2. Materials and Methods
Scintillating nanocomplexes preparation. The organic dye Rhodamine 6G (Rh6G) was purchased from Merck (Darmstadt, Germany, CAS number 989-38-8) and used as is. Au
8 superstructures were synthesized according to the procedure previously published [
9]. Briefly, after thiol-induced etching of gold nanoparticles to obtain individual Au
8 clusters, the superstructures were formed by networking of the clusters capped with 11-mercaptoundecanoic acid (MUA) ligands. The Au
8 superstructures/Rh6G complexes were spontaneously formed by mixing the two moieties in solution (see
Supplementary Materials, Figures S2–S5).
Nanocomposites preparation. The 2-Hydroxyethyl methacrylate (HEMA, Merck (Darmstadt, Germany, CAS no. 868-77-9, MW 130.14 g mol
−1) was a colorless viscous monomer in liquid phase that readily polymerizes. After the monomer purification from inhibitors, we chose 4,4′-azobis(4-cyanovaleric acid) (Merck (Darmstadt, Germany) CAS number 2638-94-0) as a free radical initiator of the polymerization reaction, that was performed in bulk at room temperature under N
2 atmosphere in sealed vials. The final composition was obtained by adding 5000 ppm of 4,4′-azobis(4-cyanovaleric acid) (CAS no. 2638-94-0) to a given quantity of HEMA (for 1.5 g of HEMA we added 7.5 mg of activator). All the samples were irradiated with a 365 nm Wood lamp for 1 h. The reaction scheme led to a transesterification process which resulted in the crosslinking of the polymer pHEMA [
10]. Given the high solubility of Rh6G in HEMA, a stock solution 10
−2 M was prepared to be employed in the samples’ series preparation. The Au
8 superstructures were dispersed in 50 μL of ultrapure water in 4 mL vials, then added to the Rh6G:HEMA solution before polymerization. This step was crucial to obtain a homogeneous dispersion of the Au
8 superstructures and it allowed the formation of the scintillating Au
8:Rh6G complexes. Prior to polymerization, the prepared solutions in the sealed vials were purified from oxygen under N
2 flux for 30 min. The solution was then irradiated with 365 nm UV light for one hour to induce the polymerization. The vials were broken to recover the samples. All composites were prepared with disc geometry with diameter 1 cm and thickness 0.2 cm. The employed crosslinking reaction led to rigid and durable samples with no appreciable residuals of unreacted monomer that can affect the material emission and scintillation properties (
Supplementary Materials) [
11].
Photoluminescence (PL) studies. Absorption spectra were recorded using a Cary Lambda 900 spectrophotometer at normal incidence with Suprasil quartz cuvettes. Steady-state PL spectra were acquired using a Varian Eclipse fluorimeter (bandwidth 1 nm) using quartz cuvettes of 1 cm optical path length. Time-resolved PL spectra were recorded under excitation by a pulsed light-emitting diode at 340 nm (3.65 eV, pulse width 80 ps; EP-LED 340, Edinburgh Instruments). The composites were excited with a pulsed laser at 405 nm (3.06 eV, pulse width 90 ps; EPL-405, Edinburgh Instruments) to avoid direct excitation of the host polymer matrix. Measurements on composites were performed on cylindrical bulk specimens of 1 cm diameter and 0.2 cm thickness.
Radioluminescence (RL) studies. Steady-state RL measurements were carried out at room temperature using a homemade apparatus featuring, as a detection system, a liquid nitrogen-cooled, back-illuminated, and UV-enhanced charge-coupled device (Jobin-Yvon Symphony II) combined with a monochromator (Jobin-Yvon Triax 180) equipped with a 100 lines/mm grating. All spectra were corrected for the spectral response of the detection system. RL excitation was obtained by unfiltered X-ray irradiation through a beryllium window, using a Philips 2274 X-ray tube with a tungsten target operated at 20 kV. At this operating voltage, a continuous X-ray spectrum is produced by a Bremsstrahlung mechanism due to the impact of electrons generated through the thermionic effect and accelerated onto a tungsten target. The dose rate was 0.2 Gy/s, evaluated by comparison with a calibrated
90Sr-
90Y beta radioactive source and using optically stimulated luminescence emission from quartz crystalline powder (100–200 μm grains) (
Figures S1 and S2).
