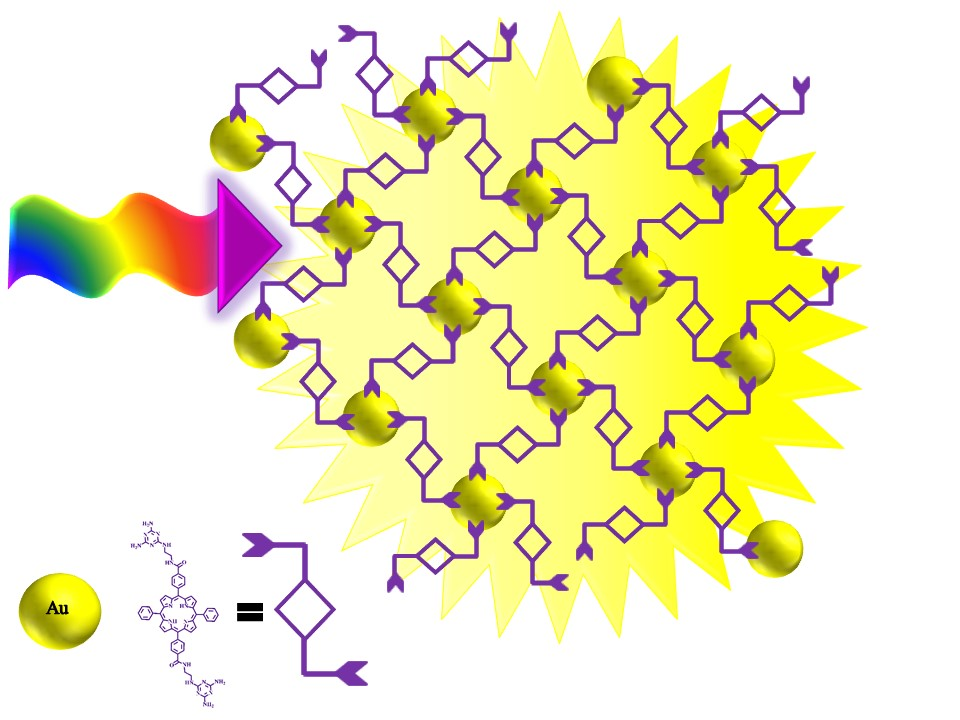
Covalently Conjugated Gold–Porphyrin Nanostructures

 ,
, 


 ,
,  ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Synthesis of Au NPs
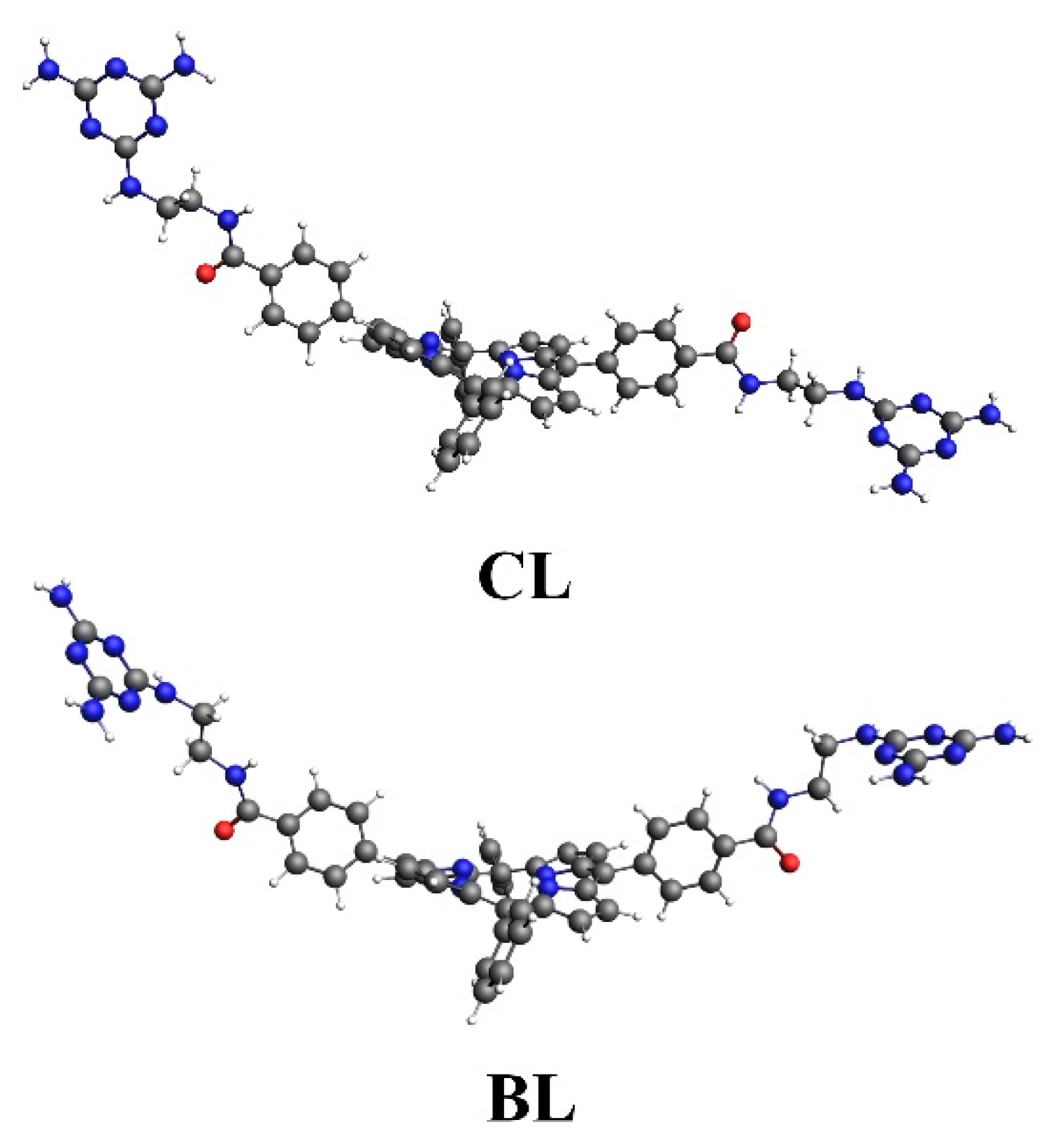
2.2. Synthesis of Di-Triazine-Porphyrin
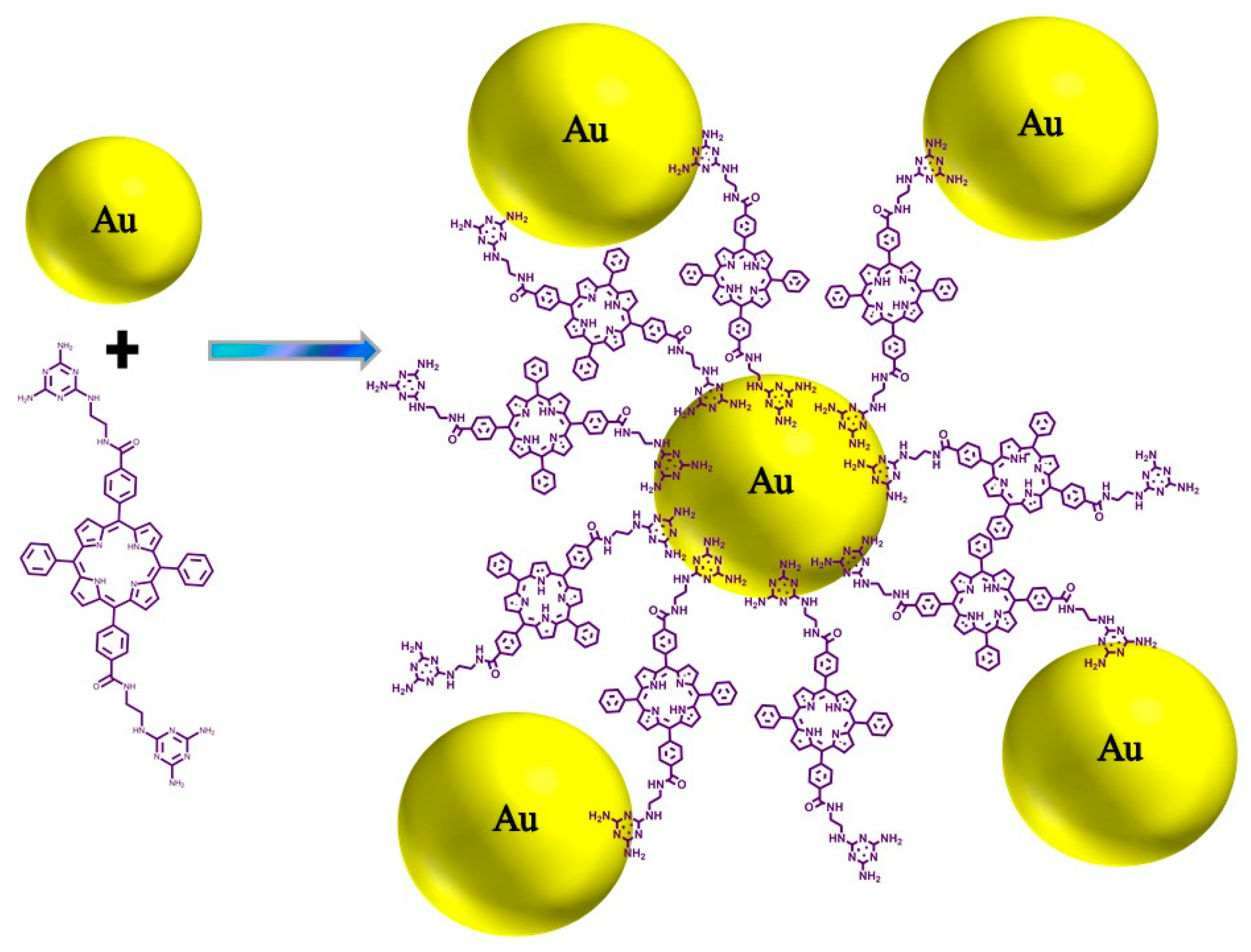
2.3. Syntheses of the Au NPs–Porphyrin Nanostructures
2.4. Transmission Electron Microscopy Measurements
2.5. X-ray Photoelectron Data
2.6. UV–vis Spectra
2.7. PL Measurements
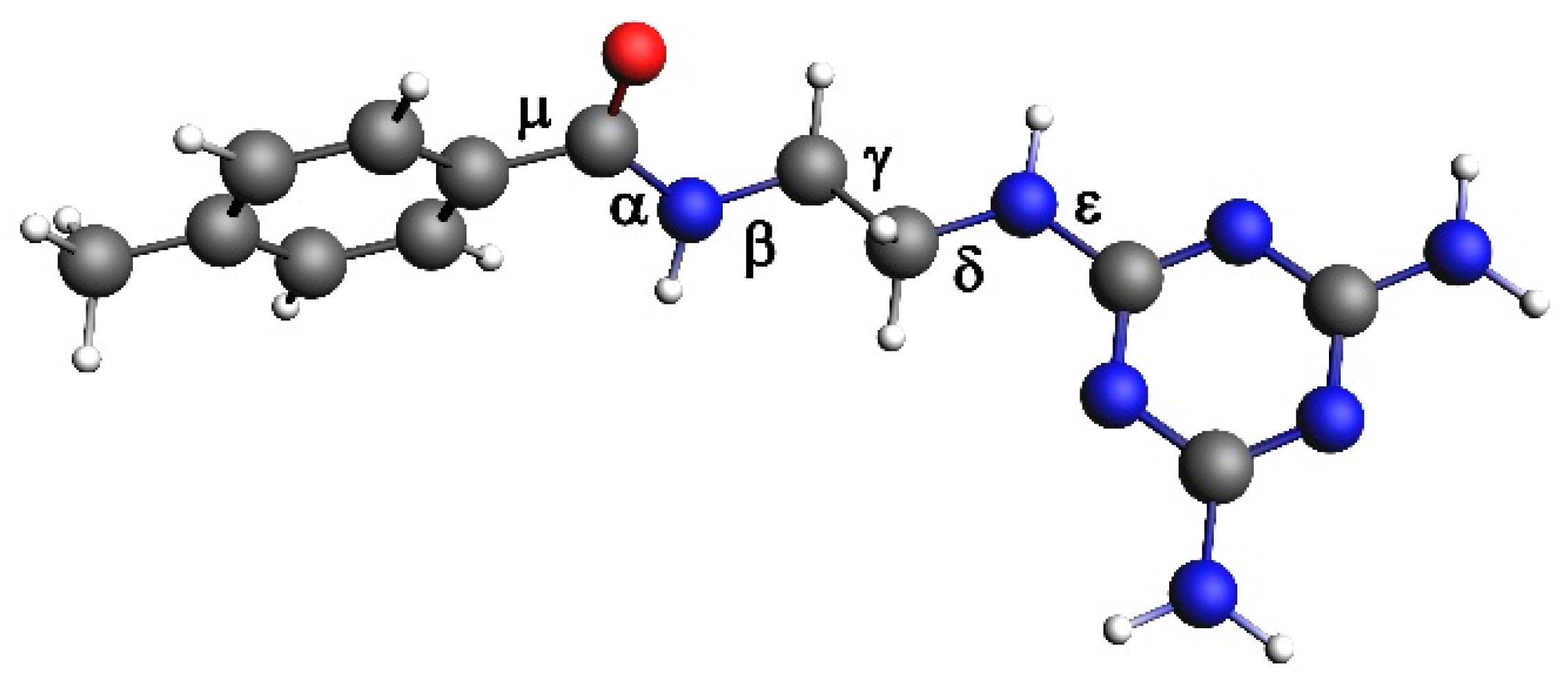
2.8. Quantum Mechanical Calculation
3. Results
3.1. Results of Quantum Mechanical Calculations
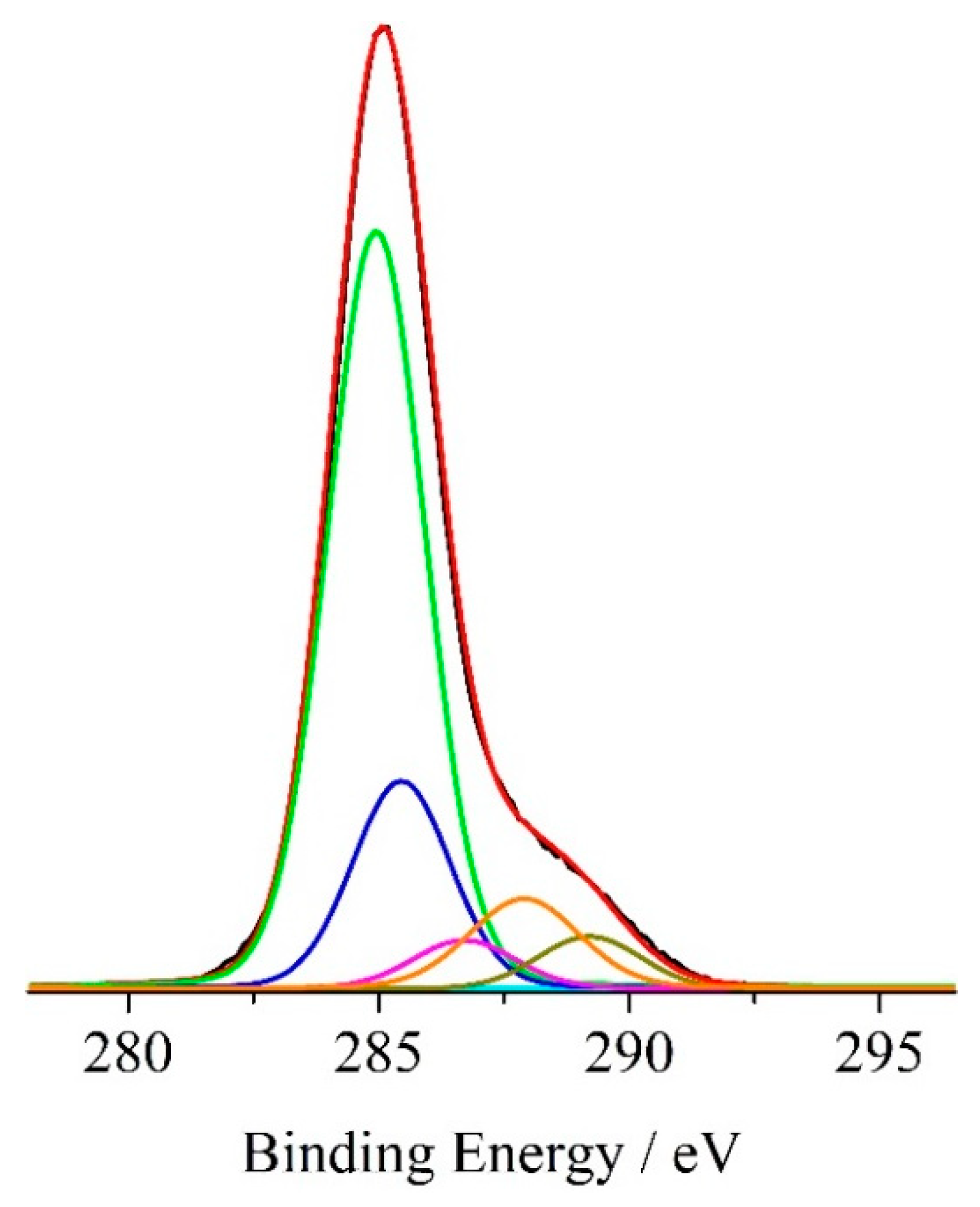
3.2. XPS Results
3.3. TEM Measurements
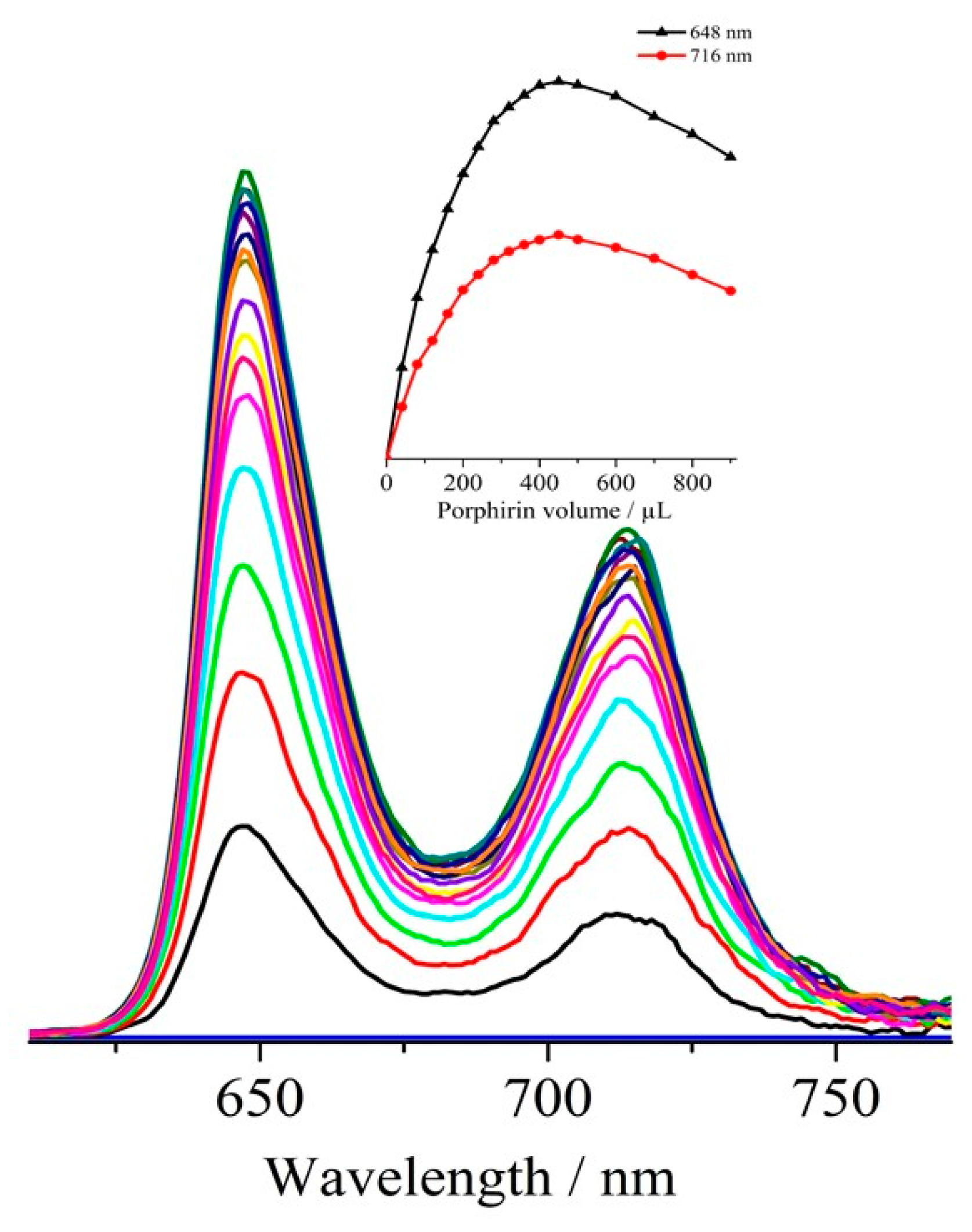
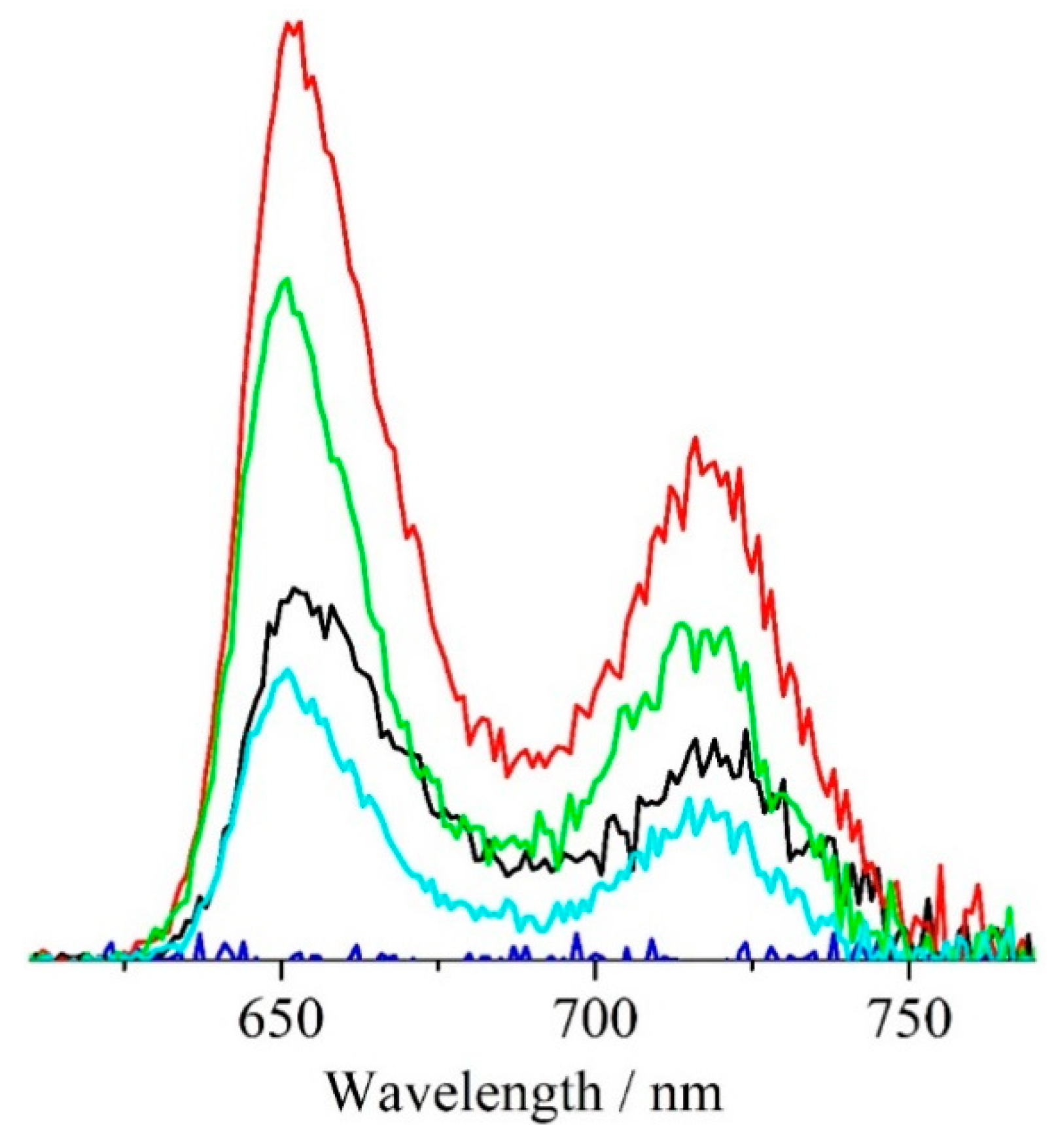
3.4. Optical Measurements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murugan, P.; Krishnamurthy, M.; Jaisankar, S.N.; Samanta, D.; Mandal, A.B. Controlled Decoration of the Surface with Macromolecules: Polymerization on a Self-Assembled Monolayer (SAM). Chem. Soc. Rev. 2015, 44, 3212–3243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Zhong, J.Q.; Lin, J.D.; Hu, W.P.; Wu, K.; Xu, G.Q.; Wee, A.T.S.; Chen, W. Towards Single Molecule Switches. Chem. Soc. Rev. 2015, 44, 2998–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Niu, L.; Wang, J.; Zhong, Y.; Bai, F.; Wang, D.; Niu, L.; Wang, J.; Zhong, Y.; Bai, F.; et al. Synthesis of Self-Assembled Porphyrin Nanoparticle Photosensitizers. ACS Nano 2018, 12, 3796–3803. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Miao, L.; Li, J.; Fu, S.; An, G.; Si, C.; Dong, Z.; Luo, Q.; Yu, S.; Xu, J.; et al. Self-Assembly of Cricoid Proteins Induced by “Soft Nanoparticles”: An Approach To Design Multienzyme-Cooperative Antioxidative Systems. ACS Nano 2015, 9, 5461–5469. [Google Scholar] [CrossRef] [PubMed]
- Kanehara, M.; Takahashi, H.; Teranishi, T. Gold(0) porphyrins on gold nanoparticles. Angew. Chem. 2008, 120, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Al-Johani, H.; Abou-Hamad, E.; Jedidi, A.; Widdifield, C.M.; Viger-Gravel, J.; Sangaru, S.S.; Gajan, D.; Anjum, D.H.; Ould-Chikh, S.; Hedhili, M.N.; et al. The structure and binding mode of citrate in the stabilization of gold nanoparticles. Nat. Chem. 2017, 9, 890–895. [Google Scholar] [CrossRef]
