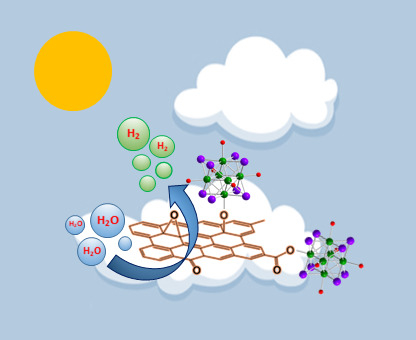
Enhanced Photocatalytic Activity and Stability in Hydrogen Evolution of Mo6 Iodide Clusters Supported on Graphene Oxide

 ,
, 
 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Instrumentation
2.3. Single-Crystal Isolation and X-ray Data Collection
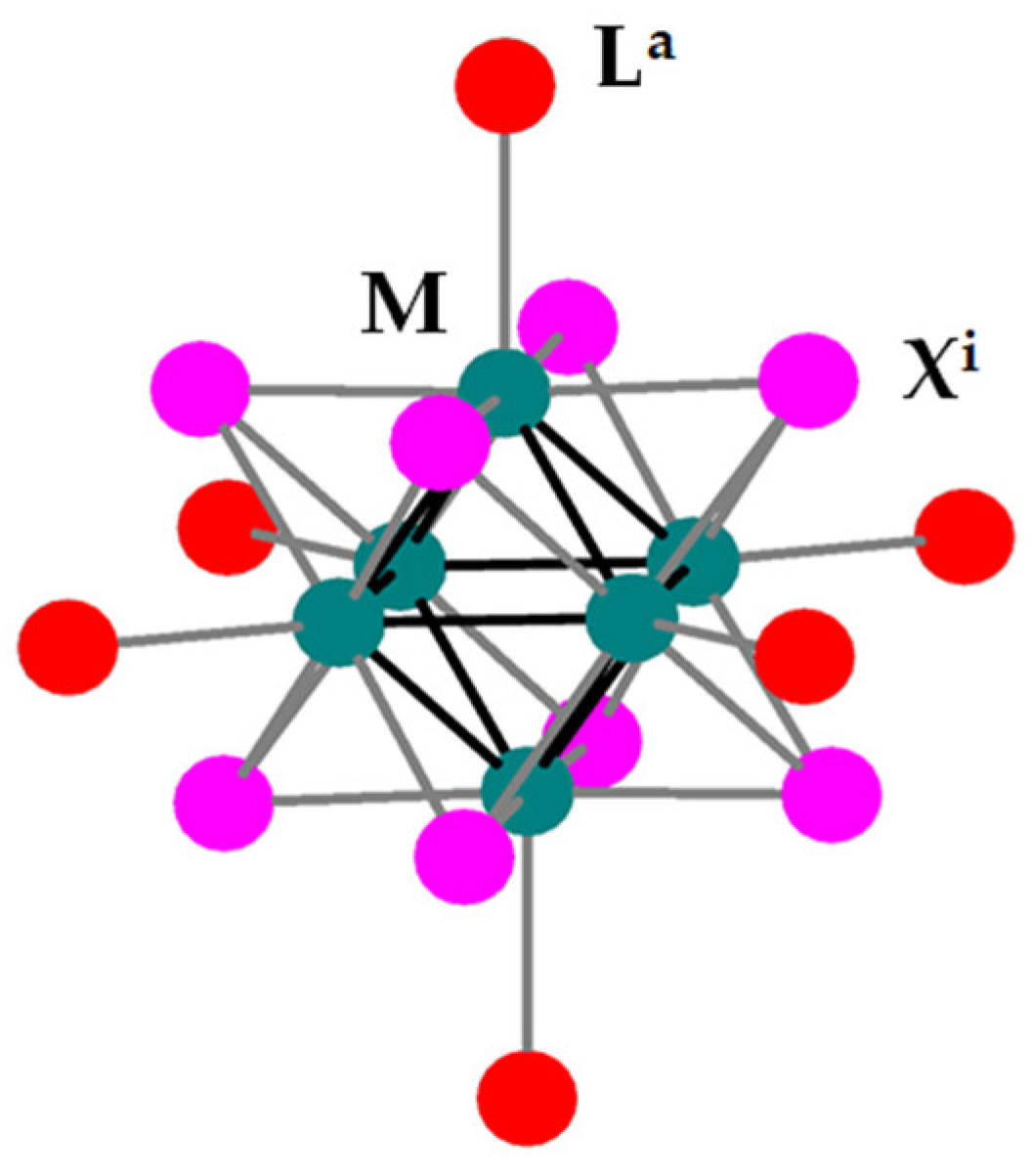
2.4. Synthesis and Characterization of (TBA)2Mo6Ii8@GO
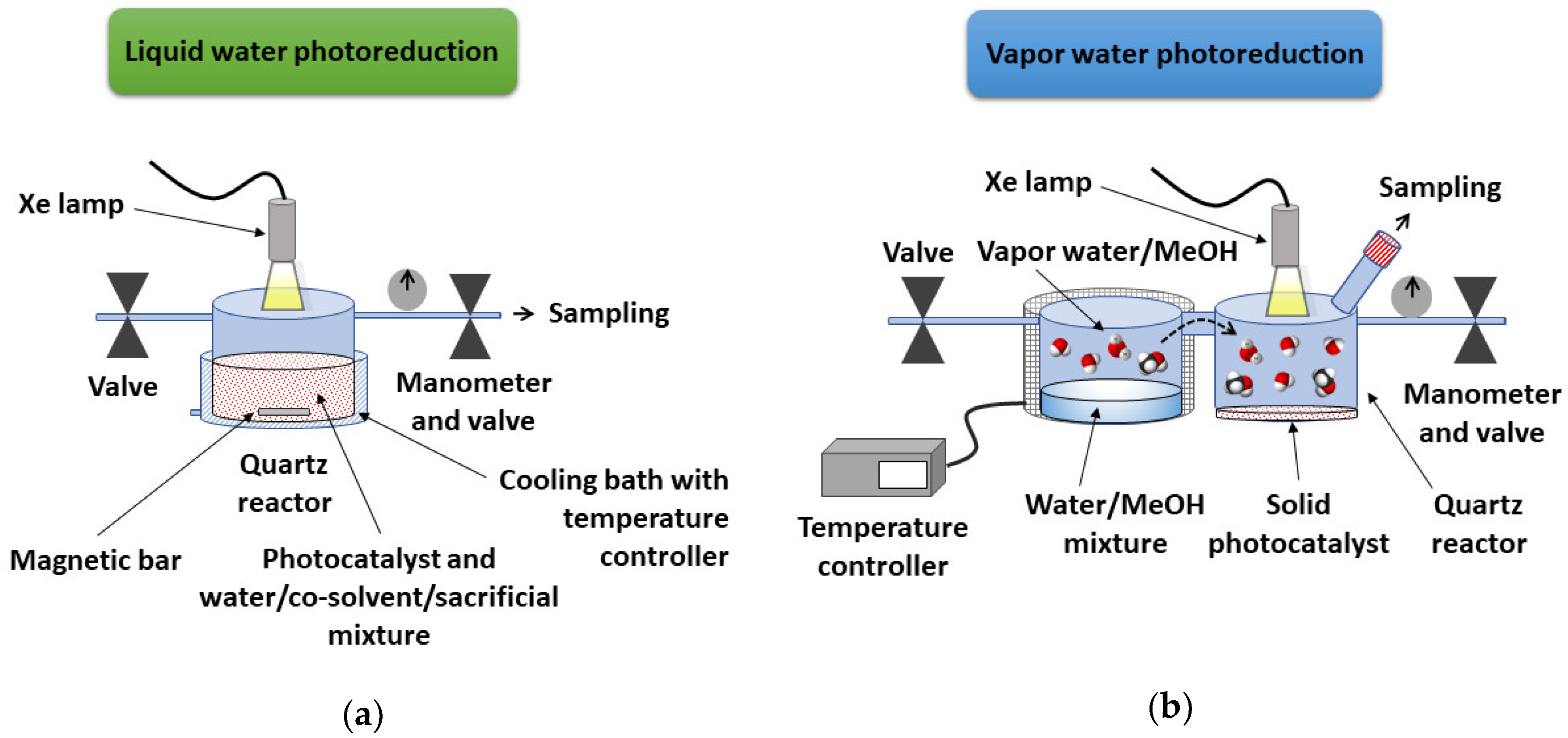
2.5. Photocatalytic H2 Evolution Procedure
3. Results and Discussion
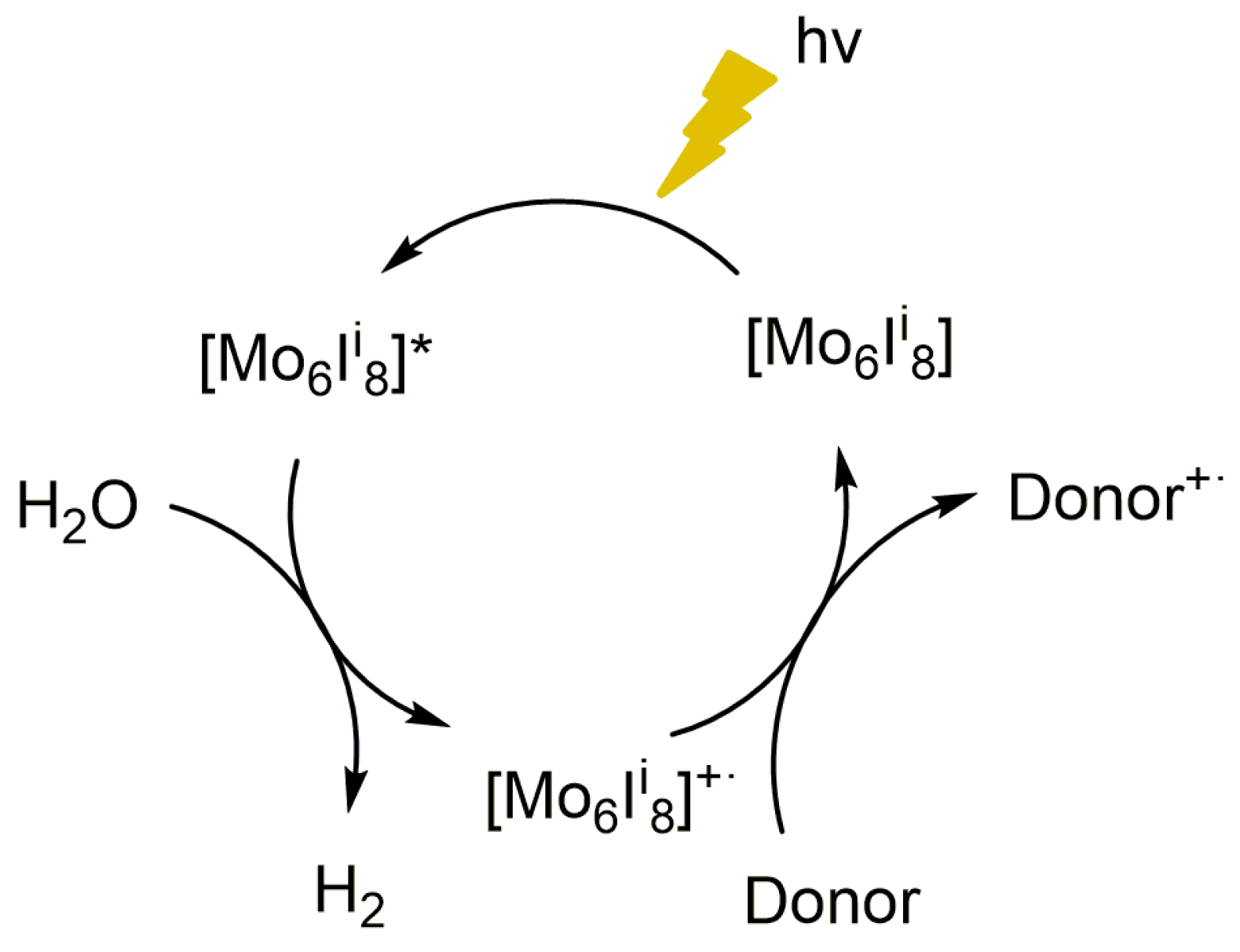
3.1. Photocatalytic Activity of the (TBA)2[Mo6Ii8(O2CCH3)a6] Compound in the Photoreduction of Water in Liquid Phase
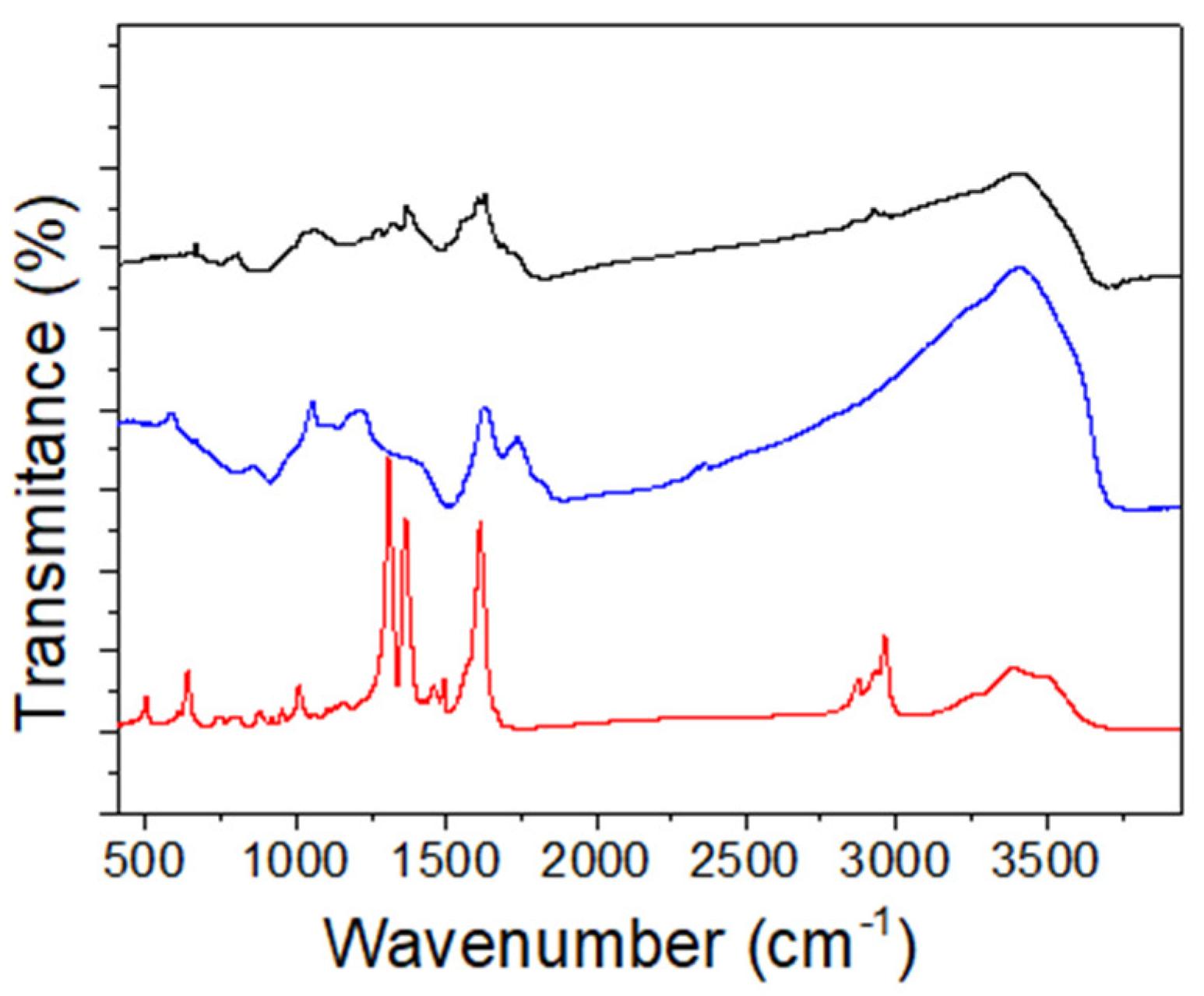
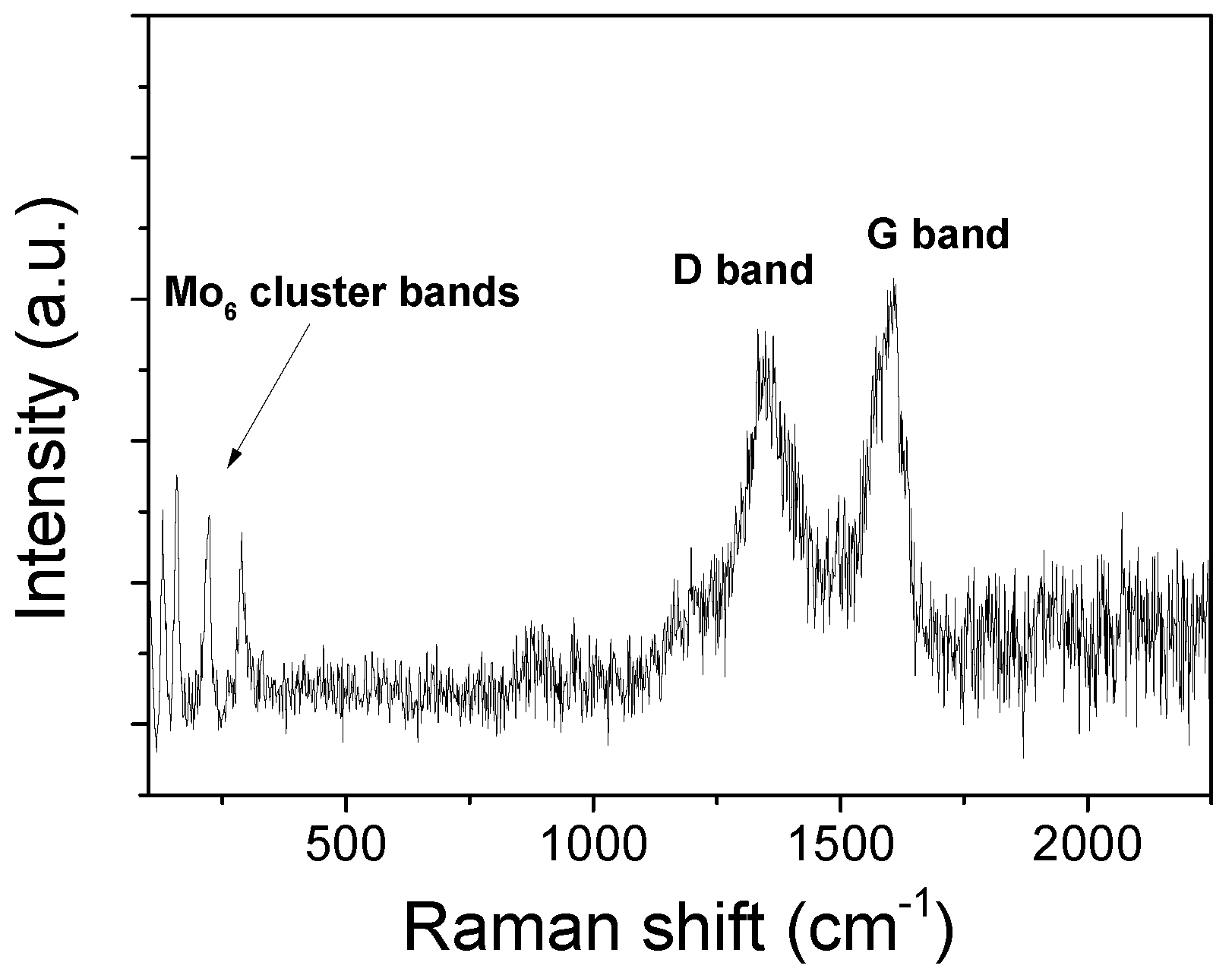
3.2. Synthesis and Characterization of the (TBA)2Mo6Ii8@GO Nanocomposite
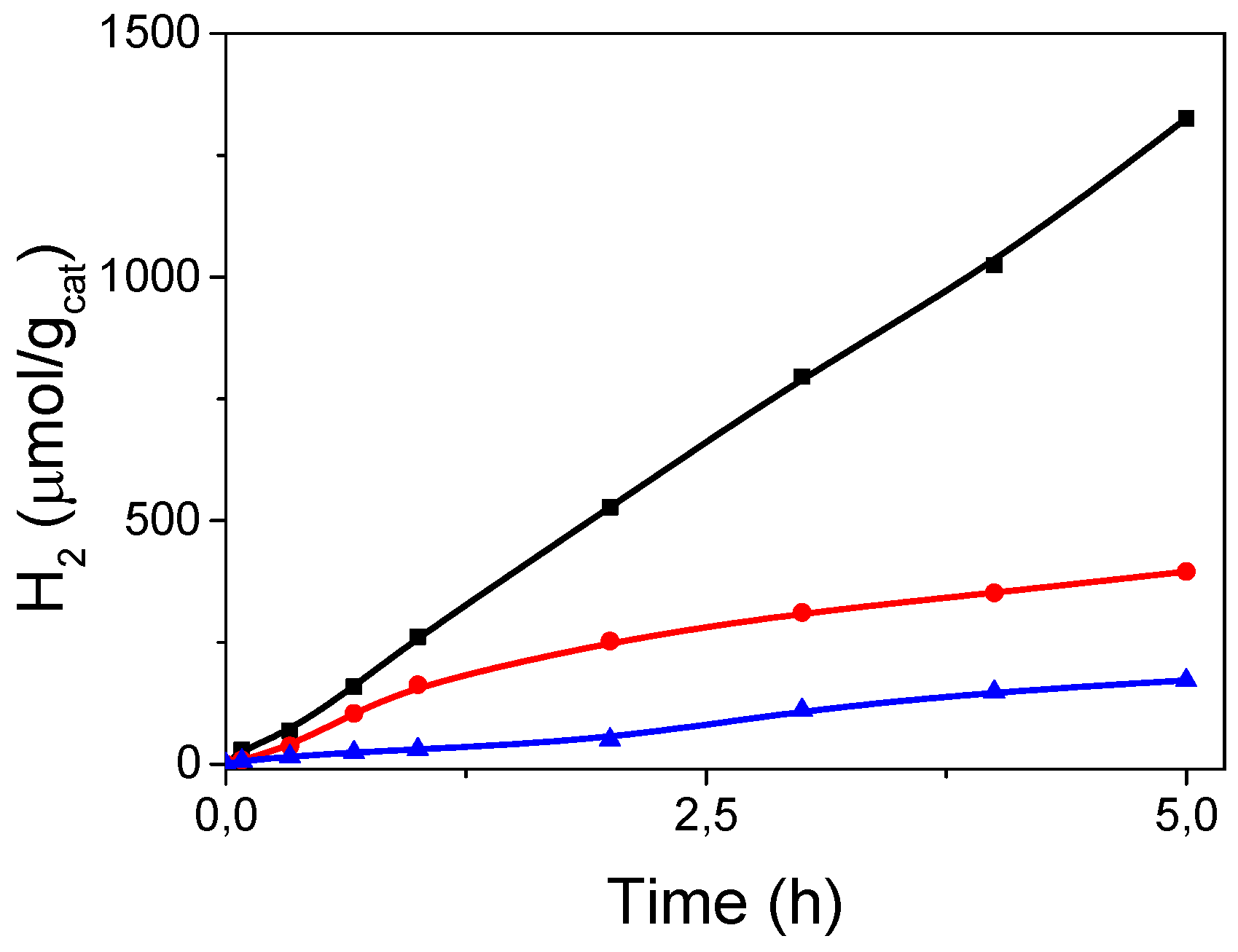
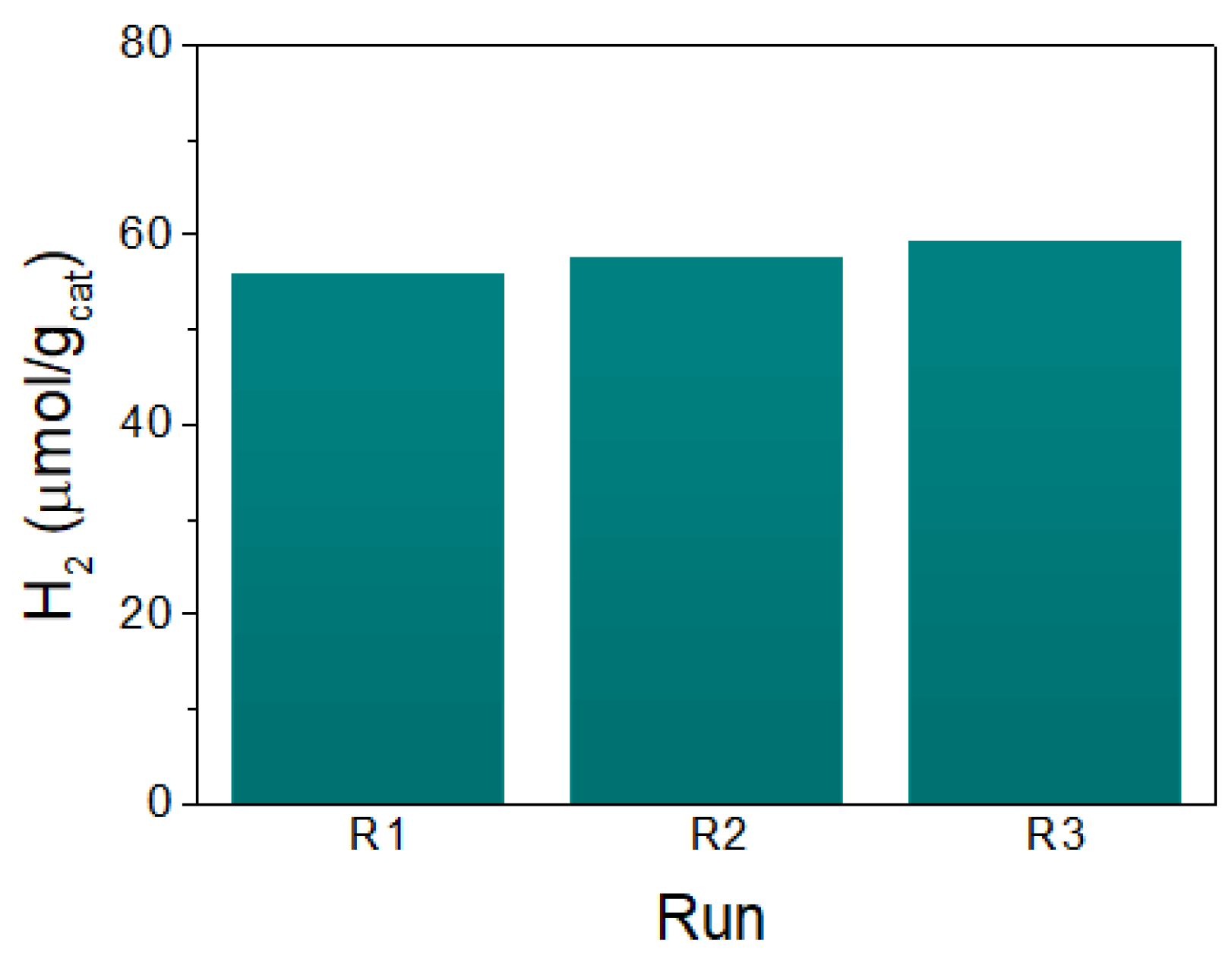
3.3. Catalytic Properties of (TBA)2Mo6Ii8@GO in the Photoreduction of Liquid Phase Water
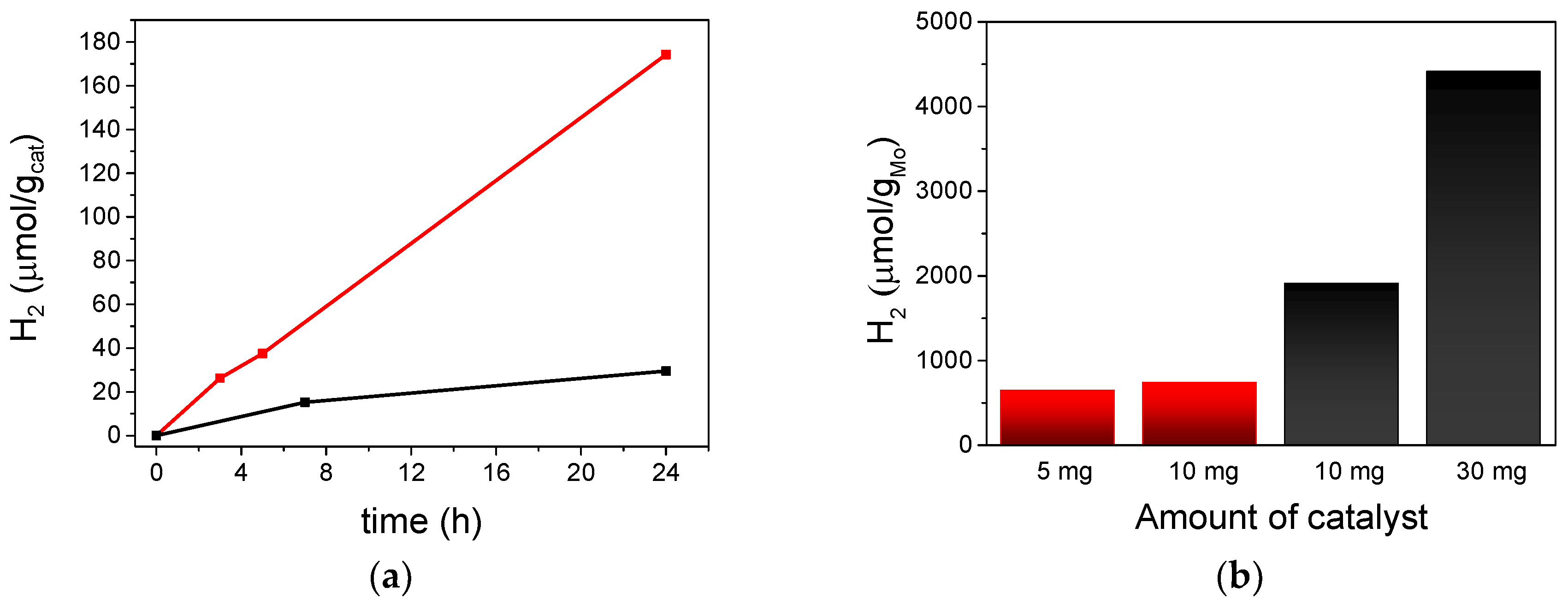
3.4. Photocatalytic Activity of Microcrystalline (TBA)2[Mo6Ii8(O2CCH3)a6] and (TBA)2Mo6Ii8@GO in the Presence of Aqueous Mixtures in Vapor Phase
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calise, F.; D’Accadia, M.D.; Santarelli, M.; Lanzini, A.; Ferrero, D.B.T. Solar Hydrogen Production, Processes, Systems and Technologies; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Ture, E. Hydrogen Production from Solar Energy. In Assessment of Hydrogen Energy for Sustainable Development; Sheffield, J.W., Sheffield, Ç., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 135–146. [Google Scholar]
- Dincer, I.; Zamfirescu, C. Sustainable Hydrogen Production; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Zou, X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef]
- Koutavarapu, R.; Venkata Reddy, C.; Babu, B.; Reddy, K.R.; Cho, M.; Shim, J. Carbon cloth/transition metals-based hybrids with controllable architectures for electrocatalytic hydrogen evolution—A review. Int. J. Hydrogen Energy 2020, 45, 7716–7740. [Google Scholar] [CrossRef]
- Babu, B.; Koutavarapu, R.; Shim, J.; Yoo, K. Enhanced visible-light-driven photoelectrochemical and photocatalytic performance of Au-SnO2 quantum dot-anchored g-C3N4 nanosheets. Sep. Purif. Technol. 2020, 240, 116652. [Google Scholar] [CrossRef]
- Volonakis, G.; Giustino, F. Surface properties of lead-free halide double perovskites: Possible visible-light photo-catalysts for water splitting. Appl. Phys. Lett. 2018, 112, 243901. [Google Scholar] [CrossRef]
- Yuan, Y.-J.; Chen, D.; Yu, Z.-T.; Zou, Z.-G. Cadmium sulfide-based nanomaterials for photocatalytic hydrogen production. J. Mater. Chem. A 2018, 6, 11606–11630. [Google Scholar] [CrossRef]