Scintillation experiments. Light yield (LY) was determined by means of amplitude spectroscopy of scintillation pulses with accordance to Ref. [
9] and compared to that of a reference BGO crystal measured under the same conditions [
12]. Scintillation pulses were excited by
239Pu α-radiation (5.2 MeV). The setup for amplitude spectroscopy consisted of a hybrid photomultiplier DEP PPO 475B, spectroscopy amplifier ORTEC 672 (shaping time set to 1 μs) and multichannel analyzer ORTEC 927TM. Ultrafast decays under pulsing X-ray excitation were measured at room temperature using picosecond (ps) X-ray tube N5084 (Hamamatsu Photonics K.K. Shizuoka, Japan) at 40 kV). The X-ray tube was driven by the ps light pulser equipped with a laser diode with a repetition rate up to 1 MHz. The signal was detected by hybrid picosecond photon detector and Fluorohub unit (Horiba Scientific, Kyoto, Japan). The setup instrumental response function FWHM was about 76 ps. The Rh6G scintillation flashes decay curves were detected from the same surface as that excited by X-rays using a 560 nm low pass optical filter (
Figure S7). The emission was monitored from the same sample’s surface where it was excited. The deconvolution procedure was applied to the decay curves to calculate true decay times and estimate the pulse rise time (SpectraSolve software package, Ames Photonics, FortWorth, TX, USA).
3. Results and Discussion
The setting-up of the equation system employed to model the evolution of the photophysical processes in the systems is based on the fundamental assumption that in a composite scintillator both the heavy sensitizers and the emitter moieties can behave as recombination centers for the high-energy free charges generated by the light–matter interaction [
13]. When ionizing radiation or high-energy particles interact with a material, the energy is mainly deposited through ionization [
14]. Considering that the created free charges can diffuse up to micrometric volumes before recombination [
15], and given that intermolecular distances of the nanosized emitters and sensitizers embedded in the composite are in the order of 10–100 nanometers for concentrations as low as 10
−5 M, we can assume the rapid diffusion limit for the charge recombination process [
16]. This means that the sensitizer and emitter charge capture rates, respectively, in first approximation are proportional to the sensitizer (
) and emitter (
) concentrations. The evolution in time of the excited species
for sensitizers and
