
Microfluidics Technology for the Design and Formulation of Nanomedicines




Abstract
:1. Introduction
2. Nano Based Drug Delivery Systems
3. Application of the Nano-Drug Delivery Systems
3.1. Nanoparticles for Nucleic Acid-Based Treatment
3.2. Nanoparticles for Cancer Cell Targeting
3.3. Nanoparticles and Angiogenesis
3.4. Nano Systems in Inflammation
4. Manufacturing Methods for Lipid Formulations
4.1. Liposomes
- Preparation of the aqueous and lipid phases;
- Primary processing involving lipid;
- Secondary processing steps (essential in some formulations and optional in others);
4.1.1. Conventional Methods
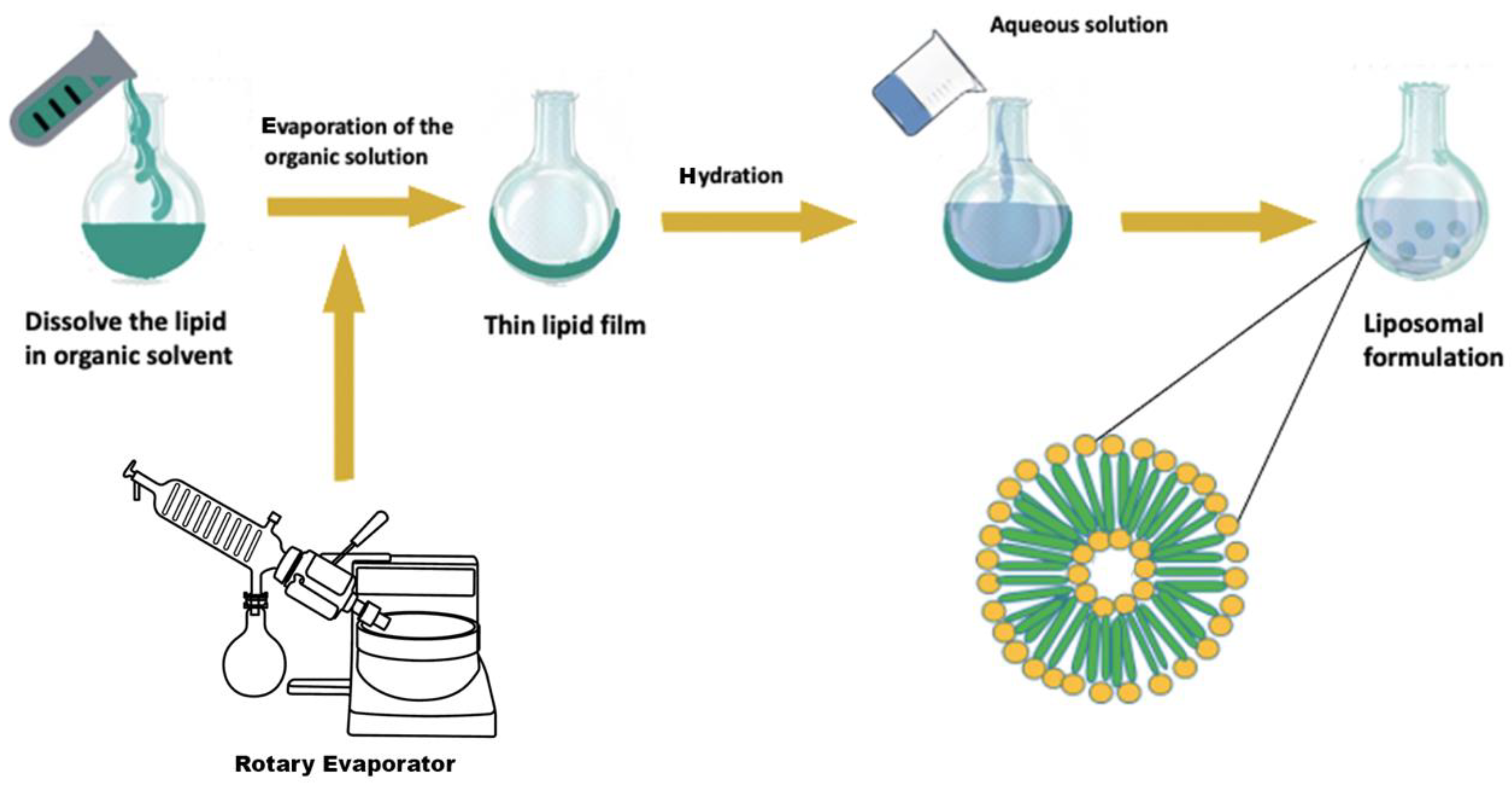
- Lipids dissolution in organic solvents;
- Drying the obtained solution;
- Hydration of the dried lipid (applying different aqueous solution);
- Separation of the liposomal vesicles;
- Quality control assays.
Film Hydration
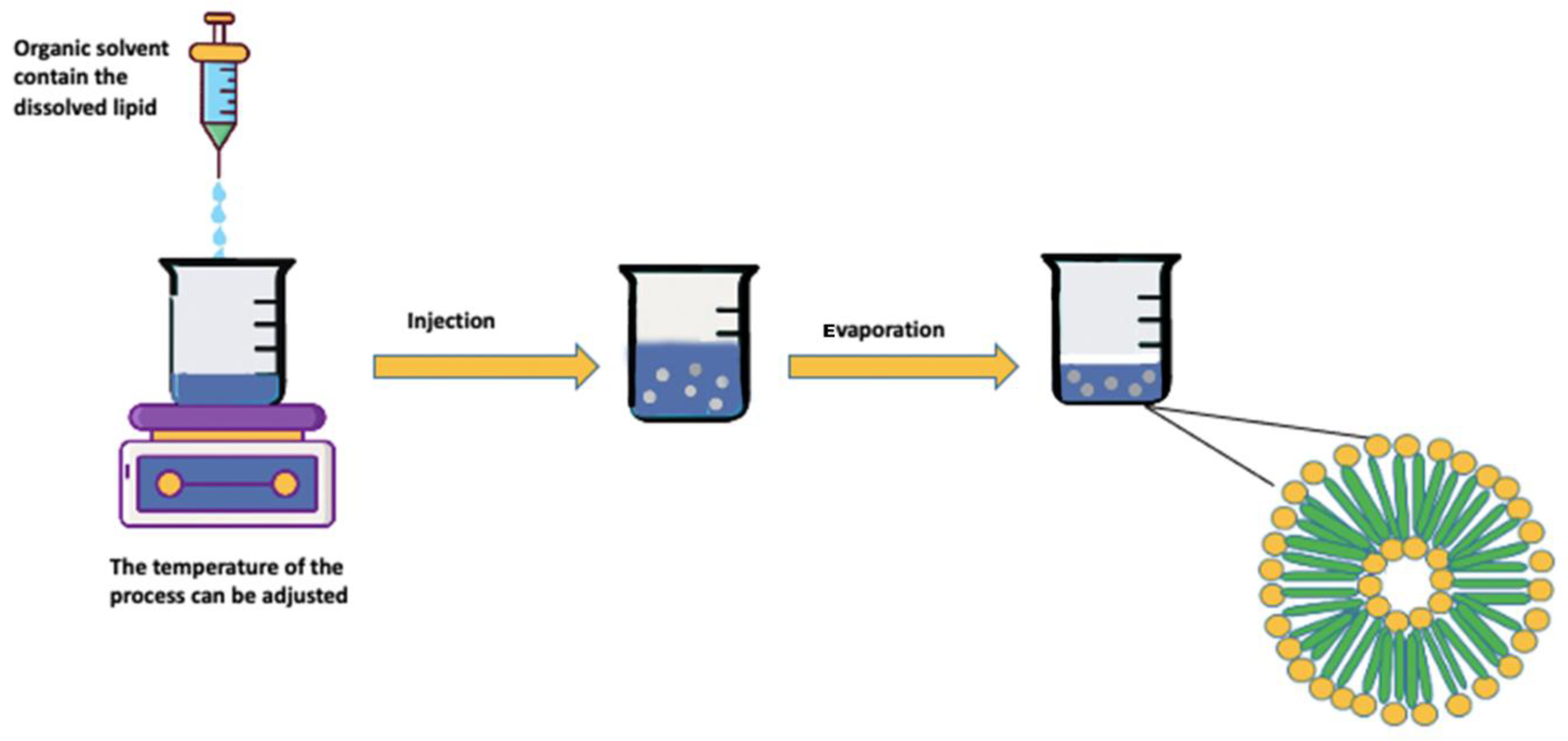
Solvent Injection
Detergent Removal
4.1.2. Re-Sizing of Lipid Suspension
Sonication
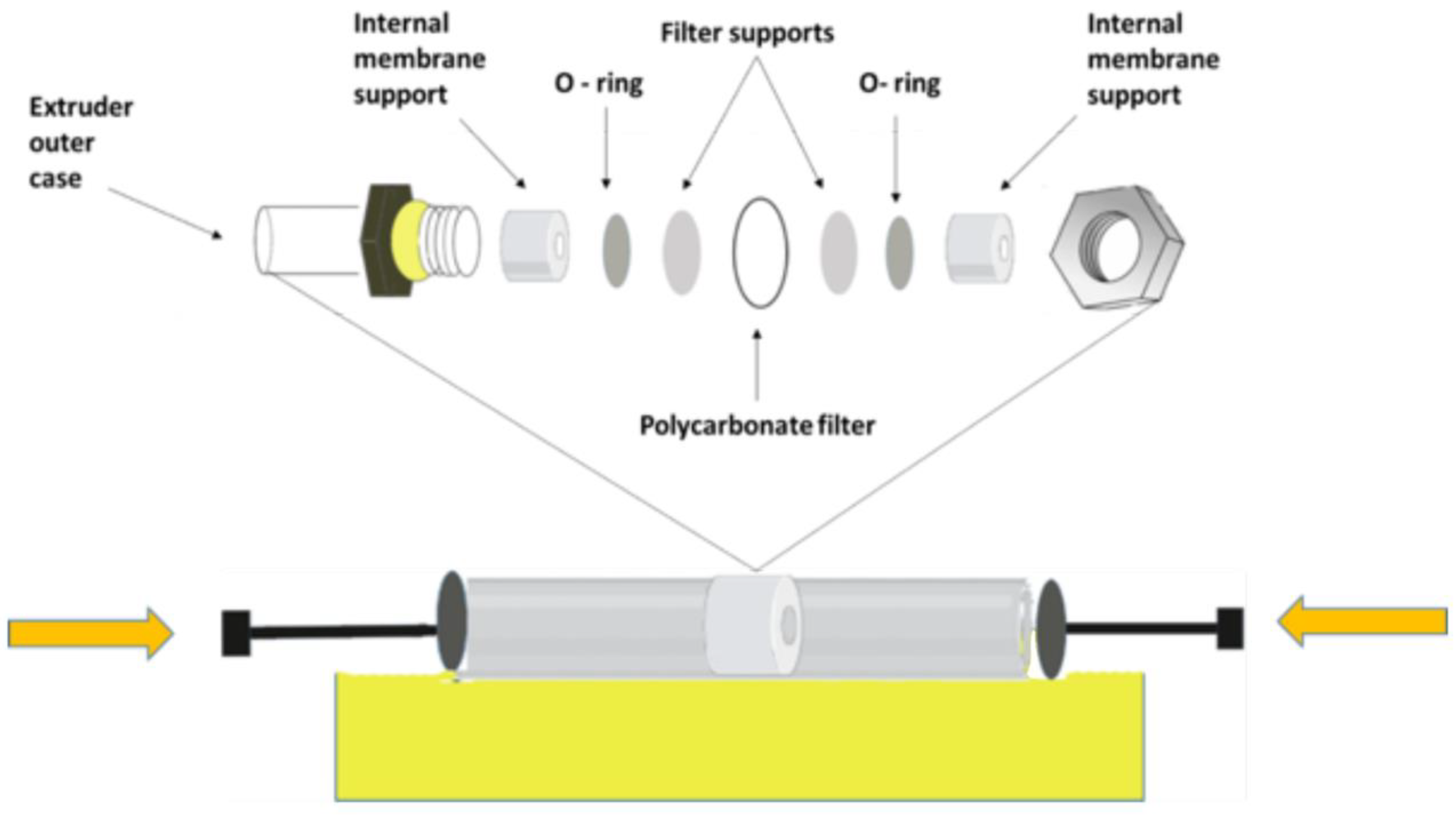
Extrusion
4.2. Solid Lipid Nanoparticles
4.2.1. High Pressure Homogenization Method
Hot Homogenization Method
Cold Homogenization Method
4.2.2. Ultrasonication or High Speed Homogenization
4.2.3. Microemulsion
5. Manufacturing Methods of Polymeric Formulations
5.1. Two-Step Emulsification Procedures
5.1.1. Emulsification-Salting Out
5.1.2. Emulsification-Solvent Diffusion
5.1.3. Emulsification-Solvent Evaporation
5.2. One Step Procedure
5.2.1. Dialysis
5.2.2. Nanoprecipitation Procedures
6. Microfluidics
6.1. Microfluidics in Nanomedicine
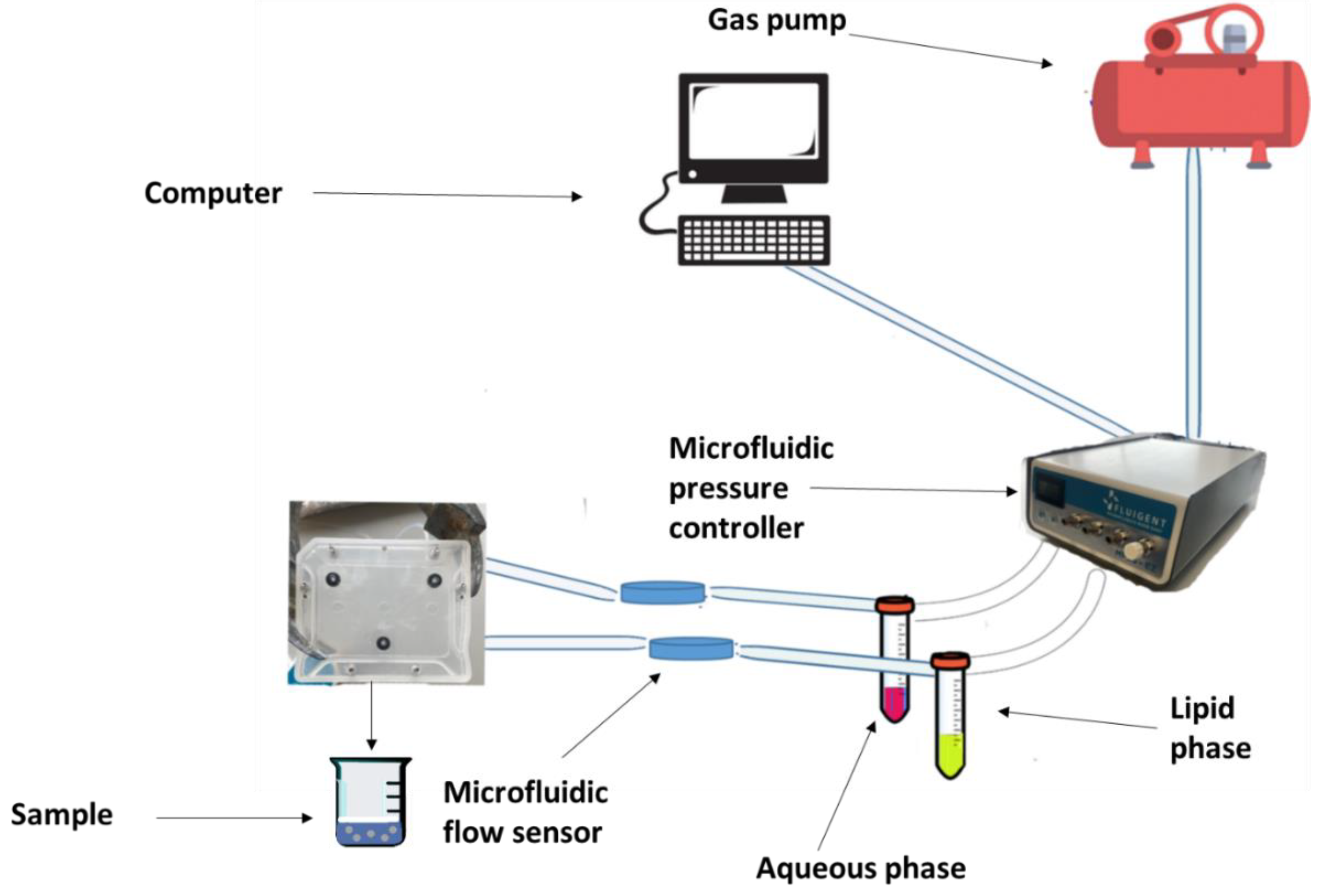
6.1.1. Microfluidics in the Manufacturing of Liposomes
6.1.2. Microfluidics in the Production of SLNs
6.1.3. Microfluidics in the Production of Polymeric Formulations
6.1.4. Lipid–Polymer Hybrid NPs
6.1.5. Microfluidics in Synthesis of Inorganic Nanoparticles
7. Conclusions & Future Predictions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shirai, Y.; Osgood, A.J.; Zhao, Y.; Yao, Y.; Saudan, L.; Yang, H.; Yu-Hung, C.; Alemany, L.B.; Sasaki, T.; Morin, J.-F. Surface-rolling molecules. J. Am. Chem. Soc. 2006, 128, 4854–4864. [Google Scholar] [CrossRef]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The History of Nanoscience and Nanotechnology: From Chemical-Physical Applications to Nanomedicine. Molecules 2019, 25, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grobert, N.; Hutton, D. Nanoscience and nanotechnologies: Opportunities and uncertainties. Lond. R. Soc. R. Acad. Eng. Rep. 2004, 46, 618. [Google Scholar]
- Thiruvengadam, M.; Rajakumar, G.; Chung, I.M. Nanotechnology: Current uses and future applications in the food industry. 3 Biotech 2018, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Noguez, C. Plasmonic Optical Properties and Applications of Metal Nanostructures. Plasmonics 2008, 3, 127–150. [Google Scholar] [CrossRef]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- De Jong, W.H.; Borm, P.J. Drug delivery and nanoparticles:applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef] [Green Version]
- Bowman, D.M. More than a decade on: Mapping today’s regulatory and policy landscapes following the publication of nanoscience and nanotechnologies: Opportunities and uncertainties. NanoEthics 2017, 11, 169–186. [Google Scholar] [CrossRef]
- Cascone, M.G.; Lazzeri, L.; Carmignani, C.; Zhu, Z. Gelatin nanoparticles produced by a simple W/O emulsion as delivery system for methotrexate. J. Mater. Sci. Mater. Med. 2002, 13, 523–526. [Google Scholar] [CrossRef]
- Kipp, J.E. The role of solid nanoparticle technology in the parenteral delivery of poorly water-soluble drugs. Int. J. Pharm. 2004, 284, 109–122. [Google Scholar] [CrossRef] [PubMed]
- LaVan, D.A.; McGuire, T.; Langer, R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003, 21, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleki, A.; Asadi, A.; Gurushankar, K.; Karimi Shayan, T.; Abedi Sarvestani, F. Importance of Nano Medicine and New Drug Therapies for Cancer. Adv. Pharm. Bull. 2021, 11, 450–457. [Google Scholar] [CrossRef]
- Abdel-Mottaleb, M.M.; Neumann, D.; Lamprecht, A. Lipid nanocapsules for dermal application: A comparative study of lipid-based versus polymer-based nanocarriers. Eur. J. Pharm. Biopharm. 2011, 79, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef] [Green Version]
- Bonadio, J.; Smiley, E.; Patil, P.; Goldstein, S. Localized, direct plasmid gene delivery in vivo: Prolonged therapy results in reproducible tissue regeneration. Nat. Med. 1999, 5, 753–759. [Google Scholar] [CrossRef]
