Oral Bioavailability Enhancement of Raloxifene with Nanostructured Lipid Carriers


Abstract
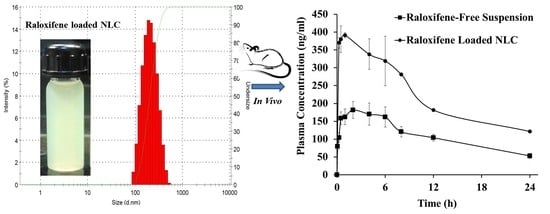
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of NLC
2.3. Formulation Optimization
2.4. Assay Method
2.5. Entrapment Efficiency
2.6. Particle Size and Zeta Potential
2.7. Differential Scanning Calorimetry
2.8. In Vitro Drug Release
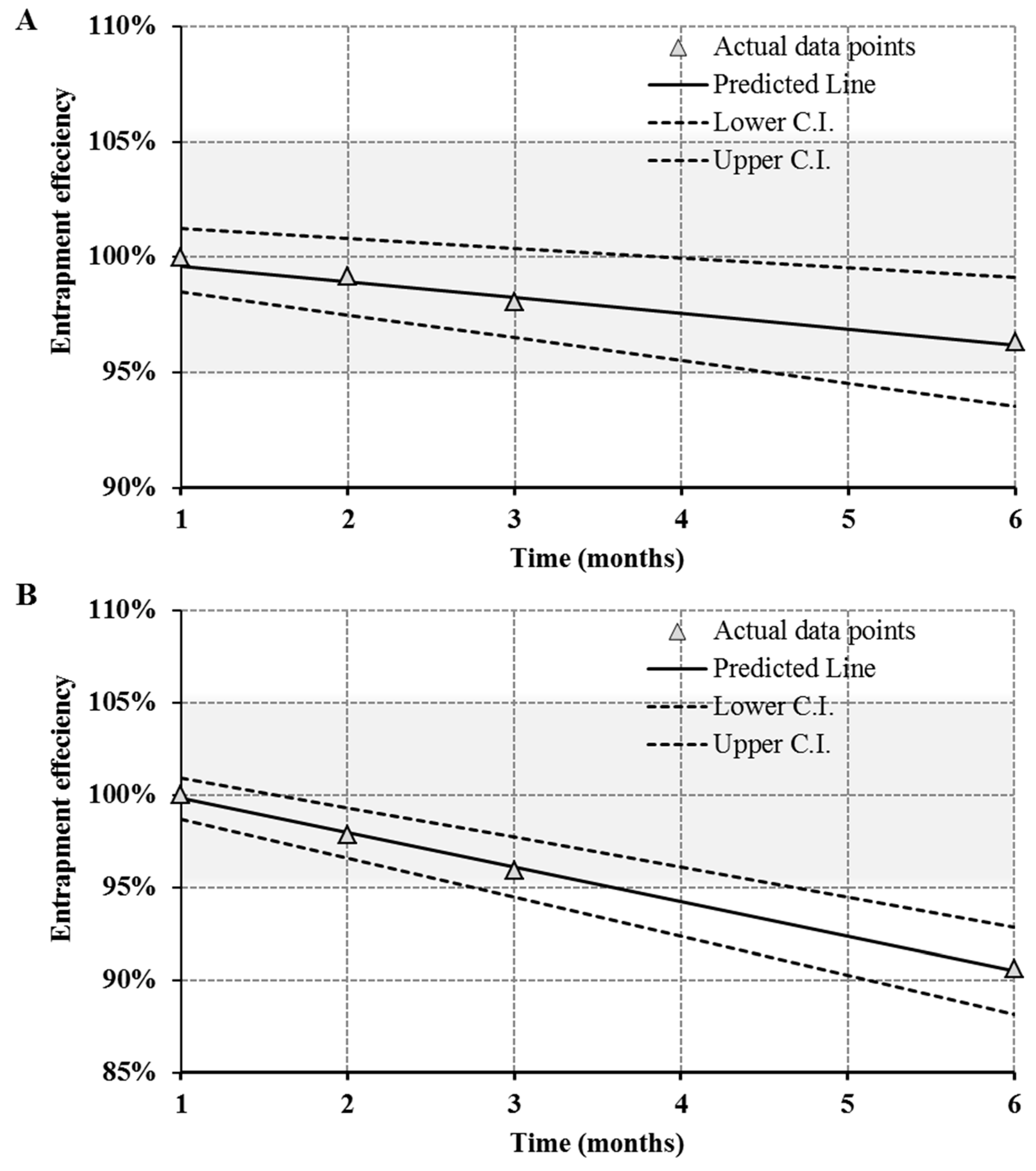
2.9. Stability Studies
2.10. In Vivo Pharmacokinetics
3. Results and Discussion
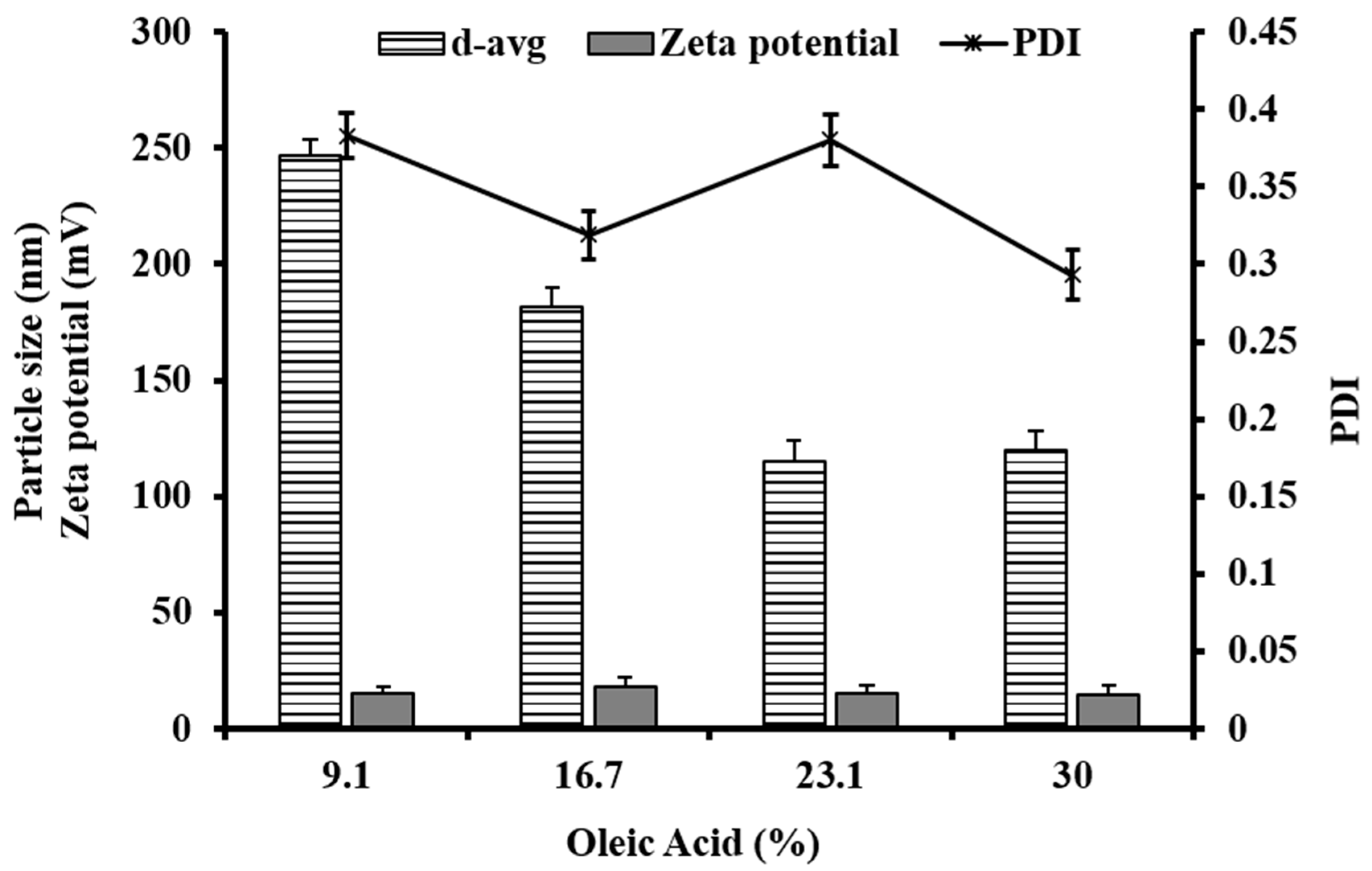
3.1. Particle Size and Polydispersity Index
3.2. Zeta Potential
