The Kinetics and Stoichiometry of Metal Cation Reduction on Multi-Crystalline Silicon in a Dilute Hydrofluoric Acid Matrix


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
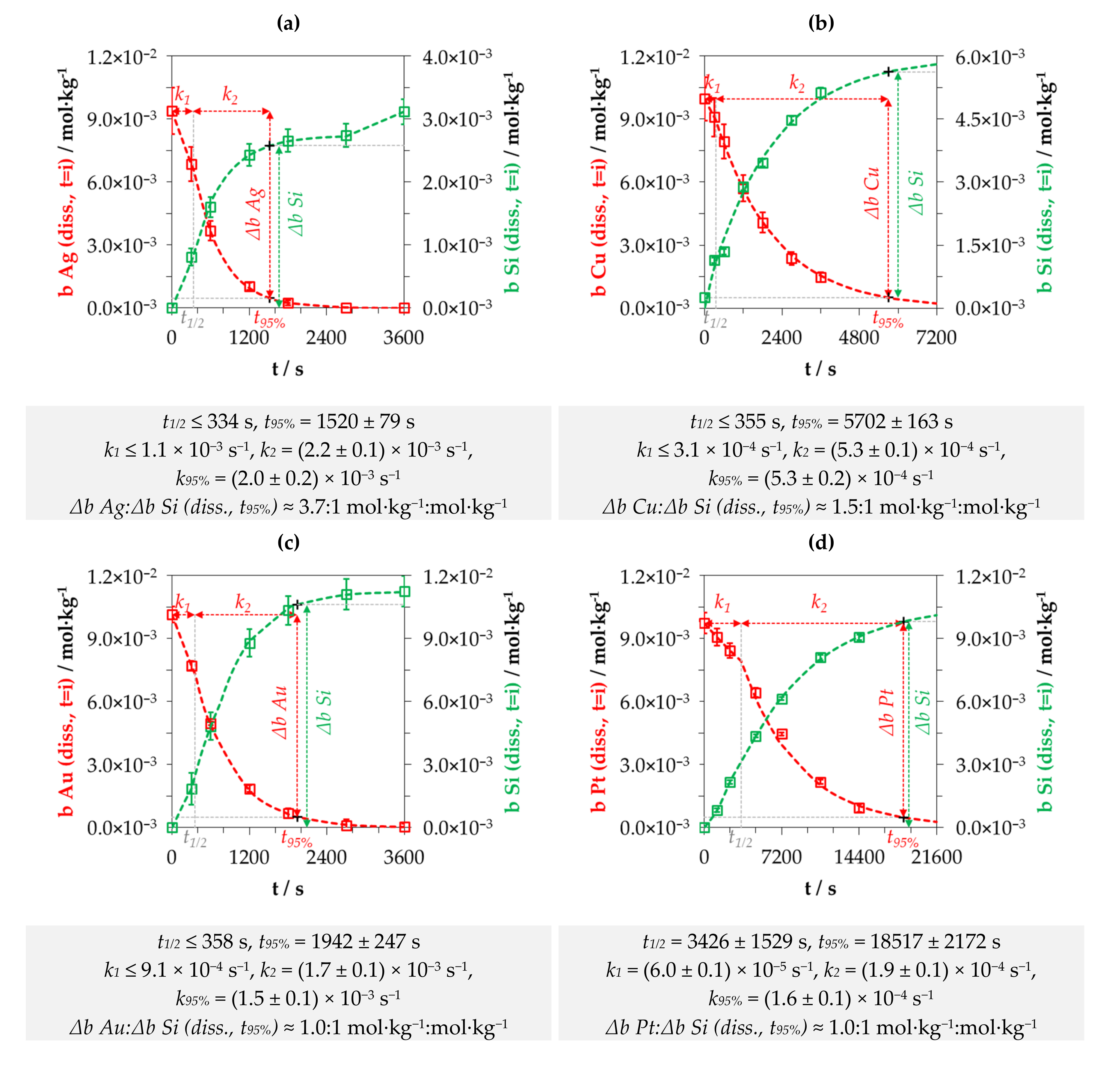
3.1. Kinetics of the Metal Depositions at One Initial Metal Ion and HF Concentration Levels
3.2. Kinetics of the Metal Depositions at Different Metal Ion and HF Concentration Levels
3.3. Discussion of Metal Deposition Kinetics
3.4. Stoichiometry of Metal Cation Reduction and Silicon Oxidation at Solitary Initial Metal Ion and HF Concentration Levels
3.5. Stoichiometry of Metal Cation Reduction and Silicon Oxidation at Different Initial Metal Ion and HF Concentration Levels
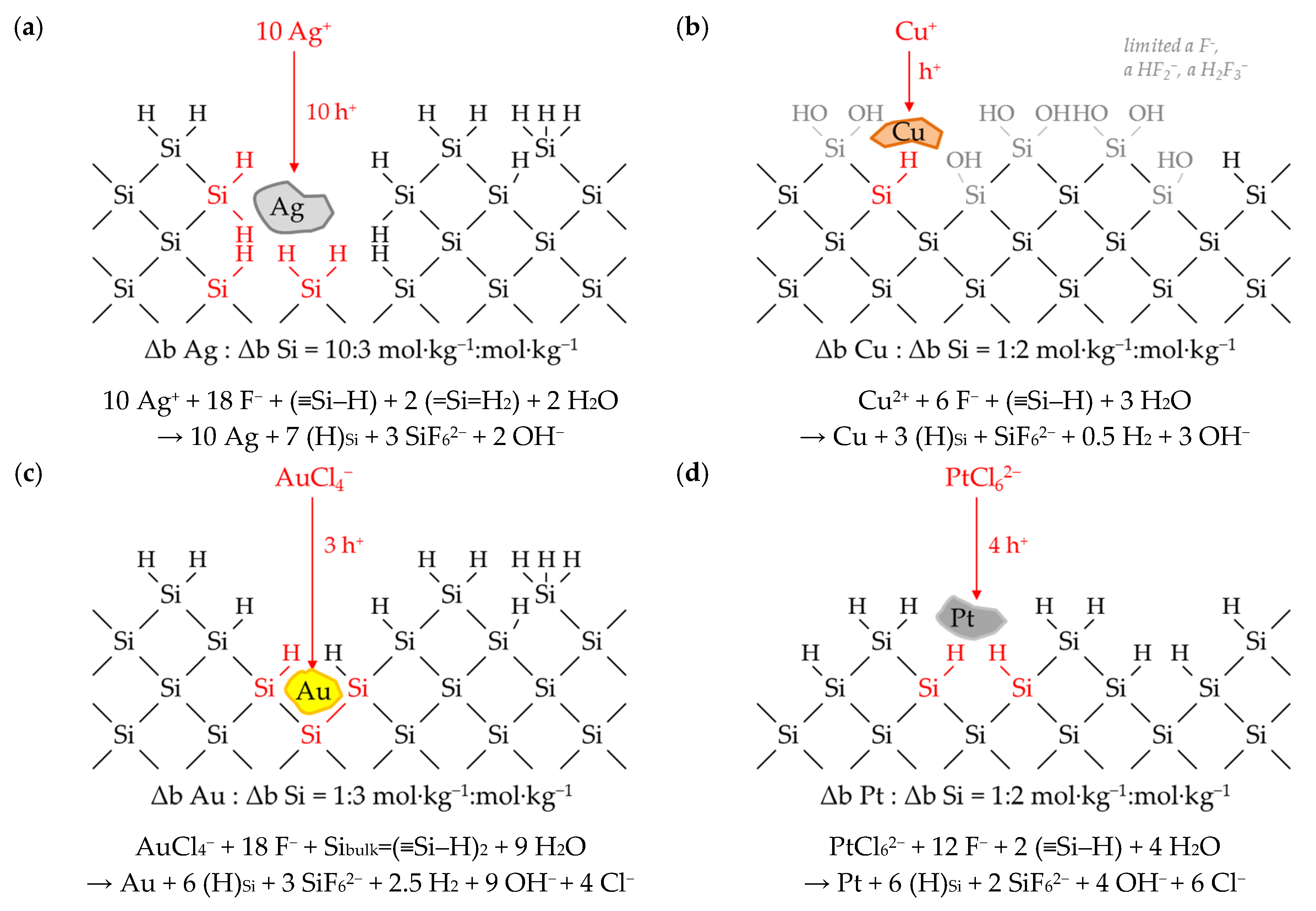
3.6. Interpretation of the Stoichiometric Findings
3.6.1. The Involvement of the Oxonium Ion (Section I)
3.6.2. The Divalent and Tetravalent Reaction Mechanism (Section II)
3.6.3. The Change from Bulk-Si Oxidation to Si–Hx Oxidation (Section III)
3.6.4. The Decrease in Δb Me:Δb Si Ratios toward a Minimum (Section IV)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Van Toan, N.; Wang, Z.; Ono, T. Metal-assisted-chemical-etching of silicon nanowires for templating 3D graphene growth towards energy storage in microsystems. J. Micromech. Microeng. 2019, 29, 055007. [Google Scholar] [CrossRef]
- Nichkalo, S.; Druzhinin, A.; Yerokhov, V.; Ostapiv, O. Fabrication and Characterization of High-Performance Anti-reflecting Nanotextured Si Surfaces for Solar Cells. In Nanooptics, Nanophotonics, Nanostructures, and Their Applications; Fesenko, O., Yatsenko, L., Eds.; NANO 2017, Springer Proceedings in Physics, 210; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Stafiniak, A.; Prazmowska, J.; Macherzynski, W.; Paszkiewicz, R. Nanostructuring of Si substrates by a metal assisted chemical etching and dewetting process. RSC Adv. 2018, 8, 31224–31230. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Han, M.; Zheng, P.; Zheng, L.; Qin, H.; Deng, L. Porous silicon templates prepared by Cu-assisted chemical etching. Mat. Lett. 2014, 118, 146–149. [Google Scholar] [CrossRef]
- Kuntyi, O.; Zozulya, G.I.; Shepida, M.V.; Nichkalo, S.I. Deposition of nanostructured metals on the surface of silicon by galvanic replacement: A mini-review. Voprosy Khimii Khimicheskoi Tekhnologii 2019, 3, 74–82. [Google Scholar]
- Nagahara, L.A.; Ohmori, T.; Hashimoto, K.; Fujishima, A. The influence of hydrofluoric-acid concentration on electroless copper deposition onto silicon. J. Electroanal. Chem. 1992, 333, 363–369. [Google Scholar] [CrossRef]