- Hamilton, A.J.; Baulcombe, D.C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 1999, 286, 950–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold Nanoparticles for Nucleic Acid Delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Draz, M.S.; Fang, B.A.; Zhang, P.; Hu, Z.; Gu, S.; Weng, K.C.; Gray, J.W.; Chen, F.F. Nanoparticle-mediated systemic delivery of siRNA for treatment of cancers and viral infections. Theranostics 2014, 4, 872–892. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Chen, A.A.; Derfus, A.M.; Khetani, S.R.; Bhatia, S.N. Quantum dots to monitor RNAi delivery and improve gene silencing. Nucleic Acids Res. 2005, 33, e190. [Google Scholar] [CrossRef] [Green Version]
- Shinde, R.R.; Bachmann, M.H.; Wang, Q.; Kasper, R.; Contag, C.H. PEG-PLA/PLGA Nanoparticles for In-Vivo RNAi Delivery; NSTI Nano Tech.: Santa Clara, CA, USA, 2007. [Google Scholar]
- Tan, W.B.; Jiang, S.; Zhang, Y. Quantum-dot based nanoparticles for targeted silencing of HER2/neu gene via RNA interference. Biomaterials 2007, 28, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Howard, K.A.; Rahbek, U.L.; Liu, X.; Damgaard, C.K.; Glud, S.Z.; Andersen, M.Ø.; Hovgaard, M.B.; Schmitz, A.; Nyengaard, J.R.; Besenbacher, F.; et al. RNA Interference in Vitro and in Vivo Using a Novel Chitosan/siRNA Nanoparticle System. Mol. Ther. 2006, 14, 476–484. [Google Scholar] [CrossRef]
- Gao, W.; Xiao, Z.; Radovic-Moreno, A.; Shi, J.; Langer, R.; Farokhzad, O.C. Progress in siRNA delivery using multifunctional nanoparticles. Methods Mol. Biol. 2010, 629, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Swierczewska, M.; Lee, S.; Chen, X. Functional nanoparticles for molecular imaging guided gene delivery. Nano Today 2010, 5, 524–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef]
- Naito, M.; Ishii, T.; Matsumoto, A.; Miyata, K.; Miyahara, Y.; Kataoka, K. A phenylboronate-functionalized polyion complex micelle for ATP-triggered release of siRNA. Angew. Chem. Int. Ed. Engl. 2012, 51, 10751–10755. [Google Scholar] [CrossRef]
- Lee, S.J.; Huh, M.S.; Lee, S.Y.; Min, S.; Lee, S.; Koo, H.; Chu, J.U.; Lee, K.E.; Jeon, H.; Choi, Y. Tumor-homing poly-siRNA/glycol chitosan self-cross-linked nanoparticles for systemic siRNA delivery in cancer treatment. Angew. Chem. Int. Ed. 2012, 51, 7203–7207. [Google Scholar] [CrossRef]
- Cousin, J.M.; Cloninger, M.J. The role of galectin-1 in cancer progression, and synthetic multivalent systems for the study of galectin-1. Int. J. Mol. Sci. 2016, 17, 1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, S.S.; Chen, G.; Stenzel, M.H. Synthesis of glycopolymers and their multivalent recognitions with lectins. Polym. Chem. 2010, 1, 1392–1412. [Google Scholar] [CrossRef]
- Kadam, R.U.; Bergmann, M.; Hurley, M.; Garg, D.; Cacciarini, M.; Swiderska, M.A.; Nativi, C.; Sattler, M.; Smyth, A.R.; Williams, P. A glycopeptide dendrimer inhibitor of the galactose-specific lectin LecA and of Pseudomonas aeruginosa biofilms. Angew. Chem. Int. Ed. 2011, 50, 10631–10635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marradi, M.; Chiodo, F.; García, I.; Penadés, S. Glyconanoparticles as multifunctional and multimodal carbohydrate systems. Chem. Soc. Rev. 2013, 42, 4728–4745. [Google Scholar] [CrossRef]
- Sharma, R.; Kottari, N.; Chabre, Y.M.; Abbassi, L.; Shiao, T.C.; Roy, R. A highly versatile convergent/divergent “onion peel” synthetic strategy toward potent multivalent glycodendrimers. Chem. Commun. 2014, 50, 13300–13303. [Google Scholar] [CrossRef]
- Liu, L.-H.; Dietsch, H.; Schurtenberger, P.; Yan, M. Photoinitiated coupling of unmodified monosaccharides to iron oxide nanoparticles for sensing proteins and bacteria. Bioconjugate Chem. 2009, 20, 1349–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, N.; Maiti, K.; Naresh, K. Multivalent glycoliposomes and micelles to study carbohydrate–protein and carbohydrate–carbohydrate interactions. Chem. Soc. Rev. 2013, 42, 4640–4656. [Google Scholar] [CrossRef]
- Vedala, H.; Chen, Y.; Cecioni, S.; Imberty, A.; Vidal, S.; Star, A. Nanoelectronic detection of lectin-carbohydrate interactions using carbon nanotubes. Nano Lett. 2011, 11, 170–175. [Google Scholar] [CrossRef]
- Besford, Q.A.; Wojnilowicz, M.; Suma, T.; Bertleff-Zieschang, N.; Caruso, F.; Cavalieri, F. Lactosylated Glycogen Nanoparticles for Targeting Prostate Cancer Cells. ACS Appl. Mater. Interfaces 2017, 9, 16869–16879. [Google Scholar] [CrossRef]
- Costantino, L.; Gandolfi, F.; Tosi, G.; Rivasi, F.; Vandelli, M.A.; Forni, F. Peptide-derivatized biodegradable nanoparticles able to cross the blood-brain barrier. J. Control. Release 2005, 108, 84–96. [Google Scholar] [CrossRef]
- Sumner, J.P.; Kopelman, R. Alexa Fluor 488 as an iron sensing molecule and its application in PEBBLE nanosensors. Analyst 2005, 130, 528–533. [Google Scholar] [CrossRef]
- Michaelis, K.; Hoffmann, M.M.; Dreis, S.; Herbert, E.; Alyautdin, R.N.; Michaelis, M.; Kreuter, J.; Langer, K. Covalent linkage of apolipoprotein e to albumin nanoparticles strongly enhances drug transport into the brain. J. Pharmacol. Exp. Ther. 2006, 317, 1246–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiniger, S.C.; Kreuter, J.; Khalansky, A.S.; Skidan, I.N.; Bobruskin, A.I.; Smirnova, Z.S.; Severin, S.E.; Uhl, R.; Kock, M.; Geiger, K.D.; et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int. J. Cancer 2004, 109, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef]