for emitters, respectively, can be therefore described by
where
and
are the free charges capture rate constants for the sensitizer and emitter, respectively, and
is a loss rate constant that is determined by the matrix characteristics. The rates
and
are the spontaneous recombination rates of the sensitizer and emitter excited states, respectively.
is the total number of electron/hole pairs generated in the host polymer. In general, the
value varies according to the system composition, because both
and
can determine the initial density of free charges created through the light–matter interaction.
In steady state conditions, the integrated radioluminescence intensity of the system is given therefore by
where
is the instrumental detection efficiency,
is the quantum yield of the emitter fluorescence and
is the yield of the sensitizer-to-emitter energy transfer. The parameters
and
are defined as the sensitizer and emitter charge capture yields, respectively, by
The sensitization efficiency can be now explicitly expressed as
where
is the relative increment of free charges density due to the presence of the heavy component with respect the unsensitized system
loaded with the same emitter concentration. The parameter
ranges from 0, where the charges are lost by mechanisms competitive to emitter luminescence, to 1, where the total additional energy is properly exploited to activate the emitter luminescence. Therefore, the
value indicates the effectiveness of the radioluminescence sensitization as a function of the system composition and the characteristic parameters of the employed materials. The calculated value of
as function of the emitter concentration, which sets both
and
, is shown in
Figure 1b for the ideal case where
= 0, i.e., without free charge loss due to the matrix. By considering a composite scintillator with fixed composition, three main scenarios can be identified:
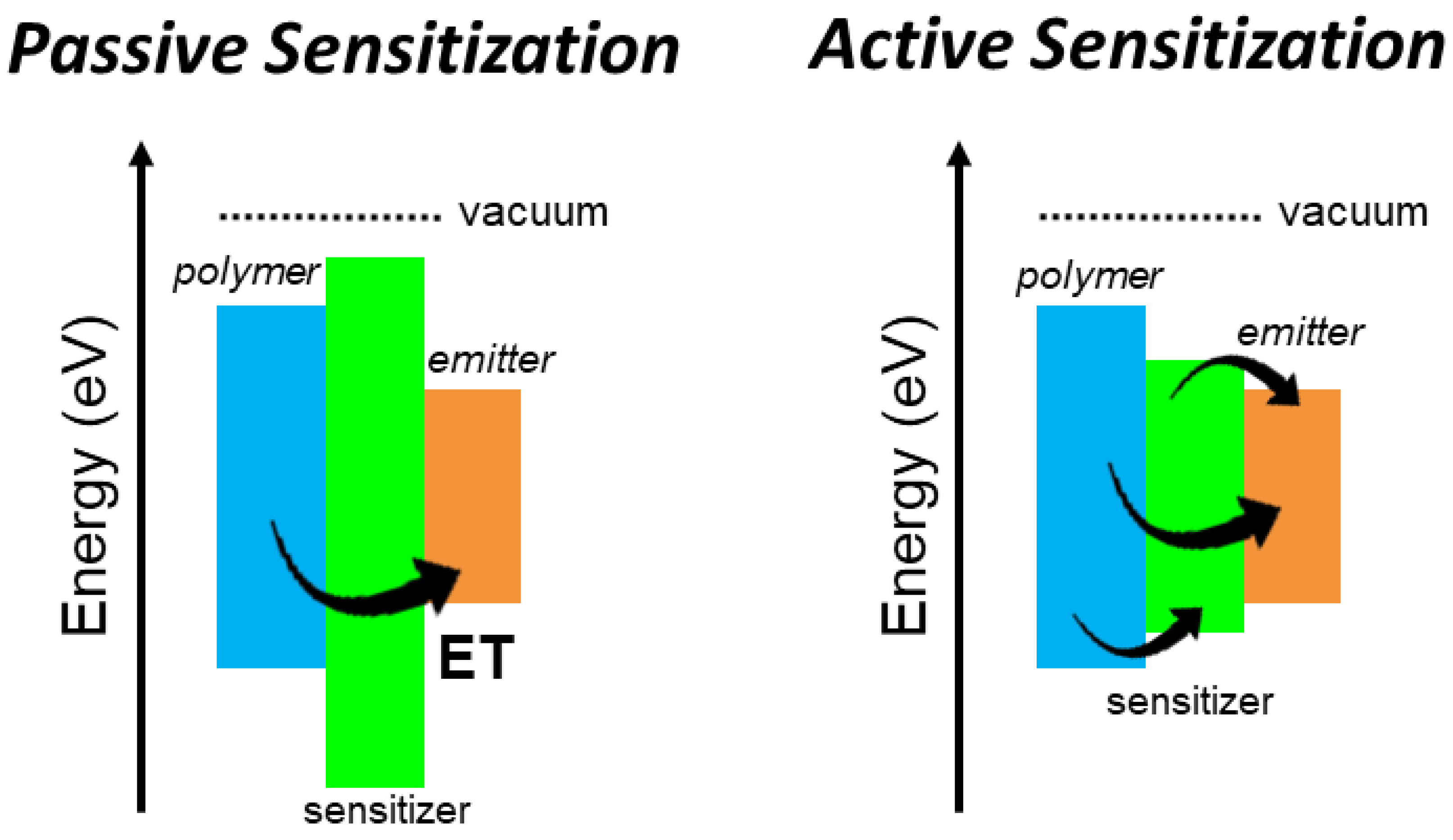
I. The limit case where
, where the sensitizers’ free charges capture ability is negligible with respect to the emitter one independently from
. Despite its luminescence properties, we are in a situation where the sensitizer moiety acts as a passive sensitization component. Any rise of
will induce a simple growth of
value because of the increment of the overall material density. Notably, this type of sensitization can be effective only with a complete energy transfer from the matrix to the emitters (
= 1,
= 0), thus obtaining
(
Figure 1b, dotted line) [
17,
18]. However, this condition is generally difficult to achieve.