- Homma, T.; Wade, C.P.; Chidsey, C.E.D. Nucleation of trace copper on the H-Si (111) surface in aqueous fluoride solutions. J. Phys. Chem. B 1998, 102, 7919–7923. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Shen, F.-Y.; Huang, F.-S. Gold nanowires fabricated by immersion plating. Nanotechnology 2008, 19, 195302. [Google Scholar] [CrossRef]
- Milazzo, R.G.; Mio, A.M.; D’Arrigo, G.; Grimaldi, M.G.; Spinella, C.; Rimini, E. Coalescence of silver clusters by immersion in diluted HF solution. J. Chem. Phys. 2015, 143, 024306. [Google Scholar] [CrossRef]
- Smith, Z.R.; Smith, R.L.; Collins, S.D. Mechanism of nanowire formation in metal assisted chemical etching. Electrochim. Acta 2013, 92, 139–147. [Google Scholar] [CrossRef]
- Norga, G.J.; Platero, M.; Black, K.A.; Reddy, A.J.; Michel, J.; Kimerling, L.C. Mechanism of copper deposition on silicon from dilute hydrofluoric acid solution. J. Electrochem. Soc. 1997, 144, 2801–2810. [Google Scholar] [CrossRef]
- Lee, M.K.; Wang, H.D.; Wang, J.J. A Cu seed layer for Cu deposition on silicon. Solid State Electron. 1997, 41, 695–702. [Google Scholar] [CrossRef]
- Srinivasan, R.; Suni, I.I. Kinetic analysis of Au deposition from aqueous HF onto Si (111) by surface second harmonic generation. J. Electrochem. Soc. 1999, 146, 570–573. [Google Scholar] [CrossRef]
- Gorji, S.G.; Razak, K.A.; Cheong, K.Y. Gold nanoparticles deposited on linker-free silicon substrate and embedded in aluminum Schottky contact. J. Colloid Interface Sci. 2013, 408, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Chyan, O.M.R.; Chen, J.-J.; Chien, H.Y.; Sees, J.; Hall, L. Copper deposition on HF etched silicon surfaces: Morphological and kinetic studies. J. Electrochem. Soc. 1996, 143, 92–96. [Google Scholar] [CrossRef]
- Brahiti, N.; Bouanik, S.-A.; Hadjersi, T. Metal-assisted electroless etching of silicon in aqueous NH4HF2 solution. Appl. Surf. Sci. 2012, 258, 5628–5637. [Google Scholar] [CrossRef]
- Scudiero, L.; Fasasi, A.; Griffiths, P.R. Characterization of a controlled electroless deposition of copper thin film on germanium and silicon surfaces. Appl. Surf. Sci. 2011, 257, 4422–4427. [Google Scholar] [CrossRef]
- Peng, K.Q.; Hu, J.J.; Yan, Y.J.; Wu, Y.; Fang, H.; Xu, Y.; Lee, S.T.; Zhu, J. Fabrication of single-crystalline silicon nanowires by scratching a silicon surface with catalytic metal particles. Adv. Funct. Mater. 2006, 16, 387–394. [Google Scholar] [CrossRef]
- Huang, Z.; Geyer, N.; Werner, P.; de Boor, J.; Gösele, U. Metal-assisted chemical etching of silicon: A review. Adv. Mater. 2011, 23, 285–308. [Google Scholar] [CrossRef]