- Rademakers, T.; Horvath, J.M.; van Blitterswijk, C.A.; LaPointe, V.L. Oxygen and nutrient delivery in tissue engineering: Approaches to graft vascularization. J. Tissue Eng. Regen. Med. 2019, 13, 1815–1829. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2020, 9, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Xu, X.; Ma, Y.; Zhang, S.; Zhang, S. RGD peptide-based non-viral gene delivery vectors targeting integrin αvβ3 for cancer therapy. J. Drug Target. 2019, 27, 1–11. [Google Scholar] [CrossRef]
- Majumder, P. Integrin-mediated delivery of drugs and nucleic acids for anti-angiogenic cancer therapy: Current landscape and remaining challenges. Bioengineering 2018, 5, 76. [Google Scholar] [CrossRef] [Green Version]
- Bharali, D.J.; Rajabi, M.; Mousa, S.A. Chapter 11—Application of Nanotechnology to Target Tumor Angiogenesis in Cancer Therapeutics. In Anti-Angiogenesis Strategies in Cancer Therapeutics; Mousa, S.A., Davis, P.J., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 165–178. [Google Scholar] [CrossRef]
- Li, L.; Wartchow, C.A.; Danthi, S.N.; Shen, Z.; Dechene, N.; Pease, J.; Choi, H.S.; Doede, T.; Chu, P.; Ning, S. A novel antiangiogenesis therapy using an integrin antagonist or anti–Flk-1 antibody coated 90Y-labeled nanoparticles. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H. iRGD: A promising peptide for cancer imaging and a potential therapeutic agent for various cancers. J. Oncol. 2019, 2019, 9367845. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, Z.; Cai, P.; Liu, J.; Liao, L.-D.; Hong, M.; Chen, X.; Thakor, N.; Liu, B. Conjugated polymer and drug co-encapsulated nanoparticles for chemo-and photo-thermal combination therapy with two-photon regulated fast drug release. Nanoscale 2015, 7, 3067–3076. [Google Scholar] [CrossRef]
- Murphy, E.A.; Majeti, B.K.; Barnes, L.A.; Makale, M.; Weis, S.M.; Lutu-Fuga, K.; Wrasidlo, W.; Cheresh, D.A. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 9343–9348. [Google Scholar] [CrossRef] [Green Version]
- Rebbaa, A.; Chu, F.; Davis, F.B.; Davis, P.J.; Mousa, S.A. Novel function of the thyroid hormone analog tetraiodothyroacetic acid: A cancer chemosensitizing and anti-cancer agent. Angiogenesis 2008, 11, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Bergh, J.J.; Dier, E.; Rebbaa, A.; O’Connor, L.J.; Yalcin, M.; Aljada, A.; Dyskin, E.; Davis, F.B.; Lin, H.-Y. Tetraiodothyroacetic acid, a small molecule integrin ligand, blocks angiogenesis induced by vascular endothelial growth factor and basic fibroblast growth factor. Angiogenesis 2008, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Sudha, T.; Darwish, N.H.; Davis, P.J.; Mousa, S.A. Synthesis of MR-49, a deiodinated analog of tetraiodothyroacetic acid (tetrac), as a novel pro-angiogenesis modulator. Bioorganic Med. Chem. Lett. 2016, 26, 4112–4116. [Google Scholar] [CrossRef] [PubMed]
- Bharali, D.J.; Yalcin, M.; Davis, P.J.; Mousa, S.A. Tetraiodothyroacetic acid-conjugated PLGA nanoparticles: A nanomedicine approach to treat drug-resistant breast cancer. Nanomedicine 2013, 8, 1943–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz-Neu, K.; Kostka, S.L.; Dinges, S.; Iwakura, Y.; Udey, M.C.; von Stebut, E. IL-1 signalling is dispensable for protective immunity in Leishmania-resistant mice. Exp. Dermatol. 2011, 20, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Dewamitta, S.R.; Nomura, T.; Kawamura, I.; Hara, H.; Tsuchiya, K.; Kurenuma, T.; Shen, Y.; Daim, S.; Yamamoto, T.; Qu, H.; et al. Listeriolysin O-dependent bacterial entry into the cytoplasm is required for calpain activation and interleukin-1 alpha secretion in macrophages infected with Listeria monocytogenes. Infect. Immun. 2010, 78, 1884–1894. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.W.; O’Brien, R.; Mackintosh, C.G.; Clark, R.G.; Griffin, J.F. Immunoregulatory cytokines are associated with protection from immunopathology following Mycobacterium avium subspecies paratuberculosis infection in red deer. Infect. Immun. 2011, 79, 2089–2097. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tan, T.; Gao, L.; Zhao, W.; Wang, P. Preparation of azithromycin nanosuspensions by high pressure homogenization and its physicochemical characteristics studies. Drug Dev. Ind. Pharm. 2007, 33, 569–575. [Google Scholar] [CrossRef]
- Suri, S.S.; Fenniri, H.; Singh, B. Nanotechnology-based drug delivery systems. J. Occup. Med. Toxicol. 2007, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Plasencia, C.; Hou, Y.; Neamati, N. Synthesis and biological evaluation of dimeric RGD peptide− paclitaxel conjugate as a model for integrin-targeted drug delivery. J. Med. Chem. 2005, 48, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- R Rama, A.; Jimenez-Lopez, J.; Cabeza, L.; Jimenez-Luna, C.; C Leiva, M.; Perazzoli, G.; Hernandez, R.; Zafra, I.; Ortiz, R.; Melguizo, C. Last advances in nanocarriers-based drug delivery systems for colorectal cancer. Curr. Drug Deliv. 2016, 13, 830–838. [Google Scholar] [CrossRef] [PubMed]