II. If
, the deposited energy is equally shared among the two populations. In this case
, thus the effectiveness of the sensitizer-to-emitters energy transfer becomes crucial to recover the energy stored on sensitizers even in the best case with
=
= 0.5 (
Figure 1b, solid line). It is worth noting that this configuration for active sensitization can be advantageous also with a non-complete energy transfer if the
value is at least doubled with respect to the unsensitized case, to balance the charges recombination on sensitizers.
III. In the limit case where
, the energy transfer step becomes more critical, because it is the unique activation channel for the emitter’s luminescence. None of the additional charges recombine on the emitters, thus
. Therefore, a poor interaction between emitters and sensitizers can completely make useless the presence of these latter even with
= 1, because the activation of the final emission is completely determined by the energy transfer step (
Figure 1b, dashed line).
In both case II and case III, the use of highly luminescent sensitizers is strongly recommended to exploit fast non-radiative energy transfer to activate the emitter luminescence while not affecting the scintillator time response. On the contrary, radiative energy transfer can induce a delay of the scintillation emission because of the emission/re-absorption step involved. The composition and structure of the composite scintillator should be therefore optimized to achieve high non radiative transfer rates and 100% transfer yield. Some conditions are then required to reach the maximum sensitization efficacy.
The role of the sensitizer-to-emitter energy transfer was investigated in a series of composite polymeric scintillators based on a matrix of poly(2-hydroxyethyl methacrylate), poly-HEMA, in which 8-atoms gold clusters aggregated superstructures (Au
8 superstructures) [
19] were employed as sensitizers and the scintillating organic dye Rh6G was employed as final emitters.
Figure 2 shows their absorption, luminescence and radioluminescence spectra. The Au
8 superstructures were selected because of the high atomic number of gold (
Z = 79) and their large Stokes shift (~0.85 eV), with a first absorption peak in near UV spectral region at 380 nm and a long living emission, with an average lifetime of
= 165 ns (
Figure 2c), peaked at 530 nm (
Figure 2a). These features make them good sensitizers candidates because they have a higher interaction cross section with the ionizing radiation compared to organic materials and they do not have any issue related to re-absorption of scintillation light. Their radioluminescence spectrum is identical to the photoluminescence and it is insensitive to exposure to soft X-rays even for large doses (
Figure S1).
The employed emitter system is the Rh6G, which possesses a fluorescence peaked at 590 nm with lifetime of 4.2 ns (inset of
Figure 2c) and a photoluminescence quantum yield of 0.95 [
19]. Crucial for this study, its absorption and emission properties are complementary to those of the Au
8 superstructures. As showed in
Figure 2b, the dye first absorption band peaked at 525 nm is in excellent resonance with the superstructure emission, thus allowing a non-radiative energy transfer mechanism of the Förster type [
20] that can be exploited for the sensitization of the emitter luminescence. Moreover, it is worth noting that the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies of both the luminescent systems are similar (
Figure 2f, inset) and deeper than the ones of the host polymer [
21,
22,
23,
24,
25]. This indicates that the electronic affinities of both compounds are comparable and therefore we can reasonably consider valid the assumption
for the charge capture rate constants. We can therefore consider as similar the ability of both sensitizers and emitters to capture scintillation free charges.
By exploiting the interaction between COO
− and NH
+ (
Figure 2d), we realized a 1:1 stable sensitizer/emitter complex (
Figure S4). We have chosen this scintillating complex configuration because it is particularly favorable according to the cases discussed above. The proximity of the luminescent moieties guarantees indeed a unitary energy transfer yield. Therefore, by employing these sensitizer/emitter complexes the composition of the scintillator can be tuned by keeping the transfer yield constant. Consequently, the system proposed is a good model to investigate the sensitization of the scintillation discussed above. The progressive complexation of the Au
8 superstructures is monitored by following the evolution of the sensitizer-to-emitter energy transfer dynamic in solution as function of the emitter concentration.