- Li, S.; Ma, W.; Zhou, Y.; Chen, X.; Xiao, Y.; Ma, M.; Zhu, W.; Wie, F. Fabrication of porous silicon nanowires by MACE method in HF/H2O2/AgNO3 system at room temperature. Nanoscale Res. Lett. 2014, 9, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, R.G.; Mio, A.M.; D’Arrigo, G.; Smecca, E.; Alberti, A.; Fisichella, G.; Giannazzo, F.; Spinella, C.; Rimini, E. Influence of hydrofluoric acid treatment on electroless deposition of Au clusters. Beilstein J. Nanotechnol. 2017, 8, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Warren, S.; Reitzle, A.; Kazimirov, A.; Ziegler, J.C.; Bunk, O.; Cao, L.X.; Renner, F.U.; Kolb, D.M.; Bedzyk, M.J.; Zegenhagen, J. A structure study of the electroless deposition of Au on Si (111): H. Surf. Sci. 2002, 496, 287–298. [Google Scholar] [CrossRef]
- Yae, S.; Nasu, N.; Matsumoto, K.; Hagihara, T.; Fukumuro, N.; Matsuda, H. Nucleation behavior in electroless displacement deposition of metals on silicon from hydrofluoric acid solutions. Electrochim. Acta 2007, 53, 35–41. [Google Scholar] [CrossRef]
- Han, H.; Huang, Z.; Lee, W. Metal-assisted chemical etching of silicon and nanotechnology applications. Nano Today 2014, 9, 271–304. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, L.A.; Ohmori, T.; Hashimoto, K.; Fujishima, A. Effects of HF solution in the electroless deposition process on silicon surfaces. J. Vac. Sci. Technol. 1993, 11, 763–767. [Google Scholar] [CrossRef]
- dos Santos Filho, S.G.; Pasa, A.A.; Hasenack, C.M. A mechanism for electroless Cu plating onto Si. Microelectron. Eng. 1997, 33, 149–155. [Google Scholar] [CrossRef]
- Magagnin, L.; Maboudian, R.; Carraro, C. Gold deposition by galvanic displacement on semiconductor surfaces: Effect of substrate on adhesion. J. Phys. Chem. B 2002, 106, 401–407. [Google Scholar] [CrossRef]
- Morinaga, H.; Suyama, M.; Ohmi, T. Mechanism of metallic particle growth and metal-induced pitting on Si wafer surface in wet chemical processing. J. Electroanal. Chem. 1994, 141, 2834–2841. [Google Scholar] [CrossRef]
- Peng, K.Q.; Fang, H.; Hu, J.; Wu, Y.; Zhu, J.Z.; Yan, Y.; Lee, S.T. Metal-particle-induced, highly localized site-specific etching of Si and formation of single-crystalline Si nanowires in aqueous fluoride solution. Chem. Eur. J. 2006, 12, 7942–7947. [Google Scholar] [CrossRef]
