S. Isobavachalcone ameliorates cognitive deficits, and Aβ and tau pathologies in triple-transgenic mice with Alzheimer’s disease. Food Funct. 2021, 12, 7749–7761. [Google Scholar] [CrossRef]
- Ari, B.; Sahiner, M.; Demirci, S.; Sahiner, N. Poly(vinyl alcohol)-tannic Acid Cryogel Matrix as Antioxidant and Antibacterial Material. Polymers 2021, 14, 70. [Google Scholar] [CrossRef]
- Huang, H.C.; Chang, P.; Dai, X.L.; Jiang, Z.F. Protective effects of curcumin on amyloid-b-induced neuronal oxidative damage. Neurochem. Res. 2012, 37, 1584–1597. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Colombrita, C.; Ravagna, A.; Pennisi, G.; Giuffrida Stella, A.M.; Galli, F.; Butterfield, D.A. Redox regulation of heat shock protein expression in aging and neurodegenerative disorders associated with oxidative stress: A nutritional approach. Amino Acids 2003, 25, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G.; Piwocka, K. The Promise of Slow Down Ageing May Come from Curcumin. Curr. Pharm. Des. 2010, 16, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dogra, S.; Prakash, A. Corrigendum to “Protective effect of curcumin (Curcuma longa), against aluminium toxicity: Possible behavioral and biochemical alterations in rats” (Protective effect of curcumin (Curcuma longa), against aluminium toxicity: Possible behavioral and biochem. Behav. Brain Res. 2020, 380, 112415. [Google Scholar] [CrossRef] [PubMed]
- Isik, A.T.; Celik, T.; Ulusoy, G.; Ongoru, O.; Elibol, B.; Doruk, H.; Bozoglu, E.; Kayir, H.; Mas, M.R.; Akman, S. Curcumin ameliorates impaired insulin/IGF signalling and memory deficit in a streptozotocin-treated rat model. Age 2009, 31, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Rainey-Smith, S.R.; Brown, B.M.; Sohrabi, H.R.; Shah, T.; Goozee, K.G.; Gupta, V.B.; Martins, R.N. Curcumin and cognition: A randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br. J. Nutr. 2016, 115, 2106–2113. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.P.; Nyunt, M.S.Z.; Gao, Q.; Gwee, X.; Chua, D.Q.L.; Yap, K.B. Curcumin-Rich Curry Consumption and Neurocognitive Function from 4.5-Year Follow-Up of Community-Dwelling Older Adults (Singapore Longitudinal Ageing Study). Nutrients 2022, 14, 1189. [Google Scholar] [CrossRef]
- Yu, X.L.; Li, Y.N.; Zhang, H.; Su, Y.J.; Zhou, W.W.; Zhang, Z.P.; Wang, S.W.; Xu, P.X.; Wang, Y.J.; Liu, R.T. Rutin inhibits amylin-induced neurocytotoxicity and oxidative stress. Food Funct. 2015, 6, 3296–3306. [Google Scholar] [CrossRef]
- Xu, P.X.; Wang, S.W.; Yu, X.L.; Su, Y.J.; Wang, T.; Zhou, W.W.; Zhang, H.; Wang, Y.J.; Liu, R.T. Rutin improves spatial memory in Alzheimer’s disease transgenic mice by reducing Aβ oligomer level and attenuating oxidative stress and neuroinflammation. Behav. Brain Res. 2014, 264, 173–180. [Google Scholar] [CrossRef]
- Pan, R.Y.; Ma, J.; Kong, X.X.; Wang, X.F.; Li, S.S.; Qi, X.L.; Yan, Y.H.; Cheng, J.; Liu, Q.; Jin, W.; et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci. Adv. 2020, 5, eaau6328. [Google Scholar] [CrossRef] [Green Version]