- Kaminker, R.; Lahav, M.; Motiei, L.; Vartanian, M.; Popovitz-Biro, R.; Iron, M.A.; van der Boom, M.E. Molecular Structure-Function Relations of the Optical Properties and Dimensions of Gold Nanoparticle Assemblies. Angew. Chem. 2010, 122, 1240–1243. [Google Scholar] [CrossRef]
- Altman, M.; Shukla, A.D.; Zubkov, T.; Evmenenko, G.; Dutta, P.; van der Boom, M.E. Controlling Structure from the Bottom-Up: Structural and Optical Properties of Layer-by-Layer Assembled Palladium Coordination-Based Multilayers. J. Am. Chem. Soc. 2006, 128, 7374–7382. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine; World Scientific Publishing Company: London, UK, 2012; Volume 18, p. 430. [Google Scholar] [CrossRef]
- Auwärter, W.; Seufert, K.; Bischoff, F.; Ecija, D.; Vijayaraghavan, S.; Joshi, S.; Klappenberger, F.; Samudrala, N.; Barth, J.V. A Surface-Anchored Molecular Four-Level Conductance Switch Based on Single Proton Transfer. Nat. Nanotechnol. 2012, 7, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, J.S.; Bocian, D.F. Molecules for Charge Based Information Storage. Acc. Chem. Res. 2011, 44, 638–650. [Google Scholar] [CrossRef]
- Liu, Z.; Yasseri, A.A.; Lindsey, J.S.; Bocian, D.F. Molecular Memories That Survive Silicon Device Processing and Real-World Operation. Science 2003, 302, 1543–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristaldi, D.A.; Motta, A.; Millesi, S.; Gupta, T.; Chhatwalb, M.; Gulino, A. Long Range Order in Si(100) Surfaces Engineered with Porphyrin Nanostructures. J. Mater. Chem. C 2013, 1, 4979–4984. [Google Scholar] [CrossRef]
- Bastús, N.G.; Comenge, J.; Puntes, V. Kinetically Controlled Seeded Growth Synthesis of Citrate-Stabilized Gold Nanoparticles of up to 200 nm: Size Focusing versus Ostwald Ripening. Langmuir 2011, 27, 11098–11105. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Willner, I. Integrated Nanoparticle-Biomolecule Hybrid Systems: Synthesis, Properties, and Applications. Angew. Chem. Int. Ed. 2004, 43, 6042–6108. [Google Scholar] [CrossRef]
- Klajn, R.; Bishop, K.J.M.; Fialkowski, M.; Paszewski, M.; Campbell, C.J.; Gray, T.P.; Grzybowski, B.A. Plastic and Moldable Metals by Self-Assembly of Sticky Nanoparticle Aggregates. Science 2007, 316, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Prasad, B.L.V.; Sorensen, C.M.; Klabunde, K.J. Gold nanoparticle Superlattices. Chem. Soc. Rev. 2008, 37, 1871–1883. [Google Scholar] [CrossRef]