3.3. Entrapment Efficiency
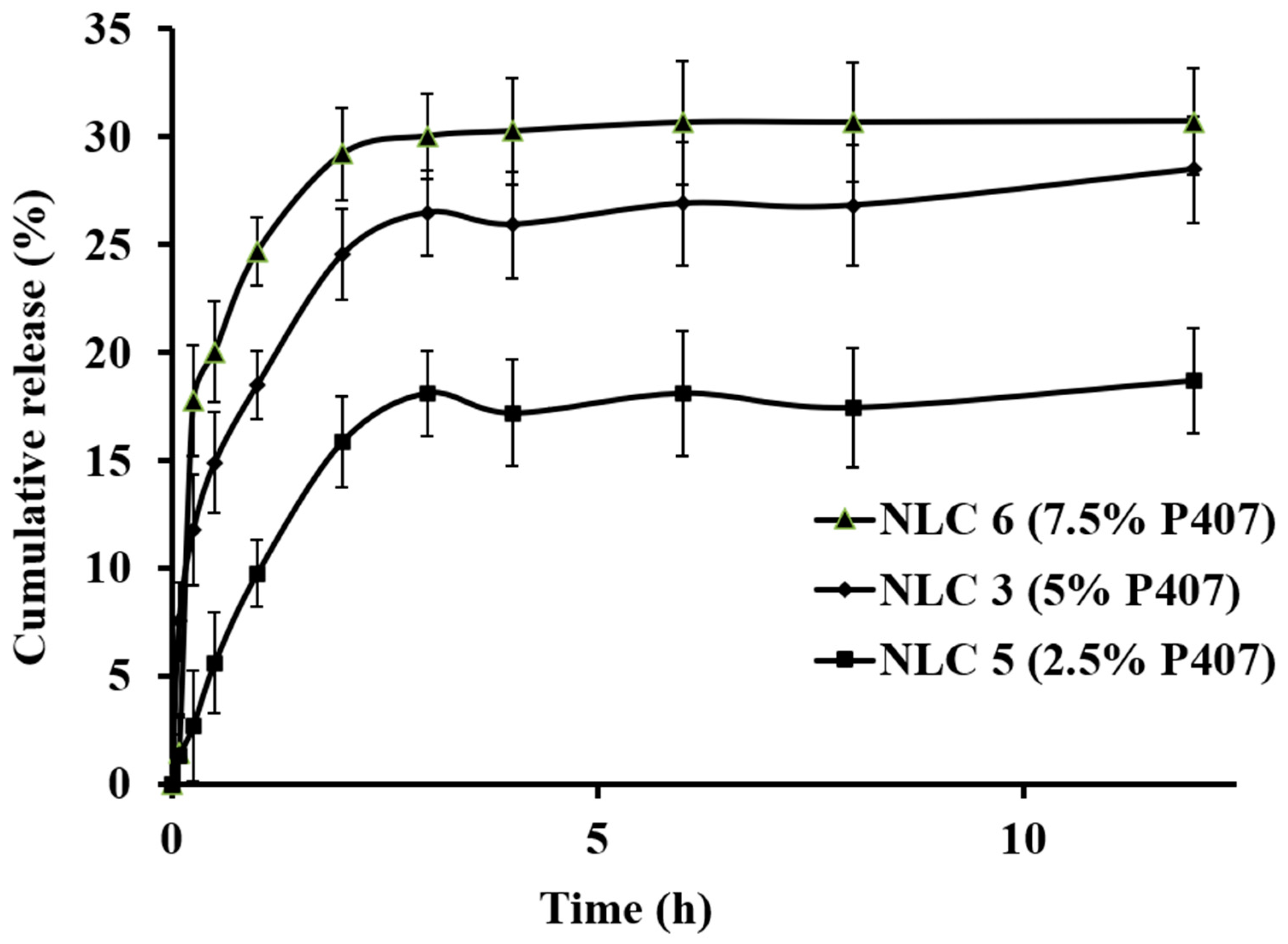
3.4. In Vitro Drug Release
3.5. Differential Scanning Calorimetry
3.6. Pharmacokinetics
3.7. Stability Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prevention and management of osteoporosis. World Health Organ Tech. Rep. Ser. 2003, 921, 1–164.