- Cordier, S.; Grasset, F.; Molard, Y.; Amela-Cortes, M.; Boukherroub, R.; Ravaine, S.; Mortier, M.; Ohashi, N.; Saito, N.; Haneda, H. Inorganic Molybdenum Octahedral Nanosized Cluster Units, Versatile Functional Building Block for Nanoarchitectonics. J. Inorg. Organomet. Polym. Mater. 2015, 25, 189–204. [Google Scholar] [CrossRef]
- Nguyen, N.T.K.; Renaud, A.; Dierre, B.; Bouteille, B.; Wilmet, M.; Dubernet, M.; Ohashi, N.; Grasset, F.; Uchikoshi, T. Extended Study on Electrophoretic Deposition Process of Inorganic Octahedral Metal Clusters: Advanced Multifunctional Transparent Nanocomposite Thin Films. Bull. Chem. Soc. Jpn. 2018, 91, 1763–1774. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.K.N.; Grasset, F.; Cordier, S.; Amela-Cortes, M.; Matsui, Y.; Ohashi, N.; Shirahata, N.; Uchikoshi, T. Preparation and characterization of hollow silica nanocomposite functionalized with UV absorbable molybdenum cluster. Adv. Powder Technol. 2020, 31, 895–903. [Google Scholar] [CrossRef]
- Renaud, A.; Nguyen, T.K.N.; Grasset, F.; Raissi, M.; Guillon, V.; Delabrouille, F.; Dumait, N.; Jouan, P.-Y.; Cario, L.; Jobic, S.; et al. Preparation by electrophoretic deposition of molybdenum iodide cluster-based functional nanostructured photoelectrodes for solar cells. Electrochim. Acta 2019, 317, 737–745. [Google Scholar] [CrossRef]
- Daigre, G.; Cuny, J.; Lemoine, P.; Amela-Cortes, M.; Paofai, S.; Audebrand, N.; Le Gal La Salle, A.; Quarez, E.; Joubert, O.; Naumov, N.G.; et al. Metal Atom Clusters as Building Blocks for Multifunctional Proton-Conducting Materials: Theoretical and Experimental Characterization. Inorg. Chem. 2018, 57, 9814–9825. [Google Scholar] [CrossRef]
- Renaud, A.; Grasset, F.; Dierre, B.; Uchikoshi, T.; Ohashi, N.; Takei, T.; Planchat, A.; Cario, L.; Jobic, S.; Odobel, F.; et al. Inorganic Molybdenum Clusters as Light-Harvester in All Inorganic Solar Cells: A Proof of Concept. ChemistrySelect 2016, 1, 2284–2289. [Google Scholar] [CrossRef]
- Vorotnikov, Y.A.; Efremova, O.A.; Vorotnikova, N.A.; Brylev, K.A.; Edeleva, M.V.; Tsygankova, A.R.; Smolentsev, A.I.; Kitamura, N.; Mironov, Y.V.; Shestopalov, M.A. On the synthesis and characterisation of luminescent hybrid particles: Mo6 metal cluster complex/SiO2. RSC Adv. 2016, 6, 43367–43375. [Google Scholar] [CrossRef] [Green Version]
- Nerambourg, N.; Aubert, T.; Neaime, C.; Cordier, S.; Mortier, M.; Patriarche, G.; Grasset, F. Multifunctional hybrid silica nanoparticles based on [Mo6Br14]2− phosphorescent nanosized clusters, magnetic γ-Fe2O3 and plasmonic gold nanoparticles. J. Colloid Interface Sci. 2014, 424, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechézelles, J.-F.; Aubert, T.; Grasset, F.; Cordier, S.; Barthou, C.; Schwob, C.; Maître, A.; Vallée, R.A.L.; Cramail, H.; Ravaine, S. Fine tuning of emission through the engineering of colloidal crystals. Phys. Chem. Chem. Phys. 2010, 12, 11993–11999. [Google Scholar] [CrossRef] [PubMed]