- Shevchenko, E.V.; Talapin, D.V.; Kotov, N.A.; O’Brien, S.; Murray, C.B. Structural Diversity in Binary Nanoparticle Superlattices. Nature 2006, 439, 55–59. [Google Scholar] [CrossRef]
- Macfarlane, R.J.; Lee, B.; Jones, M.R.; Harris, N.; Schatz, G.C.; Mirkin, C.A. Nanoparticle Superlattice Engineering with DNA. Science 2011, 334, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Kalsin, A.M.; Fialkowski, M.; Paszewski, M.; Smoukov, S.K.; Bishop, K.J.M.; Grzybowski, B.A. Electrostatic Self-Assembly of Binary Nanoparticle Crystals with a Diamond-Like Lattice. Science 2006, 312, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Osberg, K.D.; Macfarlane, R.J.; Langille, M.R.; Mirkin, C.A. Templated Techniques for the Synthesis and Assembly of Plasmonic Nanostructures. Chem. Rev. 2011, 111, 3736–3827. [Google Scholar] [CrossRef] [PubMed]
- Manna, L.; Milliron, D.J.; Meisel, A.; Scher, E.C.; Alivisatos, A.P. Controlled Growth of Tetrapod-Branched Inorganic Nanocrystals. Nat. Mat. 2003, 2, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Alivisatos, A.P. Colloidal Nanocrystal Synthesis and the Organic–Inorganic Interface. Nature 2005, 437, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, C.; Aroca, R.F. Plasmon-enhanced fluorescence spectroscopy. Chem. Soc. Rev. 2017, 46, 3962. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhou, C.; Yu, M.; Liu, J. Different Sized Luminescent Gold Nanoparticles. Nanoscale 2012, 4, 4073–4083. [Google Scholar] [CrossRef] [PubMed]
- Trapani, M.; Castriciano, M.A.; Romeo, A.; De Luca, G.; Machado, N.; Howes, B.D.; Smulevich, G.; Scolaro, L.M. Nanohybrid Assemblies of Porphyrin and Au10 Cluster Nanoparticles. Nanomaterials 2019, 9, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Li, Z.; An, W.; Liu, L.; Cui, W. Tuning the Supramolecular Structures of Metal-Free Porphyrin via Surfactant Assisted Self-Assembly to Enhance Photocatalytic Performance. Nanomaterials 2019, 9, 1321. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhou, Y.; Ding, L.; Yu, G.; Leng, Y.; Lai, W.; Xiong, Y.; Chen, X. Supramolecular Recognition-Mediated Layer-by-Layer Self-Assembled Gold Nanoparticles for Customized Sensitivity in Paper-Based Strip Nanobiosensors. Small 2019, 15, 1903861. [Google Scholar] [CrossRef]