- Francis, R.M. An Atlas of Osteoporosis: The Encyclopedia of Visual Medicine Series. J. R. Soc. Med. 1993, 86, 682. [Google Scholar]
- Kanis, J. Assessment of Osteoporosis at the Primary Health Care Level; Technical Report; World Health Organization Collaborating Centre for Metabolic Bone Diseases: Geneva, Switzerland, 2008; pp. 6–9. [Google Scholar]
- Johnell, O.; Kanis, J.A. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. 2006, 17, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Thiebaud, D.; Secrest, R.J. Selective estrogen receptor modulators: Mechanism of action and clinical experience. Focus on raloxifene. Reprod. Fertil. Dev. 2001, 13, 331–336. [Google Scholar] [CrossRef]
- Lufkin, E.G.; Whitaker, M.D.; Nickelsen, T.; Argueta, R.; Caplan, R.H.; Knickerbocker, R.K.; Riggs, B.L. Treatment of established postmenopausal osteoporosis with raloxifene: A randomized trial. J. Bone Miner. Res. 1998, 13, 1747–1754. [Google Scholar] [CrossRef]
- Pinsky, P.F.; Miller, E.A.; Heckman-Stoddard, B.M.; Minasian, L. Breast Cancer Characteristics and Survival among Users versus Nonusers of Raloxifene. Cancer Prev. Res. 2020, 13, 83–90. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Gluck, O.; Maricic, M. Skeletal and nonskeletal effects of raloxifene. Curr. Osteoporos. Rep. 2003, 1, 123–128. [Google Scholar] [CrossRef]
- Trdan Lusin, T.; Mrhar, A.; Stieger, B.; Kullak-Ublick, G.A.; Marc, J.; Ostanek, B.; Zavratnik, A.; Kristl, A.; Berginc, K.; Delic, K.; et al. Influence of hepatic and intestinal efflux transporters and their genetic variants on the pharmacokinetics and pharmacodynamics of raloxifene in osteoporosis treatment. Transl. Res. 2012, 160, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Ravi, P.R.; Aditya, N.; Kathuria, H.; Malekar, S.; Vats, R. Lipid nanoparticles for oral delivery of raloxifene: Optimization, stability, in vivo evaluation and uptake mechanism. Eur. J. Pharm. Biopharm. 2014, 87, 114–124. [Google Scholar] [CrossRef]
- Findlay, S.M. Drug Delivery Markets—An Outlook. Available online: https://www.frost.com/sublib/display-market-insight.do?id=134287829 (accessed on 26 March 2016).
- Lu, R.; Liu, S.; Wang, Q.; Li, X. Enhanced bioavailability of raloxifene hydrochloride via dry suspensions prepared from drug/HP-beta-cyclodextrin inclusion complexes. Pharmazie 2015, 70, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, T.; Li, W.; Sun, H.; Lu, D.; Wu, B.; Zhang, X. Glucose-Based Mesoporous Carbon Nanospheres as Functional Carriers for Oral Delivery of Amphiphobic Raloxifene: Insights into the Bioavailability Enhancement and Lymphatic Transport. Pharm. Res. 2016, 33, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Velpula, A.; Jukanti, R.; Janga, K.Y.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Proliposome powders for enhanced intestinal absorption and bioavailability of raloxifene hydrochloride: Effect of surface charge. Drug Dev. Ind. Pharm. 2013, 39, 1895–1906. [Google Scholar] [CrossRef]
- Tran, T.H.; Poudel, B.K.; Marasini, N.; Woo, J.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development of raloxifene-solid dispersion with improved oral bioavailability via spray-drying technique. Arch. Pharm. Res. 2013, 36, 86–93. [Google Scholar] [CrossRef]