- Mani, R.J.; Mittal, K.; Katare, D.P. Protective Effects of Quercetin in Zebrafish Model of Alzheimer’s Disease. Asian J. Pharm. 2018, 12, 660–666. [Google Scholar]
- Rifaai, R.A.; Mokhemer, S.A.; Saber, E.A.; El-Aleem, S.A.A.; El-Tahawy, N.F.G. Neuroprotective effect of quercetin nanoparticles: A possible prophylactic and therapeutic role in alzheimer’s disease. J. Chem. Neuroanat. 2020, 107, 101795. [Google Scholar] [CrossRef] [PubMed]
- Sabogal-Guáqueta, A.M.; Muñoz-Manco, J.I.; Ramírez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gómez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef]
- Rich, G.; Buchweitz, M.; Winterbone, M.; Kroon, P.; Wilde, P. Towards an Understanding of the Low Bioavailability of Quercetin: A Study of Its Interaction with Intestinal Lipids. Nutrients 2017, 9, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzystyniak, A.; Wesierska, M.; Petrazzo, G.; Gadecka, A.; Dudkowska, M.; Zmijewska, A.B.; Mosieniak, G.; Figiel, I.; Wlodarczyk, J.; Sikora, E. Combination of dasatinib and quercetin improves cognitive abilities in aged male Wistar rats, alleviates inflammation and changes hippocampal synaptic plasticity and histone H3 methylation profile. Aging 2022, 14, 572–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, M.M.; Garbarino, V.R.; Marques Zilli, E.; Petersen, R.C.; Kirkland, J.L.; Tchkonia, T.; Musi, N.; Seshadri, S.; Craft, S.; Orr, M.E. Senolytic Therapy to Modulate the Progression of Alzheimer’s Disease (SToMP-AD): A Pilot Clinical Trial. J. Prev. Alzheimer’s Dis. 2022, 9, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.; Loureiro, J.A.; Pereira, M.C. Caffeic acid for the prevention and treatment of Alzheimer’s disease: The effect of lipid membranes on the inhibition of aggregation and disruption of Aβ fibrils. Int. J. Biol. Macromol. 2021, 190, 853–861. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of alzheimer’s disease involves Nrf2/HO-1 pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jin, M.; Pi, R.; Zhang, J.; Chen, M.; Ouyang, Y.; Liu, A.; Chao, X.; Liu, P.; Liu, J.; et al. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci. Lett. 2013, 535, 146–151. [Google Scholar] [CrossRef]
- Kumar, M.; Kaur, D.; Bansal, N. Caffeic acid phenethyl ester (CAPE) prevents development of STZ-ICV induced dementia in rats. Pharmacogn. Mag. 2017, 13, 10. [Google Scholar] [CrossRef]
- Kumar, M.; Bansal, N. Caffeic acid phenethyl ester rescued streptozotocin-induced memory loss through PI3-kinase dependent pathway. Biomed. Pharmacother. 2018, 101, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S167–S174. [Google Scholar] [CrossRef] [PubMed]
- Sahiner, M.; Sahiner, N.; Sagbas, S.; Fullerton, M.L.; Blake, D.A. Fabrication of Biodegradable Poly(naringin) Particles with Antioxidant Activity and Low Toxicity. ACS Omega 2018, 3, 17359–17367. [Google Scholar] [CrossRef]