- Cao, Z.; Zacate, S.B.; Sun, X.; Liu, J.; Hale, E.M.; Carson, W.P.; Tyndall, S.B.; Xu, J.; Liu, X.; Liu, X.; et al. Tuning Gold Nanoparticles with Chelating Ligands for Highly Efficient Electrocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 12675–12679. [Google Scholar] [CrossRef]
- Kim, Y.; Kang, B.; Ahn, H.-Y.; Seo, J.; Nam, K.T. Plasmon Enhanced Fluorescence Based on Porphyrin-Peptoid Hybridized Gold Nanoparticle Platform. Small 2017, 13, 1700071. [Google Scholar] [CrossRef]
- Ikeda, K.; Takahashi, K.; Masuda, T.; Uosaki, K. Plasmonic Enhancement of Photoinduced Uphill Electron Transfer in a Molecular Monolayer System. Angew. Chem. Int. Ed. 2011, 50, 1280–1284. [Google Scholar] [CrossRef]
- Hasobe, T.; Imahori, H.; Kamat, P.V.; Ahn, T.K.; Kim, S.K.; Kim, D.; Fujimoto, A.; Hirakawa, T.; Fukuzumi, S. Photovoltaic Cells Using Composite Nanoclusters of Porphyrins and Fullerenes with Gold Nanoparticles. J. Am. Chem. Soc. 2005, 127, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- Imahori, H.; Fujimoto, A.; Kang, S.; Hotta, H.; Yoshida, K.; Umeyama, T.; Matano, Y.; Isoda, S.; Isosomppi, M.; Tkachenko, N.V.; et al. Host–Guest Interactions in the Supramolecular Incorporation of Fullerenes into Tailored Holes on Porphyrin-Modified Gold Nanoparticles in Molecular Photovoltaics. Chem. Eur. J. 2005, 11, 7265–7275. [Google Scholar] [CrossRef] [PubMed]
- Imahori, H.; Fujimoto, A.; Kang, S.; Hotta, H.; Yoshida, K.; Umeyama, T.; Matano, Y.; Isoda, S. Molecular Photoelectrochemical Devices: Supramolecular Incorporation of C60 Molecules into Tailored Holes on Porphyrin-Modified Gold Nanoclusters. Adv. Mat. 2005, 17, 1727–1730. [Google Scholar] [CrossRef]
- Shankar, S.; Orbach, M.; Kaminker, R.; Lahav, M.; van der Boom, M.E. Gold Nanoparticle Assemblies on Surfaces: Reactivity Tuning through Capping-Layer and Cross-Linker Design. Chem. Eur. J. 2016, 22, 1728–1734. [Google Scholar] [CrossRef]
- Boterashvili, M.; Shirman, T.; Popovitz-Biro, R.; Wen, Q.; Lahav, M.; van der Boom, M.E. Nanocrystallinity and Direct Cross-Linkage as Key-Factors for the Assembly of Gold Nanoparticle-Superlattices. Chem. Commun. 2016, 52, 8079–8082. [Google Scholar] [CrossRef] [Green Version]