- García-Pinel, B.; Porras-Alcalá, C.; Ortega-Rodríguez, A.; Sarabia, F.; Prados, J.; Melguizo, C.; López-Romero, J.M. Lipid-Based Nanoparticles: Application and Recent Advances in Cancer Treatment. Nanomaterials 2019, 9, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoriadis, G. Fate of injected liposomes: Observations on entrapped solute retention, vesicle clearance and tissue distribution in vivo. In Liposomes as Drug Carriers: Recent Trends and Progress; Wiley: New York, NY, USA, 1988; pp. 3–18. [Google Scholar]
- Lurquin, P.F. Incorporation of genetic material into liposomes and transfer to cells. In Liposome Technology, 2nd ed.; Gregoriadis, G., Ed.; CRC Press: Boca Raton, FL, USA, 1993; Volume 2, pp. 129–139. [Google Scholar]
- Gregoriadis, G. Liposome Technology: Volume II; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Thompson, A.K. Structure and Properties of Liposomes Prepared from Milk Phospholipids: A Thesis Presented in Partial Fulfilment of the Requirements for the Degree of Doctor of Philosophy in Food Technology. Ph.D. Thesis, Massey University, Palmerston North, New Zealand, 2005. [Google Scholar]
- Shah, S.; Dhawan, V.; Holm, R.; Nagarsenker, M.S.; Perrie, Y. Liposomes: Advancements and innovation in the manufacturing process. Adv. Drug Deliv. Rev. 2020, 154–155, 102–122. [Google Scholar] [CrossRef]
- Salim, M.; Minamikawa, H.; Sugimura, A.; Hashim, R. Amphiphilic designer nano-carriers for controlled release: From drug delivery to diagnostics. MedChemComm 2014, 5, 1602–1618. [Google Scholar] [CrossRef]
- Alam, S.; Mattern-Schain, S.; Best, M. Targeting and triggered release using lipid-based supramolecular assemblies as medicinal nanocarriers. In Comprehensive Supramolecular Chemistry II; Elsevier: Oxford, UK, 2017; pp. 329–364. [Google Scholar]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297. [Google Scholar]
- Maherani, B.; Arab-Tehrany, E.; Mozafari, M.R.; Gaiani, C.; Linder, M. Liposomes: A review of manufacturing techniques and targeting strategies. Curr. Nanosci. 2011, 7, 436–452. [Google Scholar] [CrossRef]
- Machado, A.R.; de Assis, L.M.; Machado, M.I.R.; de Souza-Soares, L.A. Importance of lecithin for encapsulation processes. Afr. J. Food Sci. 2014, 8, 176–183. [Google Scholar]
- Sharma, D.; Ali, A.A.E.; Trivedi, L.R. An Updated Review on: Liposomes as drug delivery system. PharmaTutor 2018, 6, 50–62. [Google Scholar] [CrossRef]
- Dua, J.; Rana, A.; Bhandari, A. Liposome: Methods of preparation and applications. Int. J. Pharm. Stud. Res. 2012, 3, 14–20. [Google Scholar]
- Couvreur, P.; Dubernet, C.; Puisieux, F. Controlled drug delivery with nanoparticles: Current possibilities and future trends. Eur. J. Pharm. Biopharm. 1995, 41, 2–13. [Google Scholar]
- Burgess, S. Liposome Preparation-Avanti® Polar Lipids; Sigma-Aldrich: St. Louis, MO, USA, 1998. [Google Scholar]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-H.; Chen, C.-H.; Lin, Z.-C.; Fang, J.-Y. Recent advances in oral delivery of drugs and bioactive natural products using solid lipid nanoparticles as the carriers. J. Food Drug Anal. 2017, 25, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Geszke-Moritz, M.; Moritz, M. Solid lipid nanoparticles as attractive drug vehicles: Composition, properties and therapeutic strategies. Mater. Sci. Eng. C 2016, 68, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Helgason, T.; Awad, T.S.; Kristbergsson, K.; McClements, D.J.; Weiss, J. Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SLN). J. Colloid Interface Sci. 2009, 334, 75–81. [Google Scholar] [CrossRef]
- Botto, C.; Mauro, N.; Amore, E.; Martorana, E.; Giammona, G.; Bondì, M.L. Surfactant effect on the physicochemical characteristics of cationic solid lipid nanoparticles. Int. J. Pharm. 2017, 516, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Uner, M.; Yener, G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int. J. Nanomed. 2007, 2, 289–300. [Google Scholar]
- Battaglia, L.; Gallarate, M. Lipid nanoparticles: State of the art, new preparation methods and challenges in drug delivery. Expert Opin. Drug Deliv. 2012, 9, 497–508. [Google Scholar] [CrossRef]
- Yadav, N.; Khatak, D.; Sara, U.V. Solid lipid nanoparticles—A review. Int. J. Appl. Pharm. 2013, 5, 8–18. [Google Scholar]
- Kasongo, K.W.; Müller, R.H.; Walker, R.B. The use of hot and cold high pressure homogenization to enhance the loading capacity and encapsulation efficiency of nanostructured lipid carriers for the hydrophilic antiretroviral drug, didanosine for potential administration to paediatric patients. Pharm. Dev. Technol. 2012, 17, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Triplett, M.D.; Rathman, J.F. Optimization of β-carotene loaded solid lipid nanoparticles preparation using a high shear homogenization technique. J. Nanopart. Res. 2009, 11, 601–614. [Google Scholar] [CrossRef]