- Tran, T.H.; Poudel, B.K.; Marasini, N.; Chi, S.C.; Choi, H.G.; Yong, C.S.; Kim, J.O. Preparation and evaluation of raloxifene-loaded solid dispersion nanoparticle by spray-drying technique without an organic solvent. Int. J. Pharm. 2013, 443, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, H.; Nangesh, J.; Parmar, M.; Patel, D. Formulation and characterization of lipid-based drug delivery system of raloxifene-microemulsion and self-microemulsifying drug delivery system. J. Pharm. Bioallied Sci. 2011, 3, 442–448. [Google Scholar] [CrossRef]
- Bummer, P.M. Physical chemical considerations of lipid-based oral drug delivery--solid lipid nanoparticles. Crit. Rev. Drug Carr. Syst. 2004, 21, 1–20. [Google Scholar] [CrossRef]
- Muller, R.H.; Runge, S.; Ravelli, V.; Mehnert, W.; Thunemann, A.F.; Souto, E.B. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN) versus drug nanocrystals. Int. J. Pharm. 2006, 317, 82–89. [Google Scholar] [CrossRef]
- Plapied, L.; Duhem, N.; des Rieux, A.; Préat, V. Fate of polymeric nanocarriers for oral drug delivery. Curr. Opin. Colloid Interface Sci. 2011, 16, 228–237. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef]
- Sanjula, B.; Shah, F.M.; Javed, A.; Alka, A. Effect of poloxamer 188 on lymphatic uptake of carvedilol-loaded solid lipid nanoparticles for bioavailability enhancement. J. Drug Target 2009, 17, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Battani, S.; Pawar, H.; Suresh, S. Evaluation of oral bioavailability and anticancer potential of raloxifene solid lipid nanoparticles. J. Nanosci. Nanotechnol. 2014, 14, 5638–5645. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, A.K.; Vuddanda, P.R.; Karunanidhi, P.; Singh, S.K.; Singh, S. Development and evaluation of solid lipid nanoparticles of raloxifene hydrochloride for enhanced bioavailability. Biomed. Res. Int. 2013, 2013, 584549. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.H.; Ramasamy, T.; Cho, H.J.; Kim, Y.I.; Poudel, B.K.; Choi, H.G.; Yong, C.S.; Kim, J.O. Formulation and optimization of raloxifene-loaded solid lipid nanoparticles to enhance oral bioavailability. J. Nanosci. Nanotechnol. 2014, 14, 4820–4831. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Xiong, S.B.; Yang, H.; Yin, X.D.; Chao, R.B. Solid lipid nanoparticles of mitoxantrone for local injection against breast cancer and its lymph node metastases. Eur. J. Pharm. Sci. 2006, 28, 86–95. [Google Scholar] [CrossRef]
- Severino, P.; Andreani, T.; Macedo, A.S.; Fangueiro, J.F.; Santana, M.H.; Silva, A.M.; Souto, E.B. Current State-of-Art and New Trends on Lipid Nanoparticles (SLN and NLC) for Oral Drug Delivery. J. Drug Deliv. 2012, 2012, 750891. [Google Scholar] [CrossRef]
- Yang, S.C.; Lu, L.F.; Cai, Y.; Zhu, J.B.; Liang, B.W.; Yang, C.Z. Body distribution in mice of intravenously injected camptothecin solid lipid nanoparticles and targeting effect on brain. J. Control. Release 1999, 59, 299–307. [Google Scholar] [CrossRef]
- Puro, D.; Athawale, R.; Pandya, A. Design, Optimization and Characterization of Nanostructured Lipid Carriers of Raloxifene Hydrochloride for Transdermal Delivery. Nanosci. Nanotechnol. Asia 2020, 10, 57–67. [Google Scholar] [CrossRef]
- Shah, N.V.; Seth, A.K.; Balaraman, R.; Aundhia, C.J.; Maheshwari, R.A.; Parmar, G.R. Nanostructured lipid carriers for oral bioavailability enhancement of raloxifene: Design and in vivo study. J. Adv. Res. 2016, 7, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Himanshu Kathuria, A.N.; Ravi, P.R. Raloxifene Loaded SLN and NLC: Comparison of In Vitro Properties and In Vivo Behavior after Oral Administration in Rats. In Proceedings of the 3rd Nano Today Conference, At Biopolis, Singapore, 8–11 December 2013; p. 186. [Google Scholar]