- Xi, W.; Zhang, W.; An, B.-K.; Burn, P.L.; Davis, J.J. Tunnelling Conductance of Vectorial Porphyrin Monolayers. J. Mater. Chem. 2008, 18, 3109–3120. [Google Scholar] [CrossRef]
- Ohyama, J.; Hitomi, Y.; Higuchi, Y.; Shinagawa, M.; Mukai, H.; Kodera, M.; Teramura, K.; Shishido, T.; Tanaka, T. One-Phase Synthesis of Small Gold Nanoparticles Coated by a Horizontal Porphyrin Monolayer. Chem. Commun. 2008, 6300–6302. [Google Scholar] [CrossRef]
- Ohyamaa, J.; Teramura, K.; Higuchi, Y.; Shishido, T.; Hitomi, Y.; Aoki, K.; Funabiki, T.; Kodera, M.; Kato, K.; Tanida, H.; et al. An in Situ Quick XAFS Spectroscopy Study on the Formation Mechanism of Small Gold Nanoparticles Supported by Porphyrin-Cored Tetradentate Passivants. Phys. Chem. Chem. Phys. 2011, 13, 11128–11135. [Google Scholar] [CrossRef]
- Ikeda, K.; Takahashi, K.; Masuda, T.; Kobori, H.; Kanehara, M.; Teranishi, T.; Uosaki, K. Structural Tuning of Optical Antenna Properties for Plasmonic Enhancement of Photocurrent Generation on a Molecular Monolayer System. J. Phys. Chem. C 2012, 116, 20806–20811. [Google Scholar] [CrossRef]
- Yamada, M.; Kuzume, A.; Kurihara, M.; Kubo, K.; Nishihara, H. Formation of a novel porphyrin–gold nanoparticle network film induced by IR light irradiation. Chem. Commun. 2001, 2476–2477. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, H.; Wang, J.; Li, Y.F.; Wang, J.; Sang, Y.; Xiao, S.J.; Zhan, L.; Huang, C.Z. Tetrakis(4-sulfonatophenyl)porphyrin-Directed Assembly of Gold Nanocrystals: Tailoring the Plasmon Coupling Through Controllable Gap Distances. Small 2010, 6, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Conklin, D.; Nanayakkara, S.; Park, T.-H.; Lagadec, M.F.; Stecher, J.T.; Therien, M.J.; Bonnell, D.A. Electronic Transport in Porphyrin Supermolecule-Gold Nanoparticle Assemblies. Nano Lett. 2012, 12, 2414–2419. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.J.; Dowgiallo, A.-M.; Knappenberger, K.L., Jr. Plasmonic nanoparticle networks formed using iron porphyrin molecular bridges. Phys. Chem. Chem. Phys. 2013, 15, 11840–11845. [Google Scholar] [CrossRef] [PubMed]
- Trapani, M.; Romeo, A.; Parisi, T.; Sciortino, M.T.; Patanè, S.; Villari, V.; Mazzaglia, A. Supramolecular hybrid assemblies based on gold nanoparticles, amphiphilic cyclodextrin and porphyrins with combined phototherapeutic action. RSC Adv. 2013, 3, 5607–5614. [Google Scholar] [CrossRef]