- Wang, D.; Liu, L.; Zhu, X.; Wu, W.; Wang, Y. Hesperidin Alleviates Cognitive Impairment, Mitochondrial Dysfunction and Oxidative Stress in a Mouse Model of Alzheimer’s Disease. Cell. Mol. Neurobiol. 2014, 34, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Thenmozhi, A.J.; Raja, T.R.W.; Janakiraman, U.; Manivasagam, T. Neuroprotective Effect of Hesperidin on Aluminium Chloride Induced Alzheimer’s Disease in Wistar Rats. Neurochem. Res. 2015, 40, 767–776. [Google Scholar] [CrossRef]
- Islam, F.; Javed, H.; Vaibhav, K.; Ahmed, M.E.; Khan, A.; Tabassum, R.; Islam, F.; Safhi, M.M. Effect of hesperidin on neurobehavioral, neuroinflammation, oxidative stress and lipid alteration in intracerebroventricular streptozotocin induced cognitive impairment in mice. J. Neurol. Sci. 2015, 348, 51–59. [Google Scholar] [CrossRef]
- Kean, R.J.; Lamport, D.J.; Dodd, G.F.; Freeman, J.E.; Williams, C.M.; Ellis, J.A.; Butler, L.T.; Spencer, J.P.E. Chronic consumption of flavanone-rich orange juice is associated with cognitive benefits: An 8-wk, randomized, double-blind, placebo-controlled trial in healthy older adults. Am. J. Clin. Nutr. 2015, 101, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, M.H.; Lamport, D.J.; Dodd, G.F.; Saunders, C.; Harkness, L.; Butler, L.T.; Spencer, J.P.E. Flavonoid-rich orange juice is associated with acute improvements in cognitive function in healthy middle-aged males. Eur. J. Nutr. 2016, 55, 2021–2029. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Tomata, Y.; Sugiyama, K.; Sugawara, Y.; Tsuji, I. Citrus consumption and incident dementia in elderly Japanese: The Ohsaki Cohort 2006 Study. Br. J. Nutr. 2017, 117, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, S.C.; Siddiqui, M.S.; Athar, M.; Alam, M.S. D-Limonene modulates inflammation, oxidative stress and Ras-ERK pathway to inhibit murine skin tumorigenesis. Hum. Exp. Toxicol. 2012, 31, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Liu, Q.F.; Choi, B.; Shin, C.; Lee, B.; Yuan, C.; Song, Y.J.; Yun, H.S.; Lee, I.S.; Koo, B.S.; et al. Neuroprotective effects of limonene (+) against Aβ42-induced neurotoxicity in a Drosophila model of Alzheimer’s disease. Biol. Pharm. Bull. 2020, 43, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.X.; Li, S.H.; Li, X.J. Therapeutic potential of berberine against neurodegenerative diseases. Sci. China Life Sci. 2015, 58, 564–569. [Google Scholar] [CrossRef]
- Durairajan, S.S.K.; Liu, L.F.; Lu, J.H.; Chen, L.L.; Yuan, Q.; Chung, S.K.; Huang, L.; Li, X.S.; Huang, J.D.; Li, M. Berberine ameliorates β-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol. Aging 2012, 33, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.N.; Cai, C.Z.; Wu, M.Y.; Su, H.X.; Li, M.; Lu, J.H. Neuroprotective effects of berberine in animal models of Alzheimer’s disease: A systematic review of pre-clinical studies. BMC Complement. Altern. Med. 2019, 19, 109. [Google Scholar] [CrossRef]
- Huang, M.; Jiang, X.; Liang, Y.; Liu, Q.; Chen, S.; Guo, Y. Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of β-amyloid in APP/tau/PS1 mouse model of Alzheimer’s disease. Exp. Gerontol. 2017, 91, 25–33. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.K.; Nandi, M.K.; Mishra, G.; Maurya, A.; Rai, A.; Rai, G.K.; Awasthi, R.; Sharma, B.; Kulkarni, G.T. Berberine: A Plant-derived Alkaloid with Therapeutic Potential to Combat Alzheimer’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2019, 19, 154–170. [Google Scholar] [CrossRef]