- Lander, R.; Manger, W.; Scouloudis, M.; Ku, A.; Davis, C.; Lee, A. Gaulin homogenization: A mechanistic study. Biotechnol. Prog. 2000, 16, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Peters, K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. Int. J. Pharm. 1998, 160, 229–237. [Google Scholar] [CrossRef]
- Ahlin, P.; Kristl, J.; Smid-Korbar, J. Optimization of procedure parameters and physical stability of solid lipid nanoparticles in dispersions. Acta Pharm. (Zagreb) 1998, 48, 259–267. [Google Scholar]
- Mukherjee, S.; Ray, S.; Thakur, R.S. Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian J. Pharm. Sci. 2009, 71, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Zur Mühlen, A. Feste Lipid Nanopartikel mit Prolongierter Wirkstoffliberation: Herstellung, Langzeitstabilität, Charakterisierung, Freisetzungsverhalten und-Mechanismen (Solid Lipid Nanoparticles with Prolonged Drug Release: Production, Long-Term Stability, Characterisation, Release Properties and Mechanisms). Ph.D. Thesis, Free University of Berlin, Berlin, Germany, 1996. [Google Scholar]
- Eldem, T.; Speiser, P.; Hincal, A. Optimization of spray-dried and -congealed lipid micropellets and characterization of their surface morphology by scanning electron microscopy. Pharm. Res. 1991, 8, 47–54. [Google Scholar] [CrossRef]
- Nikam, S.; Chavan, M.; Sharma, P.H. Solid lipid nanoparticles: A lipid based drug delivery. Nanotechnology 2014, 1, 5. [Google Scholar]
- Magenheim, B.; Levy, M.Y.; Benita, S. A new in vitro technique for the evaluation of drug release profile from colloidal carriers—Ultrafiltration technique at low pressure. Int. J. Pharm. 1993, 94, 115–123. [Google Scholar] [CrossRef]
- del Pozo-Rodríguez, A.; Solinís, M.A.; Gascón, A.R.; Pedraz, J.L. Short- and long-term stability study of lyophilized solid lipid nanoparticles for gene therapy. Eur. J. Pharm. Biopharm. 2008, 71, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2012, 64, 83–101. [Google Scholar] [CrossRef]
- Gasco, M. Solid Lipid Nanospheres from Warm Micro-Emulsions: Improvements in SLN production for more efficient drug delivery. Pharm. Technol. Eur. 1997, 9, 52–58. [Google Scholar]
- Ganesan, P.; Narayanasamy, D. Lipid nanoparticles: Different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain. Chem. Pharm. 2017, 6, 37–56. [Google Scholar] [CrossRef]
- Ahlin Grabnar, P.; Kristl, J. The manufacturing techniques of drug-loaded polymeric nanoparticles from preformed polymers. J. Microencapsul. 2011, 28, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Meng, F.; Deng, C.; Zhong, Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules 2014, 15, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Marin, E.; Briceño, M.I.; Caballero-George, C. Critical evaluation of biodegradable polymers used in nanodrugs. Int. J. Nanomed. 2013, 8, 3071. [Google Scholar]
- Crucho, C.I.C.; Barros, M.T. Polymeric nanoparticles: A study on the preparation variables and characterization methods. Mater. Sci. Eng. C 2017, 80, 771–784. [Google Scholar] [CrossRef]
- Sala, M.; Miladi, K.; Agusti, G.; Elaissari, A.; Fessi, H. Preparation of liposomes: A comparative study between the double solvent displacement and the conventional ethanol injection—From laboratory scale to large scale. Colloids Surf. A Physicochem. Eng. Asp. 2017, 524, 71–78. [Google Scholar] [CrossRef]
- Ong, S.G.; Chitneni, M.; Lee, K.S.; Ming, L.C.; Yuen, K.H. Evaluation of Extrusion Technique for Nanosizing Liposomes. Pharmaceutics 2016, 8, 36. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, A.A.; Moreno-Trejo, M.B.; Meléndez-Zaragoza, M.J.; Collins-Martínez, V.; López-Ortiz, A.; Martínez-Guerra, E.; Sánchez-Domínguez, M. Spinel-type ferrite nanoparticles: Synthesis by the oil-in-water microemulsion reaction method and photocatalytic water-splitting evaluation. Int. J. Hydrog. Energy 2019, 44, 12421–12429. [Google Scholar] [CrossRef]
- Rao, J.P.; Geckeler, K.E. Polymer nanoparticles: Preparation techniques and size-control parameters. Prog. Polym. Sci. 2011, 36, 887–913. [Google Scholar] [CrossRef]
- Kozlowski, M. Polymeric Nanomaterials for Nanotherapeutics. Ed. C. Vasile, Elsevier, 1st Edition. 2018. Series Volume Editors: Cornelia Vasile, eBook ISBN: 9780128139332; Paperback ISBN: 9780128139325: Elsevier, Published Date: 2nd November 2018; 558 pages. In Micro and Nano Technologies Series. Polymers 2019, 11, 1452. [Google Scholar] [CrossRef] [Green Version]
- Crucho, C.I.; Barros, M.T. Formulation of functionalized PLGA polymeric nanoparticles for targeted drug delivery. Polymer 2015, 68, 41–46. [Google Scholar] [CrossRef]
- Allouche, J. Synthesis of organic and bioorganic nanoparticles: An overview of the preparation methods. In Nanomaterials: A Danger or a Promise? Springer: London, UK, 2013; pp. 27–74. [Google Scholar]