- Ravi, P.R.; Aditya, N.; Vats, R. Development, validation, and pharmacokinetic application of liquid chromatographic method for estimation of raloxifene hydrochloride in rabbit plasma. Acta Chromatogr. 2012, 24, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Kjellman, P.; Fredriksson, S.; Kjellman, C.; Strand, S.E.; Zandt, R.I. Size-dependent lymphatic uptake of nanoscale-tailored particles as tumor mass increases. Future Sci. OA 2015, 1, FSO60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.S.; Suzuki, K.; Cho, H.; Youn, Y.S.; Bae, Y.H. Oral Nanoparticles Exhibit Specific High-Efficiency Intestinal Uptake and Lymphatic Transport. ACS Nano 2018, 12, 8893–8900. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, M.; Su, Z.; Sun, M.; Ping, Q. Size-exclusive effect of nanostructured lipid carriers on oral drug delivery. Int. J. Pharm. 2016, 511, 524–537. [Google Scholar] [CrossRef]
- Banerjee, A.; Qi, J.; Gogoi, R.; Wong, J.; Mitragotri, S. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J. Control Release 2016, 238, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, R.H.; Radtke, M.; Wissing, S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv Rev. 2002, 54 (Suppl. 1), S131–S155. [Google Scholar] [CrossRef]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Budida, R.; Scherr, M.; Kuchenbauer, F.; Lindner, R.; Noyan, F.; Suhs, K.W.; Stangel, M.; et al. Lipid nanoparticle-mediated siRNA delivery for safe targeting of human CML in vivo. Ann. Hematol. 2019, 98, 1905–1918. [Google Scholar] [CrossRef]
- Uprit, S.; Kumar Sahu, R.; Roy, A.; Pare, A. Preparation and characterization of minoxidil loaded nanostructured lipid carrier gel for effective treatment of alopecia. Saudi. Pharm. J. 2013, 21, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Chaudhury, A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech 2011, 12, 62–76. [Google Scholar] [CrossRef] [Green Version]
- Uner, M.; Yener, G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int. J. Nanomed. 2007, 2, 289–300. [Google Scholar]
- Heiati, H.; Tawashi, R.; Shivers, R.R.; Phillips, N.C. Solid lipid nanoparticles as drug carriers. I. Incorporation and retention of the lipophilic prodrug 3′-azido-3′-deoxythymidine palmitate. Int. J. Pharm. 1997, 146, 123–131. [Google Scholar] [CrossRef]
- Pandita, D.; Ahuja, A.; Velpandian, T.; Lather, V.; Dutta, T.; Khar, R.K. Characterization and in vitro assessment of paclitaxel loaded lipid nanoparticles formulated using modified solvent injection technique. Pharmazie 2009, 64, 301–310. [Google Scholar] [PubMed]
- Pezeshki, A.; Ghanbarzadeh, B.; Mohammadi, M.; Fathollahi, I.; Hamishehkar, H. Encapsulation of Vitamin A Palmitate in Nanostructured Lipid Carrier (NLC)-Effect of Surfactant Concentration on the Formulation Properties. Adv. Pharm. Bull. 2014, 4, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Kelidari, H.R.; Saeedi, M.; Akbari, J.; Morteza-Semnani, K.; Gill, P.; Valizadeh, H.; Nokhodchi, A. Formulation optimization and in vitro skin penetration of spironolactone loaded solid lipid nanoparticles. Colloids Surf B Biointerfaces 2015, 128, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Ye, N.; Luo, Z.; Lai, W.; Cai, X.; Lin, Y. Absorption, pharmacokinetics and disposition properties of solid lipid nanoparticles (SLNs). Curr. Drug Metab. 2012, 13, 447–456. [Google Scholar] [CrossRef]