- Rao, P.V.; Gan, S.H. Cinnamon: A Multifaceted Medicinal Plant. Evid.-Based Complement. Altern. Med. 2014, 2014, 642942. [Google Scholar] [CrossRef] [Green Version]
- Frydman-Marom, A.; Levin, A.; Farfara, D.; Benromano, T.; Scherzer-Attali, R.; Peled, S.; Vassar, R.; Segal, D.; Gazit, E.; Frenkel, D.; et al. Orally administrated cinnamon extract reduces β-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS ONE 2011, 6, e16564. [Google Scholar] [CrossRef] [Green Version]
- Madhavadas, S.; Subramanian, S. Cognition enhancing effect of the aqueous extract of Cinnamomum zeylanicum on non-transgenic Alzheimer’s disease rat model: Biochemical, histological, and behavioural studies. Nutr. Neurosci. 2017, 20, 526–537. [Google Scholar] [CrossRef]
- Ghafary, S.; Najafi, Z.; Mohammadi-Khanaposhtani, M.; Nadri, H.; Edraki, N.; Ayashi, N.; Larijani, B.; Amini, M.; Mahdavi, M. Novel cinnamic acid–tryptamine hybrids as potent butyrylcholinesterase inhibitors: Synthesis, biological evaluation, and docking study. Arch. Pharm. 2018, 351, 1800115. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Ademiluyi, A.O.; Akinyemi, A.J. Inhibition of acetylcholinesterase activities and some pro-oxidant induced lipid peroxidation in rat brain by two varieties of ginger (Zingiber officinale). Exp. Toxicol. Pathol. 2012, 64, 315–319. [Google Scholar] [CrossRef]
- Cuya, T.; Baptista, L.; Celmar Costa França, T. A molecular dynamics study of components of the ginger (Zingiber officinale) extract inside human acetylcholinesterase: Implications for Alzheimer disease. J. Biomol. Struct. Dyn. 2018, 36, 3843–3855. [Google Scholar] [CrossRef]
- Mahdy, K.A.; Gouda, N.A.M.; Marrie, A.E.H.; Yassin, N.A.Z.; El-shenawy, S.M.A.; Farrag, A.R.H.; Ibrahim, B.M.M. Protective Effect of Ginger (Zingiber officinale) on Alzheimer’s disease Induced in Rats. J. Neuroinfect. Dis. 2014, 5, 2. [Google Scholar]
- Sakr, S.A.; Mahran, H.A.; Lamfon, H.A. Protective effect of ginger (Zingiber officinale) on Alzheimer ’disease Induced rats. J. Med. Plants Res. 2011, 5, 133–140. [Google Scholar]
- Manochkumar, J.; Doss, C.G.P.; El-Seedi, H.R.; Efferth, T.; Ramamoorthy, S. The neuroprotective potential of carotenoids in vitro and in vivo. Phytomedicine 2021, 91, 153676. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.J.; Vishwanathan, R.; Johnson, M.A.; Hausman, D.B.; Davey, A.; Scott, T.M.; Green, R.C.; Miller, L.S.; Gearing, M.; Woodard, J.; et al. Relationship between serum and brain carotenoids, α-tocopherol, and retinol concentrations and cognitive performance in the oldest old from the georgia centenarian study. J. Aging Res. 2013, 2013, 951786. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.S.; Shin, M.; Kim, S.; Lee, S.B. Recent advances in studies on the therapeutic potential of dietary carotenoids in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2018, 2018, 4120458. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Stahl, W.; Griffiths, H.R. Nutritional cognitive neuroscience of aging: Focus on carotenoids and cognitive frailty. Redox Biol. 2021, 44, 101996. [Google Scholar] [CrossRef]