- Ganachaud, F.; Katz, J.L. Nanoparticles and nanocapsules created using the Ouzo effect: Spontaneous emulsification as an alternative to ultrasonic and high-shear devices. ChemPhysChem 2005, 6, 209–216. [Google Scholar] [CrossRef]
- Konan, Y.N.; Gurny, R.; Allémann, E. Preparation and characterization of sterile and freeze-dried sub-200 nm nanoparticles. Int. J. Pharm. 2002, 233, 239–252. [Google Scholar] [CrossRef]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.A.; Patel, D.M.; Patel, J.K.; Patel, D.H. Solvent Emulsification Evaporation and Solvent Emulsification Diffusion Techniques for Nanoparticles. In Emerging Technologies for Nanoparticle Manufacturing; Patel, J.K., Pathak, Y.V., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 287–300. [Google Scholar] [CrossRef]
- Kudr, J.; Haddad, Y.; Richtera, L.; Heger, Z.; Cernak, M.; Adam, V.; Zitka, O. Magnetic nanoparticles: From design and synthesis to real world applications. Nanomaterials 2017, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Moinard-Chécot, D.; Chevalier, Y.; Briançon, S.; Beney, L.; Fessi, H. Mechanism of nanocapsules formation by the emulsion-diffusion process. J. Colloid Interface Sci. 2008, 317, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Bouchemal, K. Methods for the preparation and manufacture of polymeric nanoparticles. Pharm. Res. 2009, 26, 1025–1058. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiao, Y.; Wang, Y.; Zhou, C.; Zhang, Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Fornaguera, C.; Feiner-Gracia, N.; Calderó, G.; García-Celma, M.; Solans, C. Galantamine-loaded PLGA nanoparticles, from nano-emulsion templating, as novel advanced drug delivery systems to treat neurodegenerative diseases. Nanoscale 2015, 7, 12076–12084. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-J.; Jeong, Y.-I.; Jang, M.-K.; Park, Y.-H.; Nah, J.-W. Effect of solvent on the preparation of surfactant-free poly (DL-lactide-co-glycolide) nanoparticles and norfloxacin release characteristics. Int. J. Pharm. 2000, 207, 99–108. [Google Scholar] [CrossRef]
- Chronopoulou, L.; Fratoddi, I.; Palocci, C.; Venditti, I.; Russo, M.V. Osmosis based method drives the self-assembly of polymeric chains into micro-and nanostructures. Langmuir 2009, 25, 11940–11946. [Google Scholar] [CrossRef] [PubMed]
- Mora-Huertas, C.; Fessi, H.; Elaissari, A. Influence of process and formulation parameters on the formation of submicron particles by solvent displacement and emulsification–diffusion methods: Critical comparison. Adv. Colloid Interface Sci. 2011, 163, 90–122. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Jallouli, Y.; Kroubi, M.; Yuan, X.-B.; Feng, W.; Kang, C.-S.; Pu, P.-Y.; Betbeder, D. Characterization of endocytosis of transferrin-coated PLGA nanoparticles by the blood–brain barrier. Int. J. Pharm. 2009, 379, 285–292. [Google Scholar] [CrossRef]
- Chacon, M.; Berges, L.; Molpeceres, J.; Aberturas, M.; Guzman, M. Optimized preparation of poly D, L (lactic-glycolic) microspheres and nanoparticles for oral administration. Int. J. Pharm. 1996, 141, 81–91. [Google Scholar] [CrossRef]
- Xie, H.; Smith, J.W. Fabrication of PLGA nanoparticles with a fluidic nanoprecipitation system. J. Nanobiotechnology 2010, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Xue, L.; Zhang, H.; Lin, J. A Review on Micromixers. Micromachines 2017, 8, 274. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, E.; Dorati, R.; Pisani, S.; Conti, B.; Bergamini, G.; Modena, T.; Genta, I. The Microfluidic Technique and the Manufacturing of Polysaccharide Nanoparticles. Pharmaceutics 2018, 10, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Song, Y. Microfluidic synthesis of nanohybrids. Small 2017, 13, 1604084. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, H.; Fontana, F.; Hirvonen, J.T.; Santos, H.A. Current developments and applications of microfluidic technology toward clinical translation of nanomedicines. Adv. Drug Deliv. Rev. 2018, 128, 54–83. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Yang, S.; Ho, Y.P.; Grigsby, C.L.; Leong, K.W.; Huang, T.J. Shape-controlled synthesis of hybrid nanomaterials via three-dimensional hydrodynamic focusing. ACS Nano 2014, 8, 10026–10034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, M.; Valencia, P.M.; Rodriguez, M.I.; Langer, R.; Farokhzad, O.C.; Karnik, R. Synthesis of size-tunable polymeric nanoparticles enabled by 3D hydrodynamic flow focusing in single-layer microchannels. Adv. Mater. 2011, 23, H79–H83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greener, J.; Li, W.; Ren, J.; Voicu, D.; Pakharenko, V.; Tang, T.; Kumacheva, E. Rapid, cost-efficient fabrication of microfluidic reactors in thermoplastic polymers by combining photolithography and hot embossing. Lab Chip 2010, 10, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, H.; Zhang, J.; Phan, H.P.; Nguyen, N.T. Flexible Microfluidics: Fundamentals, Recent Developments, and Applications. Micromachines 2019, 10, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Bhushan, B. Transparent, wear-resistant, superhydrophobic and superoleophobic poly(dimethylsiloxane) (PDMS) surfaces. J. Colloid Interface Sci. 2017, 488, 118–126. [Google Scholar] [CrossRef]