- Grasset, F.; Dorson, F.; Cordier, S.; Molard, Y.; Perrin, C.; Marie, A.-M.; Sasaki, T.; Haneda, H.; Bando, Y.; Mortier, M. Water-in-Oil Microemulsion Preparation and Characterization of Cs2[Mo6X14]@SiO2 Phosphor Nanoparticles Based on Transition Metal Clusters (X = Cl, Br, and I). Adv. Mater. 2008, 20, 143–148. [Google Scholar] [CrossRef]
- Robin, M.; Kuai, W.; Amela-Cortes, M.; Cordier, S.; Molard, Y.; Mohammed-Brahim, T.; Jacques, E.; Harnois, M. Epoxy Based Ink as Versatile Material for Inkjet-Printed Devices. ACS Appl. Mater. Interfaces 2015, 7, 21975–21984. [Google Scholar] [CrossRef]
- Dybtsev, D.; Serre, C.; Schmitz, B.; Panella, B.; Hirscher, M.; Latroche, M.; Llewellyn, P.L.; Cordier, S.; Molard, Y.; Haouas, M.; et al. Influence of [Mo6Br8F6]2− Cluster Unit Inclusion within the Mesoporous Solid MIL-101 on Hydrogen Storage Performance. Langmuir 2010, 26, 11283–11290. [Google Scholar] [CrossRef]
- Vorotnikov, Y.A.; Pozmogova, T.N.; Solovieva, A.O.; Miroshnichenko, S.M.; Vorontsova, E.V.; Shestopalova, L.V.; Mironov, Y.V.; Shestopalov, M.A.; Efremova, O.A. Luminescent silica mesoparticles for protein transduction. Mater. Sci. Eng. C 2019, 96, 530–538. [Google Scholar] [CrossRef]
- Elistratova, J.; Mukhametshina, A.; Kholin, K.; Nizameev, I.; Mikhailov, M.; Sokolov, M.; Khairullin, R.; Miftakhova, R.; Shammas, G.; Kadirov, M.; et al. Interfacial uploading of luminescent hexamolybdenum cluster units onto amino-decorated silica nanoparticles as new design of nanomaterial for cellular imaging and photodynamic therapy. J. Colloid Interface Sci. 2019, 538, 387–396. [Google Scholar] [CrossRef]
- Cheplakova, A.M.; Solovieva, A.O.; Pozmogova, T.N.; Vorotnikov, Y.A.; Brylev, K.A.; Vorotnikova, N.A.; Vorontsova, E.V.; Mironov, Y.V.; Poveshchenko, A.F.; Kovalenko, K.A.; et al. Nanosized mesoporous metal–organic framework MIL-101 as a nanocarrier for photoactive hexamolybdenum cluster compounds. J. Inorg. Biochem. 2017, 166, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Neaime, C.; Amela-Cortes, M.; Grasset, F.; Molard, Y.; Cordier, S.; Dierre, B.; Mortier, M.; Takei, T.; Takahashi, K.; Haneda, H.; et al. Time-gated luminescence bioimaging with new luminescent nanocolloids based on [Mo6I8(C2F5COO)6]2− metal atom clusters. Phys. Chem. Chem. Phys. 2016, 18, 30166–30173. [Google Scholar] [CrossRef] [PubMed]
- Solovieva, A.O.; Vorotnikov, Y.A.; Trifonova, K.E.; Efremova, O.A.; Krasilnikova, A.A.; Brylev, K.A.; Vorontsova, E.V.; Avrorov, P.A.; Shestopalova, L.V.; Poveshchenko, A.F.; et al. Cellular internalisation, bioimaging and dark and photodynamic cytotoxicity of silica nanoparticles doped by {Mo6I8}4+ metal clusters. J. Mater. Chem. B 2016, 4, 4839–4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, T.; Cabello-Hurtado, F.; Esnault, M.-A.; Neaime, C.; Lebret-Chauvel, D.; Jeanne, S.; Pellen, P.; Roiland, C.; Le Polles, L.; Saito, N.; et al. Extended Investigations on Luminescent Cs2[Mo6Br14]@SiO2 Nanoparticles: Physico-Structural Characterizations and Toxicity Studies. J. Phys. Chem. C 2013, 117, 20154–20163. [Google Scholar] [CrossRef]