- Scheeler, S.P.; Ullrich, S.; Kudera, S.; Pacholski, C. Fabrication of porous silicon by metal-assisted etching using highly ordered gold nanoparticle arrays. Nanoscale Res. Lett. 2012, 7, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Lajvardi, M.; Eshghi, H.; Izadifard, M.; Ghazi, M.E.; Goodarzi, A. Effects of silver and gold catalytic activities on the structural and optical properties of silicon nanowires. Phys. E 2016, 75, 136–143. [Google Scholar] [CrossRef]
- Dawood, M.K.; Tripathy, S.; Dolmanan, S.B.; Ng, T.H.; Tan, H.; Lam, J. Influence of catalytic gold and silver metal nanoparticles on structural, optical, and vibrational properties of silicon nanowires synthesized by metal-assisted chemical etching. J. Appl. Phys. 2012, 112, 073509. [Google Scholar] [CrossRef]
- Geyer, N.; Fuhrmann, B.; Huang, Z.; de Boor, J.; Leipner, H.S.; Werner, P. Model for the mass transport during metal-assisted chemical etching with contiguous metal films as catalysts. J. Phys. Chem. C 2012, 116, 13446–13451. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, J.; Huang, X.; Qian, Q.; He, Y.; Chi, L.; Wang, Y. Controllable and facile fabrication of gold nanostructures for selective metal-assisted etching of silicon. Small 2014, 10, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.; Kulakci, M.; Turan, R.; Unalan, H.E. Effect of electroless etching parameters on the growth and reflection properties of silicon nanowires. Nanotechnology 2011, 22, 155606. [Google Scholar] [CrossRef] [PubMed]
- daRosa, C.P.; Iglesia, E.; Maboudian, R. Dynamics of copper deposition onto silicon by galvanic displacement. J. Electrochem. Soc. 2008, 155, D244–D250. [Google Scholar] [CrossRef] [Green Version]
- Torcheux, L.; Mayeux, A.; Chemla, M. Electrochemical coupling effects on the corrosion of silicon samples in HF solutions. J. Electrochem. Soc. 1995, 142, 2037. [Google Scholar] [CrossRef]
- Jeske, M.; Schultze, J.W.; Thönissen, M.; Münder, H. Electrodeposition of metals into porous silicon. Thin Solid Film. 1995, 255, 63–66. [Google Scholar] [CrossRef]
- Xia, X.H.; Ashruf, C.M.A.; French, P.J.; Kelly, J.J. Galvanic cell formation in silicon/metal contacts: The effect on silicon surface morphology. Chem. Mater. 2000, 12, 1671–1678. [Google Scholar] [CrossRef]
- Kolasinski, K.W.; Barclay, W.B. The stoichiometry of electroless silicon etching in solutions of V2O5 and HF. Angew. Chem. Int. Ed. 2013, 52, 6731–6734. [Google Scholar] [CrossRef]
- Tsuboi, T.; Sakka, T.; Ogata, Y.H. Metal deposition into a porous silicon layer by immersion plating: Influence of halogen ions. J. Appl. Phys. 1998, 83, 4501–4506. [Google Scholar] [CrossRef]
- daRosa, C.P.; Maboudian, R.; Iglesia, E. Copper deposition onto silicon by galvanic displacement: Effect of silicon dissolution rate. J. Electrochem. Soc. 2008, 155, E70–E78. [Google Scholar] [CrossRef]