- Arduino, I.; Liu, Z.; Rahikkala, A.; Figueiredo, P.; Correia, A.; Cutrignelli, A.; Denora, N.; Santos, H.A. Preparation of cetyl palmitate-based PEGylated solid lipid nanoparticles by microfluidic technique. Acta Biomater. 2021, 121, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lin, S.; Wang, C.; Hu, J.; Li, C.; Zhuang, Z.; Zhou, Y.; Mathies, R.A.; Yang, C.J. PMMA/PDMS valves and pumps for disposable microfluidics. Lab Chip 2009, 9, 3088–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, L.; Chen, G. Fabrication, modification, and application of poly(methyl methacrylate) microfluidic chips. Electrophoresis 2008, 29, 1801–1814. [Google Scholar] [CrossRef]
- Li, J.; Liu, C.; Qiao, H.; Zhu, L.; Chen, G.; Dai, X. Hot embossing/bonding of a poly (ethylene terephthalate)(PET) microfluidic chip. J. Micromechanics Microengineering 2007, 18, 015008. [Google Scholar] [CrossRef]
- Gencturk, E.; Mutlu, S.; Ulgen, K.O. Advances in microfluidic devices made from thermoplastics used in cell biology and analyses. Biomicrofluidics 2017, 11, 051502. [Google Scholar] [CrossRef]
- Ren, K.; Zhou, J.; Wu, H. Materials for Microfluidic Chip Fabrication. Acc. Chem. Res. 2013, 46, 2396–2406. [Google Scholar] [CrossRef]
- Becker, H.; Locascio, L.E. Polymer microfluidic devices. Talanta 2002, 56, 267–287. [Google Scholar] [CrossRef]
- Ballacchino, G.; Weaver, E.; Mathew, E.; Dorati, R.; Genta, I.; Conti, B.; Lamprou, D.A. Manufacturing of 3D-Printed Microfluidic Devices for the Synthesis of Drug-Loaded Liposomal Formulations. Int. J. Mol. Sci. 2021, 22, 8064. [Google Scholar] [CrossRef]
- Sengupta, P.; Khanra, K.; Chowdhury, A.R.; Datta, P. Lab-on-a-chip sensing devices for biomedical applications. In Bioelectronics and Medical Devices: From Materials to Devices-Fabrication, Applications and Reliability; Elsevier: Amsterdam, The Netherlands, 2019; pp. 47–95. [Google Scholar]
- Niculescu, A.G.; Chircov, C.; Bîrcă, A.C.; Grumezescu, A.M. Fabrication and Applications of Microfluidic Devices: A Review. Int. J. Mol. Sci. 2021, 22, 2011. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, W.; Zhang, L.; Ban, L.; Chen, P.; Du, W.; Feng, X.; Liu, B.-F. Chemically Edited Exosomes with Dual Ligand Purified by Microfluidic Device for Active Targeted Drug Delivery to Tumor Cells. ACS Appl. Mater. Interfaces 2017, 9, 27441–27452. [Google Scholar] [CrossRef] [PubMed]
- Jahn, A.; Vreeland, W.N.; Gaitan, M.; Locascio, L.E. Controlled vesicle self-assembly in microfluidic channels with hydrodynamic focusing. J. Am. Chem. Soc. 2004, 126, 2674–2675. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, H.; Solaimany Nazar, A.R.; Varshosaz, J. Double flow focusing microfluidic-assisted based preparation of methotrexate–loaded liposomal nanoparticles: Encapsulation efficacy, drug release and stability. Colloids Surf. A Physicochem. Eng. Asp. 2021, 614, 126166. [Google Scholar] [CrossRef]
- Weaver, E.; Uddin, S.; Cole, D.K.; Hooker, A.; Lamprou, D.A. The Present and Future Role of Microfluidics for Protein and Peptide-Based Therapeutics and Diagnostics. Appl. Sci. 2021, 11, 4109. [Google Scholar] [CrossRef]
- Guimarães Sá Correia, M.; Briuglia, M.L.; Niosi, F.; Lamprou, D.A. Microfluidic manufacturing of phospholipid nanoparticles: Stability, encapsulation efficacy, and drug release. Int. J. Pharm. 2017, 516, 91–99. [Google Scholar] [CrossRef]
- Gkionis, L.; Campbell, R.A.; Aojula, H.; Harris, L.K.; Tirella, A. Manufacturing drug co-loaded liposomal formulations targeting breast cancer: Influence of preparative method on liposomes characteristics and in vitro toxicity. Int. J. Pharm. 2020, 590, 119926. [Google Scholar] [CrossRef] [PubMed]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New developments in liposomal drug delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Capretto, L.; Mazzitelli, S.; Nastruzzi, C. Design, production and optimization of solid lipid microparticles (SLM) by a coaxial microfluidic device. J. Control. Release 2012, 160, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Anderluzzi, G.; Perrie, Y. Microfluidic manufacture of solid lipid nanoparticles: A case study on tristearin-based systems. Drug Deliv. Lett. 2020, 10, 197–208. [Google Scholar] [CrossRef]
- Shepherd, S.J.; Issadore, D.; Mitchell, M.J. Microfluidic formulation of nanoparticles for biomedical applications. Biomaterials 2021, 274, 120826. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Zhang, R.; Billingsley, M.M.; Mitchell, M.J. Biomaterials for vaccine-based cancer immunotherapy. J. Control. Release 2018, 292, 256–276. [Google Scholar] [CrossRef] [PubMed]