- Hosny, K.M.; Bahmdan, R.H.; Alhakamy, N.A.; Alfaleh, M.A.; Ahmed, O.A.; Elkomy, M.H. Physically Optimized Nano-Lipid Carriers Augment Raloxifene and Vitamin D Oral Bioavailability in Healthy Humans for Management of Osteoporosis. J. Pharm. Sci. 2020. [Google Scholar] [CrossRef]
- Souto, E.B.; Wissing, S.A.; Barbosa, C.M.; Muller, R.H. Development of a controlled release formulation based on SLN and NLC for topical clotrimazole delivery. Int. J. Pharm. 2004, 278, 71–77. [Google Scholar] [CrossRef]
- Muller, R.H.; Shegokar, R.; Keck, C.M. 20 years of lipid nanoparticles (SLN and NLC): Present state of development and industrial applications. Curr. Drug Discov. Technol. 2011, 8, 207–227. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Drug (mg) | Glyceryl Behenate (g) | Oleic Acid (g) | P 407 (% w/v, [mL]) | Water q.s. (mL) | HT (min) | ST (min) |
|---|---|---|---|---|---|---|---|
| NLC 1 | NA | 0.7 | 0.3 | 5.0 [20] | 50 | 3 | 10 |
| NLC 2 | 50 | 0.7 | 0.3 | 5.0 [20] | 50 | NA | 10 |
| NLC 3 | 50 | 0.7 | 0.3 | 5.0 [20] | 50 | 3 | 10 |
| NLC 4 | 50 | 0.7 | 0.3 | 5.0 [20] | 50 | 3 | 20 |
| NLC 5 | 50 | 0.7 | 0.3 | 2.5 [20] | 50 | 3 | 10 |
| NLC 6 | 50 | 0.7 | 0.3 | 7.5 [20] | 50 | 3 | 10 |
| NLC 7 | 50 | 0.77 | 0.23 | 5.0 [20] | 50 | 3 | 10 |
| NLC 8 | 50 | 0.83 | 0.17 | 5.0 [20] | 50 | 3 | 10 |
| NLC 9 | 50 | 0.89 | 0.09 | 5.0 [20] | 50 | 3 | 10 |
| Code | d-avg (d.nm) | PDI | d50 (d.nm) | d90 (d.nm) | d95 (d.nm) | Zeta Potential (mV) |
|---|---|---|---|---|---|---|
| NLC 1 | 124 | 0.191 | 115 | 227 | 267 | −13.2 |
| NLC 2 | 310 | 0.233 | 280 | 521 | 621 | +8.71 |
| NLC 3 | 120 | 0.293 | 97 | 209 | 270 | +14.4 |
| NLC 4 | 142 | 0.259 | 123 | 250 | 302 | +12.3 |
| NLC 5 | 132 | 0.224 | 124 | 240 | 288 | +14.8 |
| NLC 6 | 179 | 0.305 | 120 | 271 | 329 | +11.7 |
| NLC 7 | 115 | 0.380 | 173 | 492 | 659 | +15.2 |
| NLC 8 | 182 | 0.319 | 255 | 572 | 689 | +17.8 |
| NLC 9 | 247 | 0.383 | 342 | 790 | 1100 | +15.4 |
| Code | EE (%) | Free Drug (%) | CR (%) | CR (%, Excluding Free Drug) |
|---|---|---|---|---|
| NLC 3 (5% P407) | 91.71 | 8.29 | 28.48 | 20.19 |
| NLC 6 (7.5% P407) | 88.13 | 11.87 | 30.16 | 18.29 |
| NLC 5 (2.5% P407) | 94.51 | 5.49 | 18.68 | 13.19 |
| NLC 4 (5% P407) | 92.07 | 7.93 | 30.71 | 22.79 |
| Parameter | RLX Suspension | RLX-NLC |
|---|---|---|
| Cmax (ng/mL) | 181.71 ± 17.83 | 391.35 ± 32.53 * |
| Tmax (h) | 2.00 ± 0.32 | 1.00 ± 0.25 * |
| MRT (h) | 13.00 ± 1.17 | 19.09 ± 1.05 * |
| AUC(0-t) (µg.h/mL) | 2.49 ± 0.23 | 7.71 ± 0.34 * |
| t1/2 (h) | 13.21 ± 1.35 | 20.00 ± 2.13 * |
| Frel | - | 3.19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murthy, A.; Rao Ravi, P.; Kathuria, H.; Malekar, S. Oral Bioavailability Enhancement of Raloxifene with Nanostructured Lipid Carriers. Nanomaterials 2020, 10, 1085. https://doi.org/10.3390/nano10061085
Murthy A, Rao Ravi P, Kathuria H, Malekar S. Oral Bioavailability Enhancement of Raloxifene with Nanostructured Lipid Carriers. Nanomaterials. 2020; 10(6):1085. https://doi.org/10.3390/nano10061085
Chicago/Turabian StyleMurthy, Aditya, Punna Rao Ravi, Himanshu Kathuria, and Shrinivas Malekar. 2020. "Oral Bioavailability Enhancement of Raloxifene with Nanostructured Lipid Carriers" Nanomaterials 10, no. 6: 1085. https://doi.org/10.3390/nano10061085
APA StyleMurthy, A., Rao Ravi, P., Kathuria, H., & Malekar, S. (2020). Oral Bioavailability Enhancement of Raloxifene with Nanostructured Lipid Carriers. Nanomaterials, 10(6), 1085. https://doi.org/10.3390/nano10061085