- Chartier, C.; Bastide, S.; Lévy-Clément, C. Metal-assisted chemical etching of silicon in HF–H2O2. Electrochim. Acta 2008, 53, 5509–5516. [Google Scholar] [CrossRef]
- Cerruti, M.; Doerk, G.; Hernandez, G.; Carraro, C.; Maboudian, R. Galvanic deposition of Pt clusters on silicon: Effect of HF concentration and application as catalyst for silicon nanowire growth. Langmuir 2010, 26, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Ye, W.; Shi, X.; Chang, Y.; Chen, Y.; Wang, C. Comparative study of electroless copper deposition based on the seed layers of Pd, PtPd and AuPd. Appl. Surf. Sci. 2009, 255, 3713–3718. [Google Scholar] [CrossRef]
- Li, X.; Xiao, Y.; Yan, C.; Zhou, K.; Schweizer, S.L.; Sprafke, A.; Lee, J.-H.; Wehrspohn, R.B. Influence of the mobility of Pt nanoparticles on the anisotropic etching properties of silicon. Solid State Lett. 2013, 2, 22–24. [Google Scholar] [CrossRef]
- Kuznetsov, G.V.; Skryshevsky, V.A.; Vdovenkova, T.A.; Tsyganova, A.I.; Gorostiza, P.; Sanz, F. Platinum electroless deposition on silicon from hydrogen fluoride solutions. Electrical properties. J. Electrochem. Soc. 2001, 148, C528–C532. [Google Scholar] [CrossRef]
- Geyer, N.; Wollschläger, N.; Fuhrmann, B.; Tonkikh, A.; Berger, A.; Werner, P.; Jungmann, M.; Krause-Rehberg, R.; Leipner, H.S. Influence of the doping level on the porosity of silicon nanowires prepared by metal-assisted chemical etching. Nanotechnology 2015, 26, 245301–245307. [Google Scholar] [CrossRef]
- Li, G.; Jiao, J.; Seraphin, S.; Raghavan, S.; Jeon, J.S. Masking effect of copper during anisotropic etching of silicon in buffered hydrofluoric acid solutions. J. Appl. Phys. 1999, 85, 1857–1863. [Google Scholar] [CrossRef]
- Lombardi, I.; Marchionna, S.; Zangari, G.; Pizzini, S. Effect of Pt particle size and distribution on photoelectrochemical hydrogen evolution by p-Si photocathodes. Langmuir 2007, 23, 12413–12420. [Google Scholar] [CrossRef]
- Cichoszewski, J.; Reuter, M.; Schwerdt, F.; Werner, J.H. Role of catalyst concentration on metal assisted chemical etching of silicon. Electrochim. Acta 2013, 109, 333–339. [Google Scholar] [CrossRef]
- Yae, S.; Abe, M.; Fukumuro, N.; Matsuda, H. Palladium enhanced etching of n-type silicon in hydrofluoric acid solution. Electrochemistry 2008, 76, 144–146. [Google Scholar] [CrossRef] [Green Version]
- Memming, R.; Schwandt, G. Anodic dissolution of silicon in hydrofluoric acid solutions. Surf. Sci. 1966, 4, 109–124. [Google Scholar] [CrossRef]