Figure 2e shows the time resolved photoluminescence spectrum recorded at 530 nm under pulsed excitation of a dispersion of Au
8 superstructures (10
−5 M) and Rh6G solutions. According to the increment of the emitter amount, the lifetime of the residual sensitizer luminescence decreases because of the more efficient energy transfer, whose efficiency
can be directly estimated from these data (
Supplementary Materials, Supplementary Table S1). This analysis gives a highly overestimated Förster interaction radius of
= 13.5 nm, which is significantly larger than the theoretical expected length
= 4.6 nm (
Figure 2e and
Supplementary Materials). This reflects the formation of the described complexes, that results in a high transfer efficiency even at extremely low emitter concentrations. Notably, in these complexes the energy transfer is orders of magnitude faster (3.2 GHz,
Figure S5) than the spontaneous decay rate of excited superstructures (
= 6 × 10
−3 GHz) thanks to the close proximity of the interacting moieties, thus overcoming the diffusion limited kinetics for the Forster mechanism in low viscosity solutions and enhancing the transfer yield. The formation of stable complexes is further demonstrated by time resolved photoluminescence experiments that show an energy transfer rate and yield independent from the complex concentration (
Figure S5).
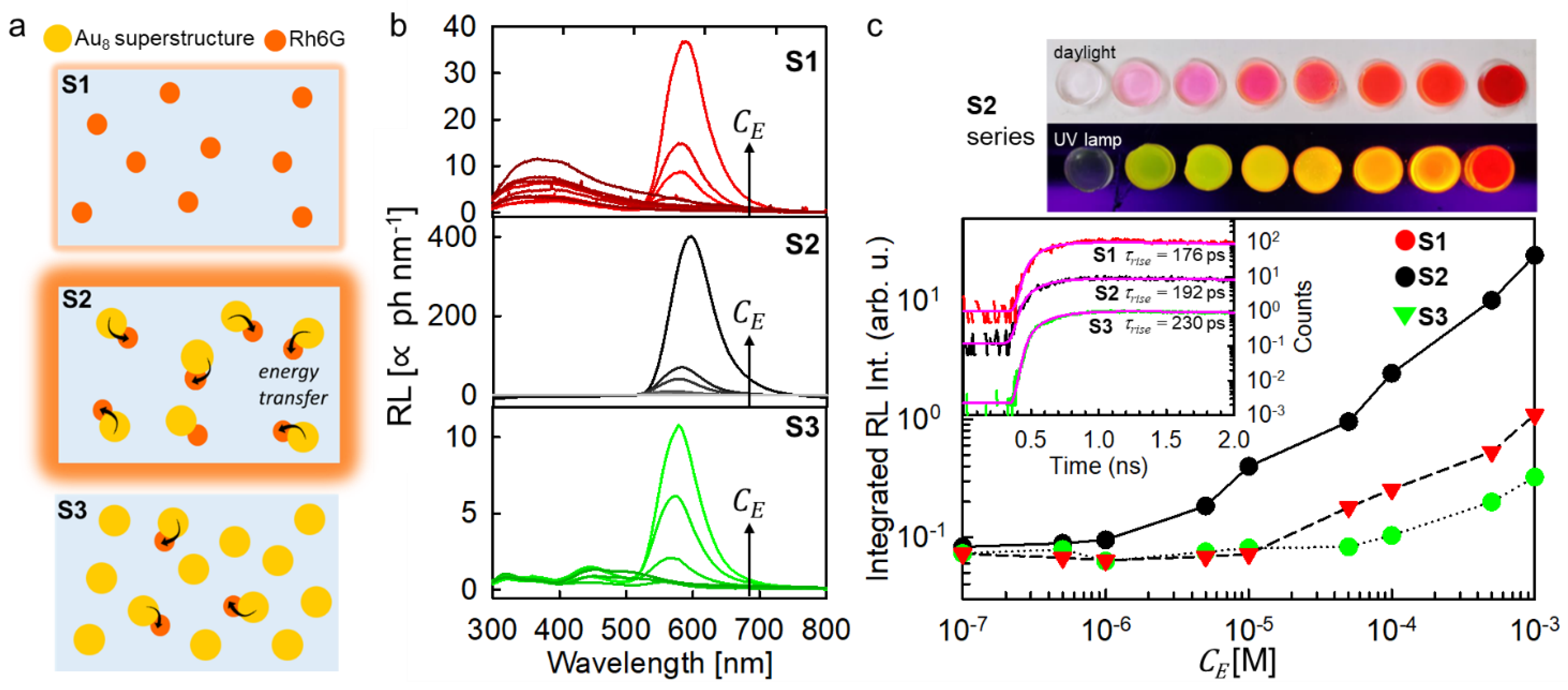
Three series of scintillating composites were fabricated with different loading level of luminescent compounds, as sketched in
Figure 3a (
Supplementary Materials). The first series (S1) contains only the emitter Rh6G, and it works as a reference unsensitized scintillator. The second series (S2) embeds the sensitizers/emitter complexes. The third one (S3) was prepared by including a large and fixed number of individual sensitizers (10
−2 M) while varying the number of scintillating complexes. In all compositions, the Rh6G keeps its emission properties (
Figure S6).