- Yuan, C.; Chen, H.; Wang, Y.; Schneider, J.A.; Willett, W.C.; Morris, M.C. Dietary carotenoids related to risk of incident Alzheimer dementia (AD) and brain AD neuropathology: A community-based cohort of older adults. Am. J. Clin. Nutr. 2021, 113, 200–208. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Gortzi, O.; Sobarzo-Sanchez, E.; Daglia, M.; Skalicka-Woźniak, K.; Nabavi, S.M. Luteolin as an anti-inflammatory and neuroprotective agent: A brief review. Brain Res. Bull. 2015, 119, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhang, J.; Guo, L.; Xu, Y.; Sun, L.; Wang, S.; Feng, Y.; Gou, L.; Zhang, L.; Liu, Y. Protective role of luteolin against cognitive dysfunction induced by chronic cerebral hypoperfusion in rats. Pharmacol. Biochem. Behav. 2014, 126, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Islam, M.N.; Ali, M.Y.; Kim, Y.M.; Park, H.J.; Sohn, H.S.; Jung, H.A. The effects of C-glycosylation of luteolin on its antioxidant, anti-Alzheimer’s disease, anti-diabetic, and anti-inflammatory activities. Arch. Pharm. Res. 2014, 37, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Rahul; Jyoti, S.; Naz, F.; Ashafaq, M.; Shahid, M.; Siddique, Y.H. Therapeutic potential of luteolin in transgenic Drosophila model of Alzheimer’s disease. Neurosci. Lett. 2019, 692, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.; Cheng, H.; Che, Z. Ameliorating effect of luteolin on memory impairment in an Alzheimer’s disease model. Mol. Med. Rep. 2016, 13, 4215–4220. [Google Scholar] [CrossRef]
- Crichton, G.E.; Bryan, J.; Murphy, K.J. Dietary Antioxidants, Cognitive Function and Dementia—A Systematic Review. Plant Foods Hum. Nutr. 2013, 68, 279–292. [Google Scholar] [CrossRef]
- Ahmed, T.; Javed, S.; Javed, S.; Tariq, A.; Šamec, D.; Tejada, S.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Resveratrol and Alzheimer’s Disease: Mechanistic Insights. Mol. Neurobiol. 2017, 54, 2622–2635. [Google Scholar] [CrossRef]
- Braidy, N.; Jugder, B.-E.; Poljak, A.; Jayasena, T.; Nabavi, S.M.; Sachdev, P.; Grant, R. Molecular Targets of Tannic Acid in Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 861–869. [Google Scholar] [CrossRef]
- Venigalla, M.; Sonego, S.; Gyengesi, E.; Münch, G. Curcumin and Apigenin—Novel and promising therapeutics against chronic neuroinflammation in Alzheimer′s disease. Neural Regen. Res. 2015, 10, 1181. [Google Scholar] [CrossRef]
- Voulgaropoulou, S.D.; van Amelsvoort, T.A.M.J.; Prickaerts, J.; Vingerhoets, C. The effect of curcumin on cognition in Alzheimer’s disease and healthy aging: A systematic review of pre-clinical and clinical studies. Brain Res. 2019, 1725, 146476. [Google Scholar] [CrossRef]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2019, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajialyani, M.; Hosein Farzaei, M.; Echeverría, J.; Nabavi, S.; Uriarte, E.; Sobarzo-Sánchez, E. Hesperidin as a Neuroprotective Agent: A Review of Animal and Clinical Evidence. Molecules 2019, 24, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddin, L.B.; Jha, N.K.; Meeran, M.F.N.; Kesari, K.K.; Beiram, R.; Ojha, S. Neuroprotective Potential of Limonene and Limonene Containing Natural Products. Molecules 2021, 26, 4535. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wang, C.; Yang, W. Role of berberine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2016, 12, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Momtaz, S.; Hassani, S.; Khan, F.; Ziaee, M.; Abdollahi, M. Cinnamon, a promising prospect towards Alzheimer’s disease. Pharmacol. Res. 2018, 130, 241–258. [Google Scholar] [CrossRef]
- Schepici, G.; Contestabile, V.; Valeri, A.; Mazzon, E. Ginger, a Possible Candidate for the Treatment of Dementias? Molecules 2021, 26, 5700. [Google Scholar] [CrossRef]
- Unger, M. Pharmacokinetic drug interactions involving Ginkgo biloba. Drug Metab. Rev. 2013, 45, 353–385. [Google Scholar] [CrossRef]
- Le Bars, P.L. A placebo-controlled, double-blind, randomized trial of an extract of Ginkgo biloba for dementia. North American EGb Study Group. JAMA J. Am. Med. Assoc. 1997, 278, 1327–1332. [Google Scholar] [CrossRef]
- von Gunten, A.; Schlaefke, S.; Überla, K. Efficacy of Ginkgo biloba extract EGb 761 ® in dementia with behavioural and psychological symptoms: A systematic review. World J. Biol. Psychiatry 2016, 17, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Savaskan, E.; Mueller, H.; Hoerr, R.; von Gunten, A.; Gauthier, S. Treatment effects of Ginkgo biloba extract EGb 761® on the spectrum of behavioral and psychological symptoms of dementia: Meta-analysis of randomized controlled trials. Int. Psychogeriatr. 2018, 30, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Ong Lai Teik, D.; Lee, X.S.; Lim, C.J.; Low, C.M.; Muslima, M.; Aquili, L. Ginseng and Ginkgo Biloba Effects on Cognition as Modulated by Cardiovascular Reactivity: A Randomised Trial. PLoS ONE 2016, 11, e0150447. [Google Scholar] [CrossRef] [PubMed]
- Yakoot, M.; Salem, A. Helmy Effect of Memo®, a natural formula combination, on Mini-Mental State Examination scores in patients with mild cognitive impairment. Clin. Interv. Aging 2013, 2013, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attia, A.; Rapp, S.R.; Case, L.D.; D’Agostino, R.; Lesser, G.; Naughton, M.; McMullen, K.; Rosdhal, R.; Shaw, E.G. Phase II study of Ginkgo biloba in irradiated brain tumor patients: Effect on cognitive function, quality of life, and mood. J. Neurooncol. 2012, 109, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.G.; Liu, M.X.; Liu, L.T.; Guan, J.; Li, H.; Wei, Y.; Hao, L.; Xiyuan Hospital of China Academy of Chinese Medical Sciences. Traditional Chinese Medicine for Preventing and Treating Alzheimer Disease and Preparation Method Thereof. Chinese Patent No. CN102078460 (A), 1 June 2011. [Google Scholar]
- Liang, S.; Zeng, Y.; Wu, Z.; Liang, S.Y.; Zeng, Y.C. Traditional Chinese Medicine Composition for Improving Memory and Mild Cognitive Impairment and Preparation Method Thereof. Chinese Patent No. CN105943888 (A), 22 June 2016. [Google Scholar]