- Bandarenka, H.; Redko, S.; Nenzi, P.; Balucan, M.; Bondarenko, V. Optimization of chemical displacement deposition of copper on porous silicon. J. Nanosci. Nanotechnol. 2012, 12, 8725–8731. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Collins, S.D. Porous silicon formation mechanisms. J. Appl. Phys. 1992, 71, R1. [Google Scholar] [CrossRef]
- Peng, K.Q.; Yan, Y.; Gao, S.; Zhu, J. Dendrite-assisted growth of silicon nanowires in electroless metal deposition. Adv. Funct. Mater. 2003, 13, 127–132. [Google Scholar] [CrossRef]
- Liu, F.M.; Green, M. Efficient SERS substrates made by electroless silver deposition into patterned silicon structures. J. Mater. Chem. 2004, 14, 1526–1532. [Google Scholar] [CrossRef]
- Knotter, M. Etching mechanism of vitreous silicon dioxide in HF-based solutions. J. Am. Chem. Soc. 2000, 122, 4345–4351. [Google Scholar] [CrossRef]
- Abramova, E.N.; Khort, A.M.; Tsygankov, V.N.; Yakovenko, A.G.; Shvets, V.I. The role of the etchant ion in the formation and growth of pores in silicon during its etching in hydrofluoric acid solutions. Dokl. Chem. 2016, 467, 61–63. [Google Scholar] [CrossRef]
- Beckmann, K.H. Investigation of the chemical properties of stain films on silicon by means of infrared spectroscopy. Surf. Sci. 1965, 3, 314–332. [Google Scholar] [CrossRef]
- Gossenberger, F.; Roman, T.; Forster-Tonigold, K.; Groß, A. Change of the work function of platinum electrodes induced by halide adsorption. Beilstein J. Nanotechnol. 2014, 5, 152–161. [Google Scholar] [CrossRef]
- Vanýsek, P. Electrochemical series. In CRC Handbook of Chemistry and Physics, 93th ed.; Haynes, W.M., Ed.; Taylor & Francis Ltd.: Milton Park, UK, 2012; pp. 2012–2013. [Google Scholar]
- Ginstrup, O. The redox system platinum(0)/platinum(II)/platinum(IV) with chloro and bromo ligands. Acta Chem. Scand. 1972, 26, 1527–1541. [Google Scholar] [CrossRef] [Green Version]
- Bromley, L.A. Thermodynamic properties of strong electrolytes in aqueous solutions. Aiche J. 1973, 19, 313–320. [Google Scholar] [CrossRef]
- Trasatti, S. Work function, electronegativity, and electrochemical behaviour of metals. II. Potentials of zero charge and “electrochemical” work functions. J. Electroanal. Chem. 1971, 33, 351–378. [Google Scholar] [CrossRef]
- Chelvayohann, M.; Mee, C.H.B. Work function measurements on (110), (100) and (111) surfaces of silver. J. Phys. C Solid State Phys. 1982, 15, 2305–2312. [Google Scholar] [CrossRef]
- Hölzl, J.; Schulte, F.K. Work functions of metals. In Solid Surface Physics; Höhler, G., Ed.; Springer: Berlin, Germany, 1979. [Google Scholar]