Figure 3b depicts the RL spectra of the composite series recorded under the same experimental conditions. The S1 series shows the UV luminescence from poly-HEMA peaked at 360 nm, which intensity decreases with the increment of the emitter concentration. This indicates a better harvesting of the energy of the interacting ionizing radiation by the included R6Gh molecules. When a concentration of
= 10
−5 M is reached, the UV emission is switched off, and the Rh6G emission appears with an intensity that gradually increases with the emitter concentration until a
= 10
−3 M is reached. A further increment of the dye concentration is not possible due to evident phase segregation of the dyes with respect to the host.
The S2 scintillator series shows a significantly better performance. No UV emission can be detected event at very low loading amounts. This suggests an enhanced ability of Au
8 superstructures to harvest the deposited energy with respect to the dye, most probably due to the resonance between the polymer luminescence and superstructures absorption that favors direct energy transfer from the matrix. The radioluminescence intensity reaches the maximum value observed in the S1 series with a
= 5 × 10
−5 M, which is more than one order of magnitude lower than the one employed for the unsensitized case, suggesting an effective sensitization of the Rh6G luminescence. With the highest loading level of
= 10
−3 M, the composite shows a LY of 80 ph MeV
−1 when excited by
239Pu α-radiation (
Supplementary Table S2) and an enhanced radioluminescence integrated intensity that indicates a sensitization efficiency of
25 (
Figure 3c). It is worth noting that in addition to the improvement of the scintillation performance, the stable structural coupling of the active moieties has several important consequences. First, it makes the energy transfer rate and yield independent from
, that is useful for the technological perspective. Indeed, considering the typical Förster radii values of 2–3 nm [
20], a huge amount
10
−2 M of the acceptor/emitter moieties should be included to maximize the transfer in disordered solid systems such has polymeric hosts, where molecular excitons cannot diffuse freely and the transfer is limited to the closest acceptor units [
26]. In other words, any configuration with separated sensitizers and emitters would result in a lower sensitization efficiency. Second, this architecture avoids emitters aggregation by enabling a better management of the scintillator composition at high loading levels pivotal for fabrication of efficient detectors with good optical quality by handling a single moiety [
27]. Third the coupling sensitizer and emitter at a fixed close distance (≤1 nm) enables a fast energy transfer. Specifically, the transfer rate of 3.2 GHz is significantly larger than the spontaneous recombination rate of the emitter
=
= 0.24 GHz. This result is pivotal to preserve the time response of the scintillator, as demonstrated by the scintillation data reported in the inset of
Figure 3c. Both the S1 and S2 composites show indeed almost identical scintillation activation dynamics, with a rise time of the pulse at around 190 ps unaffected by the fast energy transfer process involved in S2.
The crucial role of the energy transfer optimization is further highlighted by the results obtained on the S3 composite series. Here, the scintillating complexes and the isolated Au
8 superstructures were added to the composites in order to increase the amount of the Rh6G emitters by keeping the total
value as large as 10
−2 M, i.e., significantly higher than the maximum value employed in the best sample of the S2 series. In such a way, the
value should be significantly higher than for the S2 scintillators. However, the radioluminescence intensity is lower also with respect to unsensitized S1 series for any composition thus indicating that most of the deposited energy is lost by competitive channels as suggested by the presence of a residual emission from isolated Au
8 superstructures in the blue/green spectral region (
Figure 3b, bottom panel). This result can be explained considering that the large number of sensitizers amplifies
, but also makes their charges capture yield dominant with respect to the emitter one (
, case III). Therefore, considering that even in the best composition only 1 tenth of sensitizers are coupled to emitters, the poor energy transfer efficiency results in a significant lower scintillation performance despite the higher amount of deposited energy.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}