- Kirakci, K.; Kubát, P.; Fejfarová, K.; Martinčík, J.; Nikl, M.; Lang, K. X-ray Inducible Luminescence and Singlet Oxygen Sensitization by an Octahedral Molybdenum Cluster Compound: A New Class of Nanoscintillators. Inorg. Chem. 2016, 55, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Evtushok, D.V.; Melnikov, A.R.; Vorotnikova, N.A.; Vorotnikov, Y.A.; Ryadun, A.A.; Kuratieva, N.V.; Kozyr, K.V.; Obedinskaya, N.R.; Kretov, E.I.; Novozhilov, I.N.; et al. A comparative study of optical properties and X-ray induced luminescence of octahedral molybdenum and tungsten cluster complexes. Dalt. Trans. 2017, 46, 11738–11747. [Google Scholar] [CrossRef] [Green Version]
- Kirakci, K.; Zelenka, J.; Rumlová, M.; Cvačka, J.; Ruml, T.; Lang, K. Cationic octahedral molybdenum cluster complexes functionalized with mitochondria-targeting ligands: Photodynamic anticancer and antibacterial activities. Biomater. Sci. 2019, 7, 1386–1392. [Google Scholar] [CrossRef]
- Nagashima, S.; Kamiguchi, S.; Chihara, T. Catalytic Reactions over Halide Cluster Complexes of Group 5–7 Metals. Metals 2014, 4, 235–313. [Google Scholar] [CrossRef] [Green Version]
- Kamiguchi, S.; Nagashima, S.; Chihara, T. Characterization of Catalytically Active Octahedral Metal Halide Cluster Complexes. Metals 2014, 4, 84–107. [Google Scholar] [CrossRef] [Green Version]
- Barras, A.; Cordier, S.; Boukherroub, R. Fast photocatalytic degradation of rhodamine B over [Mo6Br8(N3)6]2− cluster units under sun light irradiation. Appl. Catal. B Environ. 2012, 123–124, 1–8. [Google Scholar] [CrossRef]
- Barras, A.; Das, M.R.; Devarapalli, R.R.; Shelke, M.V.; Cordier, S.; Szunerits, S.; Boukherroub, R. One-pot synthesis of gold nanoparticle/molybdenum cluster/graphene oxide nanocomposite and its photocatalytic activity. Appl. Catal. B Environ. 2013, 130–131, 270–276. [Google Scholar] [CrossRef]
- Kumar, P.; Mungse, H.P.; Cordier, S.; Boukherroub, R.; Khatri, O.P.; Jain, S.L. Hexamolybdenum clusters supported on graphene oxide: Visible-light induced photocatalytic reduction of carbon dioxide into methanol. Carbon 2015, 94, 91–100. [Google Scholar] [CrossRef]
- Beltrán, A.; Mikhailov, M.; Sokolov, M.N.; Pérez-Laguna, V.; Rezusta, A.; Revillo, M.J.; Galindo, F. A photobleaching resistant polymer supported hexanuclear molybdenum iodide cluster for photocatalytic oxygenations and photodynamic inactivation of Staphylococcus aureus. J. Mater. Chem. B 2016, 4, 5975–5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliz, M.; Puche, M.; Atienzar, P.; Concepción, P.; Cordier, S.; Molard, Y. In Situ Generation of Active Molybdenum Octahedral Clusters for Photocatalytic Hydrogen Production from Water. ChemSusChem 2016, 9, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Feliz, M.; Atienzar, P.; Amela-Cortés, M.; Dumait, N.; Lemoine, P.; Molard, Y.; Cordier, S. Supramolecular Anchoring of Octahedral Molybdenum Clusters onto Graphene and Their Synergies in Photocatalytic Water Reduction. Inorg. Chem. 2019, 58, 15443–15454. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, M.N.; Vorotnikov, Y.A.; Plotnikova, E.E.; Marchuk, M.V.; Ivanov, A.A.; Asanov, I.P.; Tsygankova, A.R.; Grayfer, E.D.; Fedorov, V.E.; Shestopalov, M.A. Hexamolybdenum Clusters Supported on Exfoliated h-BN Nanosheets for Photocatalytic Water Purification. Inorg. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, S.; Cordier, S.; Paofai, S.; Boukherroub, R.; Jain, S.L. Photoreduction of CO2 to methanol with hexanuclear molybdenum [Mo6Br14]2− cluster units under visible light irradiation. RSC Adv. 2014, 4, 10420–10423. [Google Scholar] [CrossRef]
- Prévôt, M.; Amela-Cortes, M.; Manna, S.K.; Lefort, R.; Cordier, S.; Folliot, H.; Dupont, L.; Molard, Y. Design and Integration in Electro-Optic Devices of Highly Efficient and Robust Red-NIR Phosphorescent Nematic Hybrid Liquid Crystals Containing [Mo6I8(OCOCnF2n+1)6]2− (n = 1, 2, 3) Nanoclusters. Adv. Funct. Mater. 2015, 25, 4966–4975. [Google Scholar] [CrossRef]
- Sokolov, M.N.; Mihailov, M.A.; Peresypkina, E.V.; Brylev, K.A.; Kitamura, N.; Fedin, V.P. Highly luminescent complexes [Mo6X8(n-C3F7COO)6]2− (X = Br, I). Dalt. Trans. 2011, 40, 6375–6377. [Google Scholar] [CrossRef]
- Kirakci, K.; Kubát, P.; Dušek, M.; Fejfarová, K.; Šícha, V.; Mosinger, J.; Lang, K. A Highly Luminescent Hexanuclear Molybdenum Cluster—A Promising Candidate toward Photoactive Materials. Eur. J. Inorg. Chem. 2012, 3107–3111. [Google Scholar] [CrossRef]
- Kirakci, K.; Kubat, P.; Langmaier, J.; Polivka, T.; Fuciman, M.; Fejfarova, K.; Lang, K. A comparative study of the redox and excited state properties of (nBu4N)2[Mo6X14] and (nBu4N)2[Mo6X8(CF3COO)6] (X = Cl, Br, or I). Dalt. Trans. 2013, 42, 7224–7232. [Google Scholar] [CrossRef]
- Efremova, O.A.; Shestopalov, M.A.; Chirtsova, N.A.; Smolentsev, A.I.; Mironov, Y.V.; Kitamura, N.; Brylev, K.A.; Sutherland, A.J. A highly emissive inorganic hexamolybdenum cluster complex as a handy precursor for the preparation of new luminescent materials. Dalt. Trans. 2014, 43, 6021–6025. [Google Scholar] [CrossRef] [Green Version]
- Efremova, O.A.; Vorotnikov, Y.A.; Brylev, K.A.; Vorotnikova, N.A.; Novozhilov, I.N.; Kuratieva, N.V.; Edeleva, M.V.; Benoit, D.M.; Kitamura, N.; Mironov, Y.V.; et al. Octahedral molybdenum cluster complexes with aromatic sulfonate ligands. Dalt. Trans. 2016, 45, 15427–15435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailov, M.A.; Brylev, K.A.; Abramov, P.A.; Sakuda, E.; Akagi, S.; Ito, A.; Kitamura, N.; Sokolov, M.N. Synthetic Tuning of Redox, Spectroscopic, and Photophysical Properties of {Mo6I8}4+ Core Cluster Complexes by Terminal Carboxylate Ligands. Inorg. Chem. 2016, 55, 8437–8445. [Google Scholar] [CrossRef]