- Rivière, J.C. Work function: Measurements and results. In Solid Surface Science; Green, M., Ed.; Decker: New York, NY, USA, 1969. [Google Scholar]
- Michaelson, H.B. The work function of the elements and its periodicity. J. Appl. Phys. 1977, 48, 4729. [Google Scholar] [CrossRef] [Green Version]
- Bertocchi, M.; Amato, M.; Marri, I.; Ossicini, S. Tuning the work function of Si (100) surface by halogen absorption: A DFT study. Phys. Status Solidi C 2017, 14, 1700193. [Google Scholar] [CrossRef]
- Zvyagin, I.P.; Kurova, I.A.; Nal’gieva, M.A.; Ormont, N.N. Hopping ε2 conductivity of boron-doped a-Si: H films annealed in hydrogen at high temperature. Semiconductors 2006, 40, 108–112. [Google Scholar] [CrossRef]
- Novikov, A. Experimental measurement of work function in doped silicon surfaces. Solid State Electron. 2010, 54, 8–13. [Google Scholar] [CrossRef]
- Fujimura, N.; Ohta, A.; Makihara, K.; Miyazaki, S. Evaluation of valence band top and electron affinity of SiO2 and Si-based semiconductors using X-ray photoelectron spectroscopy. Jpn. J. Appl. Phys. 2016, 55, 08PC06. [Google Scholar] [CrossRef]
- Reed, M.L.; Fedder, G.K. Micro mechanical systems 2—Photolithographic microfabrication. In Handbook of Sensors and Actuators; Fukuda, T., Menz, W., Eds.; Elsevier Science: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Messnaoui, B. Representation of excess properties and liquid composition of aqueous solutions of the HF+water system. J. Solut. Chem. 2008, 37, 715–726. [Google Scholar] [CrossRef]
- Hunt, H.; Chittum, J.F.; Ritchev, H.W. Overvoltage. In Seventy-Third Generation Meeting; The Electrochemical Society: Savannah, GA, USA, 1938; pp. 299–314. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (∙OH/O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513. [Google Scholar] [CrossRef] [Green Version]
- Zona, R.; Solar, S.; Getoff, N.; Sehested, K.; Holcman, J. Reactivity of H atoms and hydrated electrons with chlorobenzoic acids. Radiat. Phys. Chem. 2008, 77, 162–168. [Google Scholar] [CrossRef]
- Atkins, P.W.; Paula, J. Physikalische Chemie; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Giguère, P.A.; Turrell, S. The nature of hydrofluoric acid. A spectroscopic study of the proton-transfer complex H3O+∙F−. J. Am. Chem. Soc. 1980, 102, 5473–5477. [Google Scholar] [CrossRef]
- Es, F.; Kulakcı, M.; Turan, R. An alternative metal-assisted etching route for texturing silicon wafers for solar cell applications. IEEE J. Photovolt. 2016, 6, 440–446. [Google Scholar] [CrossRef]