- Karnik, R.; Gu, F.; Basto, P.; Cannizzaro, C.; Dean, L.; Kyei-Manu, W.; Langer, R.; Farokhzad, O.C. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008, 8, 2906–2912. [Google Scholar] [CrossRef]
- Abstiens, K.; Goepferich, A.M. Microfluidic manufacturing improves polydispersity of multicomponent polymeric nanoparticles. J. Drug Deliv. Sci. Technol. 2019, 49, 433–439. [Google Scholar] [CrossRef]
- Vu, H.T.; Streck, S.; Hook, S.M.; McDowell, A. Utilization of Microfluidics for the Preparation of Polymeric Nanoparticles for the Antioxidant Rutin: A Comparison with Bulk Production. Pharm. Nanotechnol. 2019, 7, 469–483. [Google Scholar] [CrossRef]
- Behnke, M.; Vollrath, A.; Klepsch, L.; Beringer-Siemers, B.; Stumpf, S.; Czaplewska, J.A.; Hoeppener, S.; Werz, O.; Schubert, U.S. Optimized Encapsulation of the FLAP/PGES-1 Inhibitor BRP-187 in PVA-Stabilized PLGA Nanoparticles Using Microfluidics. Polymers 2020, 12, 2751. [Google Scholar] [CrossRef]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, N.; Madni, A.; Li, W.; Correia, A.; Khan, M.M.; Rahim, M.A.; Santos, H.A. Microfluidic fabrication and characterization of Sorafenib-loaded lipid-polymer hybrid nanoparticles for controlled drug delivery. Int. J. Pharm. 2020, 581, 119275. [Google Scholar] [CrossRef]
- Abalde-Cela, S.; Taladriz-Blanco, P.; de Oliveira, M.G.; Abell, C. Droplet microfluidics for the highly controlled synthesis of branched gold nanoparticles. Sci. Rep. 2018, 8, 2440. [Google Scholar] [CrossRef] [PubMed]
- Kubendhiran, S.; Bao, Z.; Dave, K.; Liu, R.-S. Microfluidic synthesis of semiconducting colloidal quantum dots and their applications. ACS Appl. Nano Mater. 2019, 2, 1773–1790. [Google Scholar] [CrossRef]
- Larrea, A.; Sebastian, V.; Ibarra, A.; Arruebo, M.; Santamaria, J. Gas Slug Microfluidics: A Unique Tool for Ultrafast, Highly Controlled Growth of Iron Oxide Nanostructures. Chem. Mater. 2015, 27, 4254–4260. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticles Type | Manufacturing Method | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Lipid formulation | Film hydration |
|
| [108] |
| Solvent injection |
|
| [108] | |
| Extrusion |
|
| [109] | |
| High pressure homogenization |
|
| [108] | |
| Microemulsion |
|
| [110] | |
| Polymeric | Emulsification -salting out |
|
| [107,111] |
| Emulsification- solvent diffusion |
|
| [107,112] | |
| Emulsification- evaporation |
|
| [111] | |
| Dialysis |
|
| [111,113] | |
| Nonparticipation |
|
| [107,111] |
| Material | Tg 1 (°C) | Advantages | Disadvantages | Manufacturing Method | References |
|---|---|---|---|---|---|
| PDMS (elastomer) | −125 | Low Tg, easiness of shaping in the channels, optical transparency, resistance to water, ability to produce microscale features precisely. | hydrophobic nature, sensitive to organic solvents (e.g., strong acids, hydrocarbon, amines. | soft lithography, plasma-enhanced bonding | [137,138,139] |
| PMMA | 105 | low cost, optical transparency, attractive mechanical/chemical characteristics, and simple fabrication processes. | High Tg, Sensitive to alcohol, isopropyl alcohol and acetone, high bonding temperature, Commercial availability | solvent imprinting, hot embossing thermal bonding, injection molding and laser ablation. | [140,141] |
| PET | 69–78 | Low Tg, low rigidity, low surface energy, easiness of molding, chemically inertness, good gas and moisture barrier characteristics, recyclable. | reduced chemical resistance, require surface treatment for bonding due to the low plasma bonding strength. | hot embossing, thermal bonding | [137,142] |
| COP | 70–180 | High stability, low interaction with protein, suitable rigidity, resistance to almost all solvents including ethanol, IPA, and acetone, low water absorbency, high moisturiser barrier. | High Tg, brittleness and low heat diffusivity, not resistance to the non-polar organic solvent (e.g., hexane). | hot embossing, chemical etching, thermal bonding methods. | [143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaradat, E.; Weaver, E.; Meziane, A.; Lamprou, D.A. Microfluidics Technology for the Design and Formulation of Nanomedicines. Nanomaterials 2021, 11, 3440. https://doi.org/10.3390/nano11123440
Jaradat E, Weaver E, Meziane A, Lamprou DA. Microfluidics Technology for the Design and Formulation of Nanomedicines. Nanomaterials. 2021; 11(12):3440. https://doi.org/10.3390/nano11123440
Chicago/Turabian StyleJaradat, Eman, Edward Weaver, Adam Meziane, and Dimitrios A. Lamprou. 2021. "Microfluidics Technology for the Design and Formulation of Nanomedicines" Nanomaterials 11, no. 12: 3440. https://doi.org/10.3390/nano11123440
APA StyleJaradat, E., Weaver, E., Meziane, A., & Lamprou, D. A. (2021). Microfluidics Technology for the Design and Formulation of Nanomedicines. Nanomaterials, 11(12), 3440. https://doi.org/10.3390/nano11123440