- Riehl, L.; Seyboldt, A.; Ströbele, M.; Enseling, D.; Jüstel, T.; Westberg, M.; Ogilby, P.R.; Meyer, H.J. A ligand substituted tungsten iodide cluster: Luminescence vs. singlet oxygen production. Dalt. Trans. 2016, 45, 15500–15506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolov, M.N.; Brylev, K.A.; Abramov, P.A.; Gallyamov, M.R.; Novozhilov, I.N.; Kitamura, N.; Mikhaylov, M.A. Complexes of {W6I8}4+ Clusters with Carboxylates: Preparation, Electrochemistry, and Luminescence. Eur. J. Inorg. Chem. 2017, 2017, 4131–4137. [Google Scholar] [CrossRef]
- Mikhaylov, M.A.; Sokolov, M.N. Molybdenum Iodides–from Obscurity to Bright Luminescence. Eur. J. Inorg. Chem. 2019, 2019, 4181–4197. [Google Scholar] [CrossRef]
- Yam, K.M.; Guo, N.; Jiang, Z.; Li, S.; Zhang, C. Graphene-Based Heterogeneous Catalysis: Role of Graphene. Catalysts 2020, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, C.; Shi, G. Graphene based catalysts. Energy Environ. Sci. 2012, 5, 8848–8868. [Google Scholar] [CrossRef]
- Xiang, Q.; Yu, J.; Jaroniec, M. Graphene-based semiconductor photocatalysts. Chem. Soc. Rev. 2012, 41, 782–796. [Google Scholar] [CrossRef]
- Yeh, T.-F.; Syu, J.-M.; Cheng, C.; Chang, T.-H.; Teng, H. Graphite Oxide as a Photocatalyst for Hydrogen Production from Water. Adv. Funct. Mater. 2010, 20, 2255–2262. [Google Scholar] [CrossRef]
- Yeh, T.-F.; Cihlář, J.; Chang, C.-Y.; Cheng, C.; Teng, H. Roles of graphene oxide in photocatalytic water splitting. Mater. Today 2013, 16, 78–84. [Google Scholar] [CrossRef]
- Latorre-Sánchez, M.; Lavorato, C.; Puche, M.; Fornés, V.; Molinari, R.; Garcia, H. Visible-Light Photocatalytic Hydrogen Generation by Using Dye-Sensitized Graphene Oxide as a Photocatalyst. Chem. A Eur. J. 2012, 18, 16774–16783. [Google Scholar] [CrossRef] [PubMed]
- Lerf, A.; He, H.; Riedl, T.; Forster, M.; Klinowski, J. 13C and 1H MAS NMR studies of graphite oxide and its chemically modified derivatives. Solid State Ion. 1997, 101–103, 857–862. [Google Scholar] [CrossRef]
- Konios, D.; Stylianakis, M.M.; Stratakis, E.; Kymakis, E. Dispersion behaviour of graphene oxide and reduced graphene oxide. J. Colloid Interface Sci. 2014, 430, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kharisov, B.I.; Kharissova, O.V.; Vázquez Dimas, A.; Gómez De La Fuente, I.; Peña Méndez, Y. Review: Graphene-supported coordination complexes and organometallics: Properties and applications. J. Coord. Chem. 2016, 69, 1125–1151. [Google Scholar] [CrossRef]
- Axet, M.R.; Dechy-Cabaret, O.; Durand, J.; Gouygou, M.; Serp, P. Coordination chemistry on carbon surfaces. Coord. Chem. Rev. 2016, 308, 236–345. [Google Scholar] [CrossRef]