- Kim, Y.O.; Lee, S.W.; Kim, H.D.; Republic of Korea (Management: Rural Development Administration). Composition Having Brain Function and Congnition Enhancing Activity Comprising Ginseng Mixed Herbal Extracts, Ginsenoside Rg2 and Ginsenoside F2. Korean Patent No. KR101509056 (B1), 8 April 2015. [Google Scholar]







{kind=link}
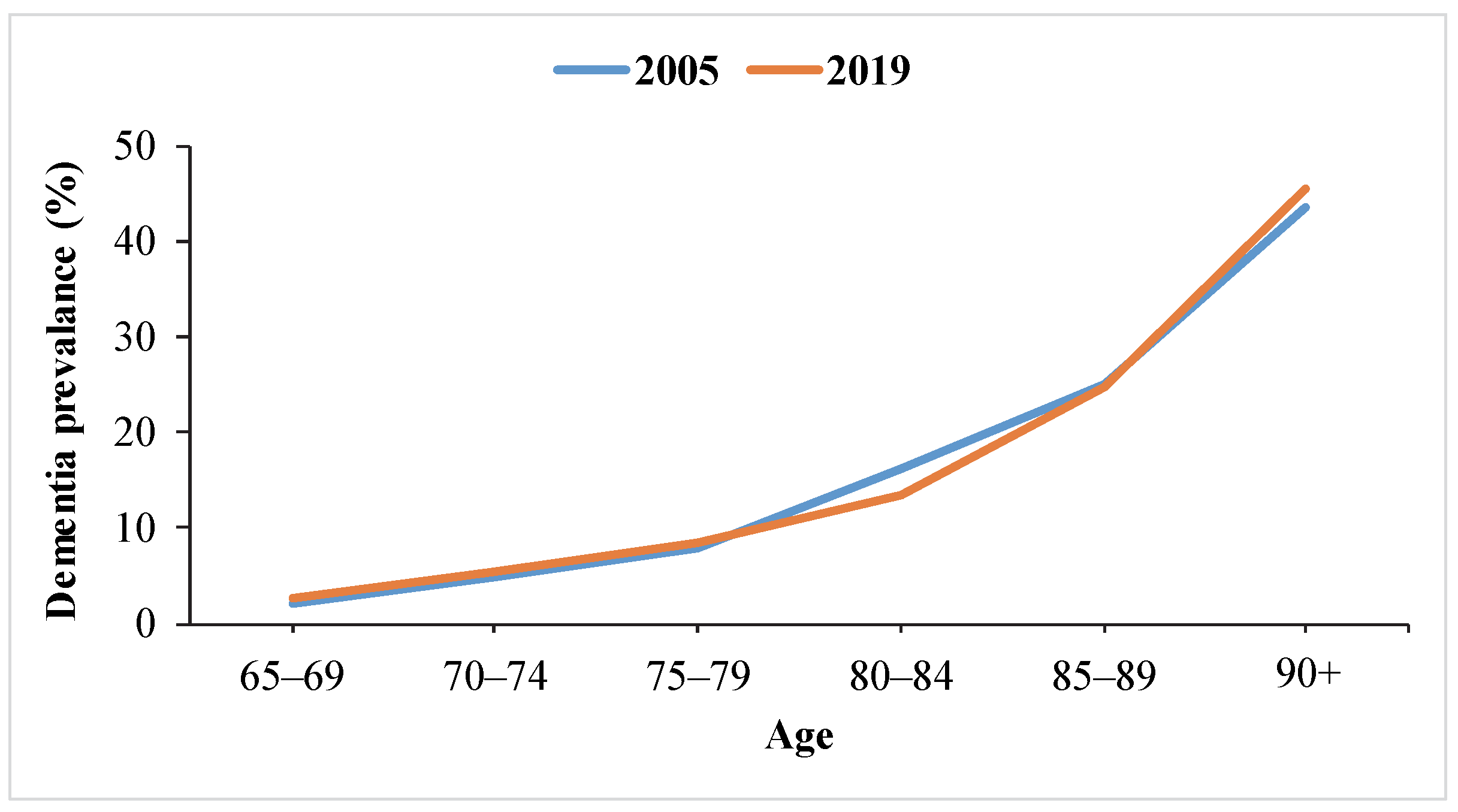{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Current Treatment Modalities in Alzheimer’s Disease | Limitations and Failures in Treatment |
|---|---|
| Acetylcholinesterase Inhibitors | Not effective in all patient groups, may cause intense gastrointestinal side effects, drowsiness, insomnia, heart rhythm disorders |
| Memantine | Clinical efficacy differs in moderate-to-severe AD and mild AD There are insufficient data on the long-term safety and benefit of starting memantine therapy early |
| Immunotherapy | The definitive results of the use of immunotherapy in AD have not been disclosed and certain immunization assessments could not be tested in humans |
| Antipsychotics and antidepressants | Some of this group of drugs can exacerbate AD sequelae Treatment may be interrupted due to drug interactions Only moderate results can be obtained with antipsychotic group drugs |
| Statins | Only certain lipid-lowering drugs can give results in treatment |
| Vitamin supplements | Antioxidant supplements have not been associated with reduced incidence of dementia in asymptomatic individuals |
| Phytocompounds | Mechanism of Action | Reference |
|---|---|---|
| Resveratrol | Reduces neuroinflammation, inhibits tauopathy and Aβ-plaque formation, decreases the level of SIRT1 levels in neurons, prevents NF-κB activation, reduces the activity of β-secretase and reduces oxidative stress in brain, prevents apoptosis | [191] |
| Tannic acid | Natural inhibitor of β-secretase (BACE1) activity, destabilizes neurotoxic Aβ fibrils, inhibits the aggregation of tau peptide and NFTs | [192] |
| Apigenin | Regulates the expression of inflammatory mediators in neurons/glial cells, protects neuroinflammation, reduces Aβ, fibrillar amyloid deposits, oxidative stress, and neuronal hyper-excitability | [193] |
| Curcumin | Improves neuronal apoptosis, restores cerebral blood flow, and reduces AchE activity, suppresses tau aggregation, reduces oxidative stress | [194] |
| Quercetin | Inhibition of Aβ aggregation and tau phosphorylation, inhibits (BACE-1) enzyme activity, competitively inhibits AChE, modulates the cell’s own antioxidant pathways | [195] |
| Hesperidin | Attenuates APP expression and suppresses the levels of Aβ and β- and γ-secretase activity, decreases AChE activity and lipid peroxidation, blocks inflammatory process, increases the anti-oxidative defense system, diminish neuro-inflammatory and apoptotic pathways | [196] |
| Limonene | Decreases AChE activity, reduces oxidative stress | [197] |
| Berberine | Reduces Aβ levels, inhibits BACE-1 activity, decreases the hyperphosphorylation of tau. Berberine retards oxidative stress and neuroinflammation in the brain | [198] |