- Hill, H.D.; Mirkin, C.A. The Bio-Barcode Assay for the Detection of Protein and Nucleic Acid Targets Using DTT-Induced Ligand Exchange. Nat. Protoc. 2006, 1, 324–336. [Google Scholar] [CrossRef]
- Gangemi, C.M.A.; Randazzo, R.; Fragalà, M.E.; Tomaselli, G.A.; Ballistreri, F.P.; Pappalardo, A.; Toscano, R.M.; Trusso Sfrazzetto, G.; Purrello, R.; D’Urso, A. Hierarchically Controlled Protonation/Aggregation of a Porphyrin–Spermine Derivative. New J. Chem. 2015, 39, 6722–6725. [Google Scholar] [CrossRef]
- Shi, H.; Nie, Q.; Yang, M.; Wang, C.; Liu, E.; Ji, Z.; Fa, J. A ratiometric fluorescence probe for melamine detection based on luminescence resonance energy transfer between the NaYF4:Yb, Er upconversion nanoparticles and gold nanoparticles. J. Photochem. Photobiol. A Chem. 2020, 389, 112259. [Google Scholar] [CrossRef]
- Deng, H.-H.; Li, G.-W.; Hong, L.; Liu, A.-L.; Chen, W.; Lin, X.-H.; Xia, X.-H. Colorimetric sensor based on dual-functional gold nanoparticles: Analyte-recognition and peroxidase-like activity. Food Chem. 2014, 14, 257–261. [Google Scholar] [CrossRef]
- Tuccitto, N.; Trusso Sfrazzetto, G.; Gangemi, C.M.A.; Ballistreri, F.P.; Toscano, R.M.; Tomaselli, G.A.; Pappalardo, A.; Marletta, G. The Memory-Driven Order–Disorder Transition of a 3D-Supramolecular Architecture Based on Calix[5]Arene and Porphyrin Derivatives. Chem. Commun. 2016, 52, 11681–11684. [Google Scholar] [CrossRef]
- Zhou, Z.; Bong, D. Small-Molecule/Polymer Recognition Triggers Aqueous-Phase Assembly and Encapsulation. Langmuir 2013, 29, 144–150. [Google Scholar] [CrossRef]
- Contino, A.; Maccarrone, G.; Fragalà, M.E.; Spitaleri, L.; Gulino, A. Conjugated Gold-Porphyrin Monolayers Assambled on Inorganic Surface. Chem. Eur. J. 2017, 23, 14937–14943. [Google Scholar] [CrossRef] [PubMed]
- Briggs, D.; Grant, J.T. Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy; IMP: Chichester, UK, 2003. [Google Scholar]
- Greczynski, G.; Hultman, L. Compromising science by ignorant instrument calibration—Need to revisit half a century of published XPS data. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]