- Axet, M.R.; Durand, J.; Gouygou, M.; Serp, P. Surface coordination chemistry on graphene and two-dimensional carbon materials for well-defined single atom supported catalysts. Adv. Organomet. Chem. 2019, 71, 53–174. [Google Scholar] [CrossRef]
- Arora, S.; Gupta, N.; Singh, V. Improved Pd/Ru metal supported graphene oxide nano-catalysts for hydrodeoxygenation (HDO) of vanillyl alcohol, vanillin and lignin. Green Chem. 2020, 22, 2018–2027. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, J.; Fan, W. Graphene-based catalysis for biomass conversion. Catal. Sci. Technol. 2015, 5, 3845–3858. [Google Scholar] [CrossRef]
- Das, V.K.; Shifrina, Z.B.; Bronstein, L.M. Graphene and graphene-like materials in biomass conversion: Paving the way to the future. J. Mater. Chem. A 2017, 5, 25131–25143. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Khatri, O.P.; Cordier, S.; Boukherroub, R.; Jain, S.L. Graphene Oxide Supported Molybdenum Cluster: First Heterogenized Homogeneous Catalyst for the Synthesis of Dimethylcarbonate from CO2 and Methanol. Chem. A Eur. J. 2015, 21, 3488–3494. [Google Scholar] [CrossRef]
- Sheldon, J.C. 76. Bromo- and iodo-molybdenum(II) compounds. J. Chem. Soc. 1962, 410–415. [Google Scholar] [CrossRef]
- Schreck, M.; Niederberger, M. Photocatalytic Gas Phase Reactions. Chem. Mater. 2019, 31, 597–618. [Google Scholar] [CrossRef]
- Lapicque, F.; Lédé, J.; Villermaux, J. Design and optimization of a reactor for high temperature dissociation of water and carbon dioxide using solar energy. Chem. Eng. Sci. 1986, 41, 677–684. [Google Scholar] [CrossRef]
- Dionigi, F.; Vesborg, P.C.K.; Pedersen, T.; Hansen, O.; Dahl, S.; Xiong, A.; Maeda, K.; Domen, K.; Chorkendorff, I. Gas phase photocatalytic water splitting with Rh2−yCryO3/GaN:ZnO in μ-reactors. Energy Environ. Sci. 2011, 4, 2937–2942. [Google Scholar] [CrossRef]
- Volostnykh, M.V.; Mikhaylov, M.A.; Sinelshchikova, A.A.; Kirakosyan, G.A.; Martynov, A.G.; Grigoriev, M.S.; Piryazev, D.A.; Tsivadze, A.Y.; Sokolov, M.N.; Gorbunova, Y.G. Hybrid organic–inorganic supramolecular systems based on a pyridine end-decorated molybdenum(ii) halide cluster and zinc(ii) porphyrinate. Dalt. Trans. 2019, 48, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Felip-León, C.; Puche, M.; Miravet, J.F.; Galindo, F.; Feliz, M. A spectroscopic study to assess the photogeneration of singlet oxygen by graphene oxide. Mater. Lett. 2019, 251, 45–51. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro Agilent Technologies, version 1.171.40.53; Rigaku Oxford Diffraction: Woodlands, TX, USA; Agilent Technologies UK Ltd.: Yarnton, UK, 2019.
- Puche, M. Nanomateriales Híbridos Basados en Complejos de Metales de Transición Anclados Sobre óxido de Grafeno. Ph.D. Thesis, Universitat Politécnica de València, Valencia, Spain, 2017. [Google Scholar]
- Montes-Navajas, P.; Asenjo, N.G.; Santamaría, R.; Menéndez, R.; Corma, A.; García, H. Surface Area Measurement of Graphene Oxide in Aqueous Solutions. Langmuir 2013, 29, 13443–13448. [Google Scholar] [CrossRef] [PubMed]