| Cinnamon | Inhibits the formation, accumulation, and toxic effects of Aβ plaques, and has potential antioxidant effects and restoration of redox balance | [199] |
| Ginger | Inhibits AChE activity, reduces Aβ levels and β- and γ-secretase activity, represses neuroinflammation and amyloid genesis, acts as a radical scavenger, prevents apoptosis | [200] |
| Development Status (Description/Study Design) | Chemical Constituents | Mechanism of Action (Outcomes) | Patent Number | Reference |
|---|---|---|---|---|
| 3 monthly age APP695V717I transgenic mice donepezil hydrochloride control group and Chinese medicine group orally for 6 months | Traditional Chinese medicine: Prepared fleece flower, ginseng, Rhizoma Acori Graminei, Coptis and Chuanxiong Rhizome | This medicine improves the impaired ability of learning and memory of APP transgenic mice, improves the content of Ach, strengthens the activity of choline acetyltransferase (CHAT), suppresses the activity of AchE | CN102078460 | [208] |
| Wistar senile rats’ donepezil hydrochloride control group and Chinese medicine group orally once a day, continuous for 8 weeks | This medicine improves space learning memory ability of old cognitive disorder rats, alleviates hippocampus neuronal damage, by influencing lipid metabolism, oxidative stress, level of inflammation and apoptosis (Aβ level, bcl-2 expresses, NF-KB) | |||
| Cognitive disorder model mouse | The traditional Chinese medicine composition: thin leaf milkwort rootbark, sweet flag rhizome, ginseng, Gastralia Tuber, paper mulberry fruit, ginkgo leaves, and borneol | This medicine can significantly improve the learning memory central brain SOD activity, reduce its neuronal cell lipofuscin content, and oxygen-free radical injury | CN105943888A | [209] |
| Four-vessel occlusion-induced cerebral ischemia rat models | Ginseng mixed herbal extracts, ginsenoside Rg2, and ginsenoside F2 | This medicine improves memory capacity of symptoms such as mild cognitive impairment or dementia, and shows excellent acetylcholinesterase inhibitory activity and antioxidant activity | KR101509056B1 | [210] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahiner, M.; Yilmaz, A.S.; Gungor, B.; Sahiner, N. A Review on Phyto-Therapeutic Approaches in Alzheimer’s Disease. J. Funct. Biomater. 2023, 14, 50. https://doi.org/10.3390/jfb14010050
Sahiner M, Yilmaz AS, Gungor B, Sahiner N. A Review on Phyto-Therapeutic Approaches in Alzheimer’s Disease. Journal of Functional Biomaterials. 2023; 14(1):50. https://doi.org/10.3390/jfb14010050
Chicago/Turabian StyleSahiner, Mehtap, Aynur Sanem Yilmaz, Buket Gungor, and Nurettin Sahiner. 2023. "A Review on Phyto-Therapeutic Approaches in Alzheimer’s Disease" Journal of Functional Biomaterials 14, no. 1: 50. https://doi.org/10.3390/jfb14010050
APA StyleSahiner, M., Yilmaz, A. S., Gungor, B., & Sahiner, N. (2023). A Review on Phyto-Therapeutic Approaches in Alzheimer’s Disease. Journal of Functional Biomaterials, 14(1), 50. https://doi.org/10.3390/jfb14010050