- Gulino, A. Structural and Electronic Characterization of Self-Asssembled Molecular Nanoarchitectures by X-ray Photoelectron Spectroscopy. Anal. Bioanal. Chem. 2013, 405, 1479–1495. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Zhao, M.; Meng, L.; Ma, L.; Wu, G.; Wang, Y.; Xie, F.; Huang, Y. Interfacially Reinforced Carbon Fiber/Epoxy Composites by Grafting Melamine onto Carbon Fibers in Supercritical Methanol. RSC Adv. 2016, 6, 29654–29662. [Google Scholar] [CrossRef]
- Dementjev, A.P.; de Graaf, A.; van de Sanden, M.C.M.; Maslakov, K.I.; Naumkin, A.V.; Serov, A.A. X-Ray Photoelectron Spectroscopy Reference Data for Identification of the C3N4 Phase in Carbon–Nitrogen films. Diam. Relat. Mat. 2000, 9, 1904–1907. [Google Scholar] [CrossRef]
- Xue, R.; Guo, H.; Wang, T.; Wang, X.; Ai, J.; Yue, L.; Wei, Y.; Yang, W. Synthesis and Characterization of a New Covalent Organic Framework Linked by –NH– Linkage. Mat. Lett. 2017, 209, 171–174. [Google Scholar] [CrossRef]
- Osadchii, D.Y.; Olivos-Suarez, A.I.; Bavykina, A.V.; Gascon, J. Revisiting Nitrogen Species in Covalent Triazine Frameworks. Langmuir 2017, 33, 14278–14285. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yao, K.X.; Teng, B.; Zhang, T.; Han, Y. A Perfluorinated Covalent Triazine-Based Framework for Highly Selective and Water–Tolerant CO2 Capture. Energy Environ. Sci. 2013, 6, 3684–3692. [Google Scholar] [CrossRef]
- Yao, C.; Li, G.; Wang, J.; Xu, Y.; Chang, L. Template-Free Synthesis of Porous Carbon from Triazine Based Polymers and Their Use in Iodine Adsorption and CO2 Capture. Sci. Rep. 2018, 8, 1867. [Google Scholar] [CrossRef] [Green Version]
- Ni, W.; Ambjörnsson, T.; Apell, S.P.; Chen, H.; Wang, J. Observing Plasmonic-Molecular Resonance Coupling on Single Gold Nanorods. Nano Lett. 2010, 10, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Ni, W.; Yang, Z.; Chen, H.; Li, L.; Wang, J. Coupling between Molecular and Plasmonic Resonances in Freestanding Dye-Gold Nanorod Hybrid Nanostructures. J. Am. Chem. Soc. 2008, 130, 6692–6693. [Google Scholar] [CrossRef]
- Hao, Y.-W.; Wang, H.-Y.; Jiang, Y.; Chen, Q.-D.; Ueno, K.; Wang, W.-Q.; Misawa, H.; Sun, H.-B. Hybrid-State Dynamics of Gold Nanorods/Dye J-Aggregates under Strong Coupling. Angew. Chem. Int. Ed. 2011, 50, 7824–7828. [Google Scholar] [CrossRef]
- Dulkeith, E.; Morteani, A.C.; Niedereichholz, T.; Klar, T.A.; Feldmann, J.; Levi, S.A.; van Veggel, F.C.J.M.; Reinhoudt, D.N.; Möller, M.; Gittins, D.I. Fluorescence Quenching of Dye Molecules near Gold Nanoparticles: Radiative and Nonradiative Effects. Phys. Rev. Lett. 2002, 89, 203002. [Google Scholar] [CrossRef] [Green Version]