- Povedailo, V.A.; Ronishenko, B.V.; Stepuro, V.I.; Tsybulsky, D.A.; Shmanai, V.V.; Yakovlev, D.L. Fluorescence Quenching of Dyes by Graphene Oxide. J. Appl. Spectrosc. 2018, 85, 605–610. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, C.; Liu, Y. Investigation on fluorescence quenching of dyes by graphite oxide and graphene. Appl. Surf. Sci. 2011, 257, 5513–5518. [Google Scholar] [CrossRef]
- De Miguel, M.; Álvaro, M.; García, H. Graphene as a Quencher of Electronic Excited States of Photochemical Probes. Langmuir 2012, 28, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Nannelli, P.; Block, B.P. Molybdenum(II) cluster compounds involving alkoxy groups. Inorg. Chem. 1968, 7, 2423–2426. [Google Scholar] [CrossRef]
- Brničević, N.; Bašic, I.; Hoxha, B.; Planinić, P.; McCarley, R.E. Molybdenum and tungsten methoxo clusters with differently bonded methoxo groups: Crystal structure of [Na(CH3OH)5]2[Mo6(μ3-Br)8(OCH3)6]. Polyhedron 2003, 22, 1553–1559. [Google Scholar] [CrossRef]
- Schoonover, J.R.; Zietlow, T.C.; Clark, D.L.; Heppert, J.A.; Chisholm, M.H.; Gray, H.B.; Sattelberger, A.P.; Woodruff, W.H. Resonance Raman Spectra of [M6X8Y6]2− Cluster Complexes (M = Mo, W; X, Y = Cl, Br, I). Inorg. Chem. 1996, 35, 6606–6613. [Google Scholar] [CrossRef]
- Gao, W.; Alemany, L.B.; Ci, L.; Ajayan, P.M. New insights into the structure and reduction of graphite oxide. Nat. Chem. 2009, 1, 403–408. [Google Scholar] [CrossRef]
- Gurunathan, S.; Woong Han, J.; Kim, J. Green chemistry approach for the synthesis of biocompatible graphene. Int. J. Nanomed. 2013, 8, 2719. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puche, M.; García-Aboal, R.; Mikhaylov, M.A.; Sokolov, M.N.; Atienzar, P.; Feliz, M. Enhanced Photocatalytic Activity and Stability in Hydrogen Evolution of Mo6 Iodide Clusters Supported on Graphene Oxide. Nanomaterials 2020, 10, 1259. https://doi.org/10.3390/nano10071259
Puche M, García-Aboal R, Mikhaylov MA, Sokolov MN, Atienzar P, Feliz M. Enhanced Photocatalytic Activity and Stability in Hydrogen Evolution of Mo6 Iodide Clusters Supported on Graphene Oxide. Nanomaterials. 2020; 10(7):1259. https://doi.org/10.3390/nano10071259
Chicago/Turabian StylePuche, Marta, Rocío García-Aboal, Maxim A. Mikhaylov, Maxim N. Sokolov, Pedro Atienzar, and Marta Feliz. 2020. "Enhanced Photocatalytic Activity and Stability in Hydrogen Evolution of Mo6 Iodide Clusters Supported on Graphene Oxide" Nanomaterials 10, no. 7: 1259. https://doi.org/10.3390/nano10071259
APA StylePuche, M., García-Aboal, R., Mikhaylov, M. A., Sokolov, M. N., Atienzar, P., & Feliz, M. (2020). Enhanced Photocatalytic Activity and Stability in Hydrogen Evolution of Mo6 Iodide Clusters Supported on Graphene Oxide. Nanomaterials, 10(7), 1259. https://doi.org/10.3390/nano10071259