- Harraz, F.A.; Sakka, T.; Ogata, Y.H. Immersion plating of nickel onto a porous silicon layer from fluoride solutions. Phys. Status Solidi A 2003, 197, 51–56. [Google Scholar] [CrossRef]
- Memming, R. Semiconductor Electrochemistry; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2001. [Google Scholar]
- Papaconstantopoulos, D.A.; Economou, E.N. Calculation of the electronic properties of hydrogenated silicon. Phys. Rev. B 1981, 24, 7233–7246. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Ag | Cu | Au | Pt | ||||
|---|---|---|---|---|---|---|---|---|---|
| f1 | b Me (diss., t = 0) valid range | mol | 7.1 × 10−7 | 5.5 × 10−3 | 2.1 × 10−5 | 8.2 × 10−3 | 3.3 × 10−7 | 4.2 × 10−7 | 6.0 × 10−5 |
| kg | 5.5 × 10−3 | 1.3 × 10−1 | 8.2 × 10−3 | 4.1 × 10−1 | 9.3 × 10−2 | 5.1 × 10−5 | 9.7 × 10−3 | ||
| (Δb Me:Δb Si)max | mol∙kg−1 | 4:1 | 5:1 | 2:1 | 3:1 | 4:3 | 1:1 | 6:5 | |
| mol∙kg−1 | |||||||||
| x | mol0.99 | 3.0 × 10−5 | 3.3 × 10−4 | 4.5 × 10−5 | 1.5 × 10−3 | 4.1 × 10−5 | 5.1 × 10−5 | 3.5 × 10−5 | |
| kg0.99 | |||||||||
| f2 | b Me (diss., t = 0) valid range | mol | 7.1 × 10−7–1.3 × 10−1 | 2.1 × 10−5–2.3 × 10−3 | 3.3 × 10−7 | 4.2 × 10−7–6.5 × 10−4 | |||
| kg | 2.4 × 10−3 | ||||||||
| (Δb Me:Δb Si)max | mol∙kg−1 | 10:3 | 2:1 | 4:3 | 6:5 | ||||
| mol∙kg−1 | |||||||||
| x | mol0.99 | 2.5 × 10−4 | 4.0 × 10−4 | 3.7 × 10−4 | 2.1 × 10−4 | ||||
| kg0.99 | |||||||||
| f3 | b Me (diss., t = 0) valid range | mol | --- | 2.3 × 10−3–4.1 × 10−1 | 2.4 × 10−3 | 6.5 × 10−4–9.7 × 10−3 | |||
| kg | 9.3 × 10−2 | ||||||||
| (Δb Me:Δb Si)min | mol∙kg−1 | --- | 1:2 | 1:3 | 1:2 | ||||
| mol∙kg−1 | |||||||||
| x | mol0.99 | --- | 3.5 × 10−3 | 9.0 × 10−3 | 1.2 × 10−3 | ||||
| kg0.99 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schönekerl, S.; Acker, J. The Kinetics and Stoichiometry of Metal Cation Reduction on Multi-Crystalline Silicon in a Dilute Hydrofluoric Acid Matrix. Nanomaterials 2020, 10, 2545. https://doi.org/10.3390/nano10122545
Schönekerl S, Acker J. The Kinetics and Stoichiometry of Metal Cation Reduction on Multi-Crystalline Silicon in a Dilute Hydrofluoric Acid Matrix. Nanomaterials. 2020; 10(12):2545. https://doi.org/10.3390/nano10122545
Chicago/Turabian StyleSchönekerl, Stefan, and Jörg Acker. 2020. "The Kinetics and Stoichiometry of Metal Cation Reduction on Multi-Crystalline Silicon in a Dilute Hydrofluoric Acid Matrix" Nanomaterials 10, no. 12: 2545. https://doi.org/10.3390/nano10122545
APA StyleSchönekerl, S., & Acker, J. (2020). The Kinetics and Stoichiometry of Metal Cation Reduction on Multi-Crystalline Silicon in a Dilute Hydrofluoric Acid Matrix. Nanomaterials, 10(12), 2545. https://doi.org/10.3390/nano10122545