- Dulkeith, E.; Ringler, M.; Klar, T.A.; Feldmann, J.; Muñoz Javier, A.; Parak, W.J. Gold Nanoparticles Quench Fluorescence by Phase Induced Radiative Rate Suppression. Nano Lett. 2005, 5, 585–589. [Google Scholar] [CrossRef]
- Nerambourg, N.; Praho, R.; Werts, M.H.V.; Blanchard-Desce, M. Distance-Dependent Fluorescence Quenching on Gold Nanoparticles Ensheathed with Layer-by-Layer Assembled Polyelectrolytes. Nano Lett. 2006, 6, 530–536. [Google Scholar] [CrossRef]
- Jennings, T.L.; Singh, M.P.; Strouse, G.F. Fluorescent Lifetime Quenching near d = 1.5 nm Gold Nanoparticles: Probing NSET Validity. J. Am. Chem. Soc. 2006, 128, 5462–5467. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kang, J.; Yang, J.; Wu, B. Distance Dependence of Fluorescence Enhancement in Au Nanoparticle@Mesoporous Silica@Europium Complex. J. Phys. Chem. C 2016, 120, 16907–16912. [Google Scholar] [CrossRef]
- Anger, P.; Bharadwaj, P.; Novotny, L. Enhancement and Quenching of Single-Molecule Fluorescence. Phys. Rev. Lett. 2006, 96, 113002. [Google Scholar] [CrossRef] [PubMed] [Green Version]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spitaleri, L.; Gangemi, C.M.A.; Purrello, R.; Nicotra, G.; Trusso Sfrazzetto, G.; Casella, G.; Casarin, M.; Gulino, A. Covalently Conjugated Gold–Porphyrin Nanostructures. Nanomaterials 2020, 10, 1644. https://doi.org/10.3390/nano10091644
Spitaleri L, Gangemi CMA, Purrello R, Nicotra G, Trusso Sfrazzetto G, Casella G, Casarin M, Gulino A. Covalently Conjugated Gold–Porphyrin Nanostructures. Nanomaterials. 2020; 10(9):1644. https://doi.org/10.3390/nano10091644
Chicago/Turabian StyleSpitaleri, Luca, Chiara M. A. Gangemi, Roberto Purrello, Giuseppe Nicotra, Giuseppe Trusso Sfrazzetto, Girolamo Casella, Maurizio Casarin, and Antonino Gulino. 2020. "Covalently Conjugated Gold–Porphyrin Nanostructures" Nanomaterials 10, no. 9: 1644. https://doi.org/10.3390/nano10091644
APA StyleSpitaleri, L., Gangemi, C. M. A., Purrello, R., Nicotra, G., Trusso Sfrazzetto, G., Casella, G., Casarin, M., & Gulino, A. (2020). Covalently Conjugated Gold–Porphyrin Nanostructures. Nanomaterials, 10(9), 1644. https://doi.org/10.3390/nano10091644