
Microemulsion Derived Titania Nanospheres: An Improved Pt Supported Catalyst for Glycerol Aqueous Phase Reforming




Abstract
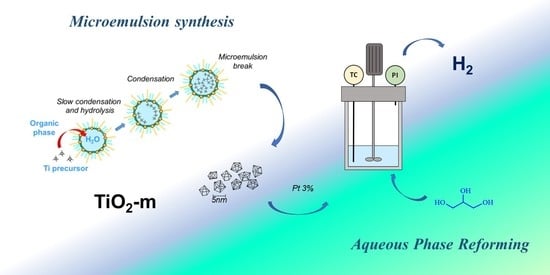
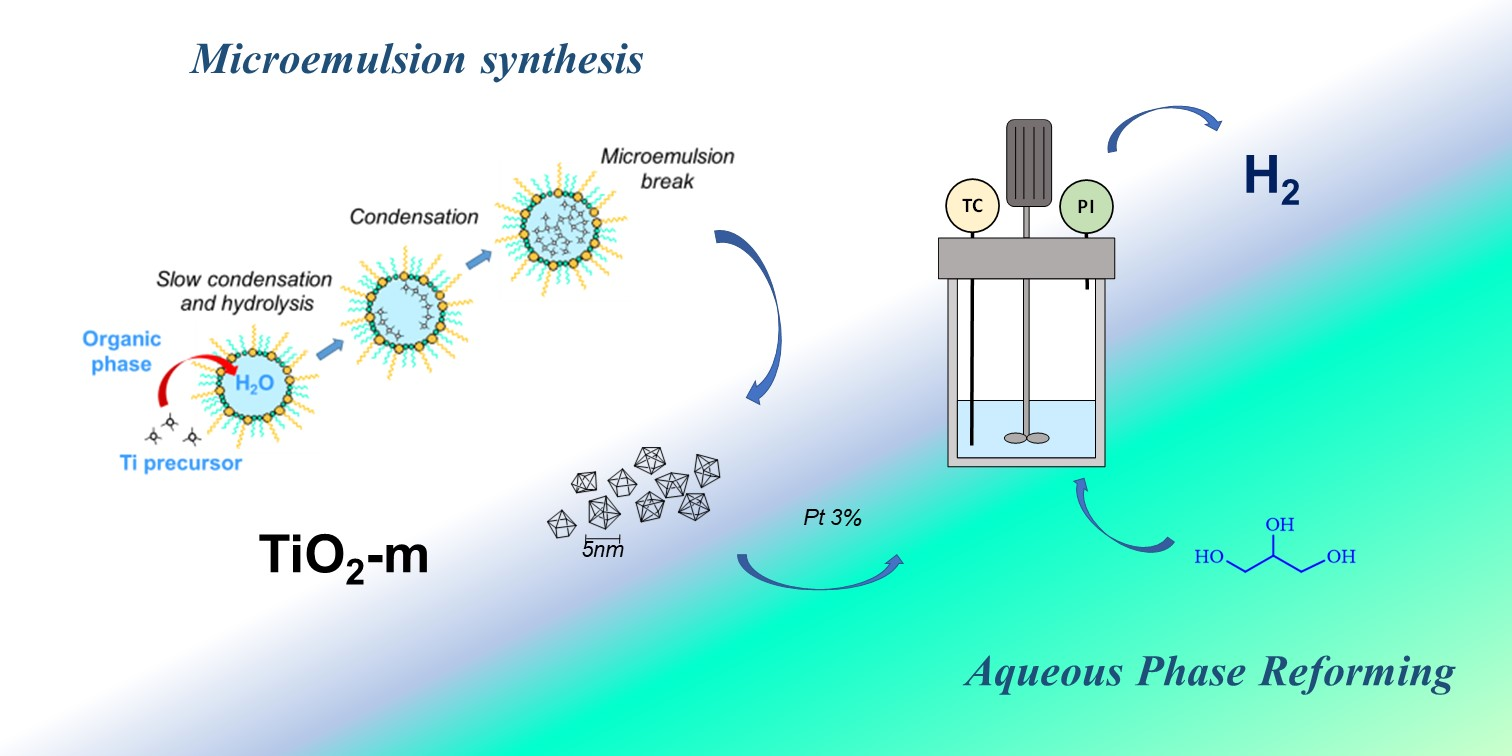
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
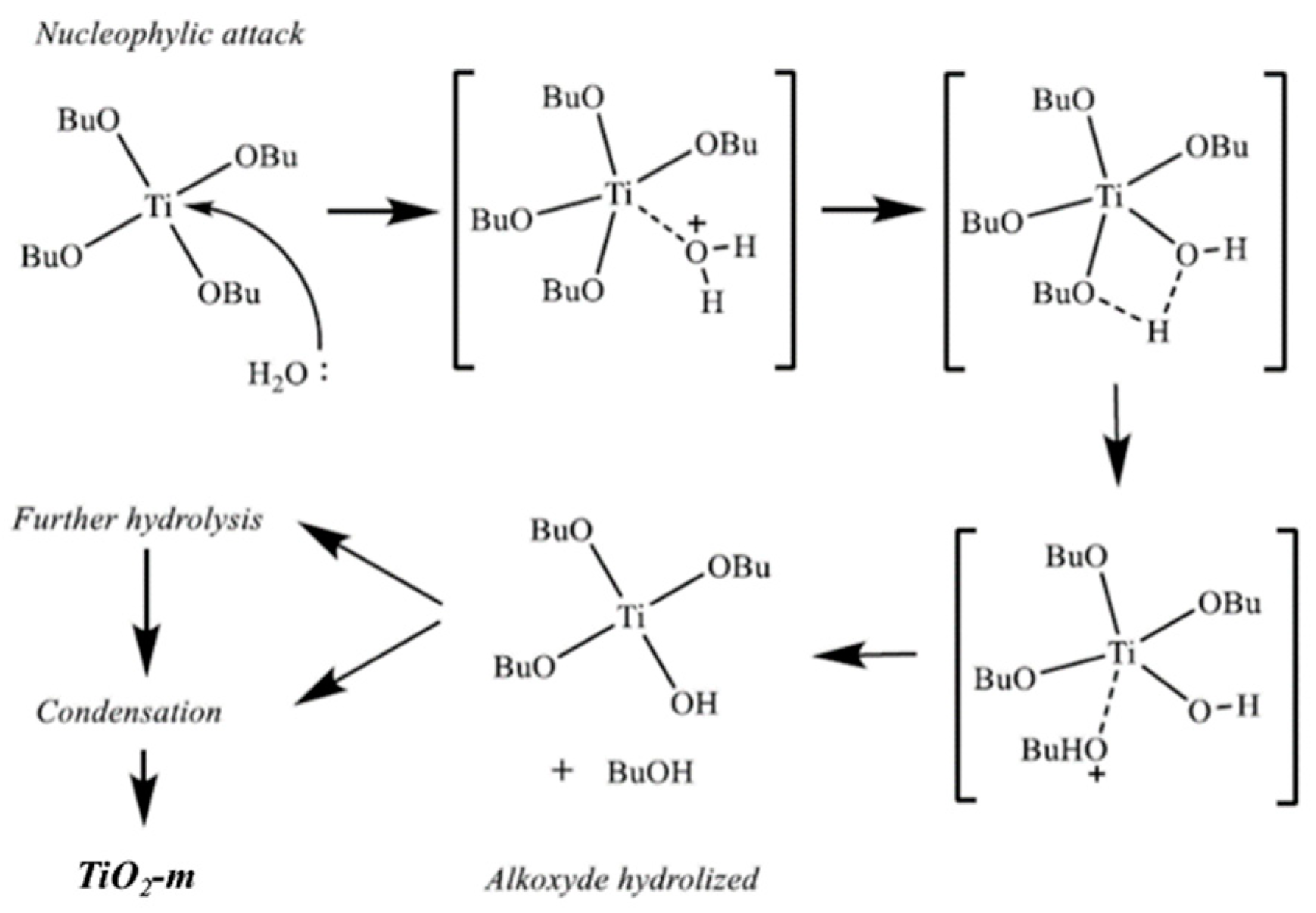{kind=link}
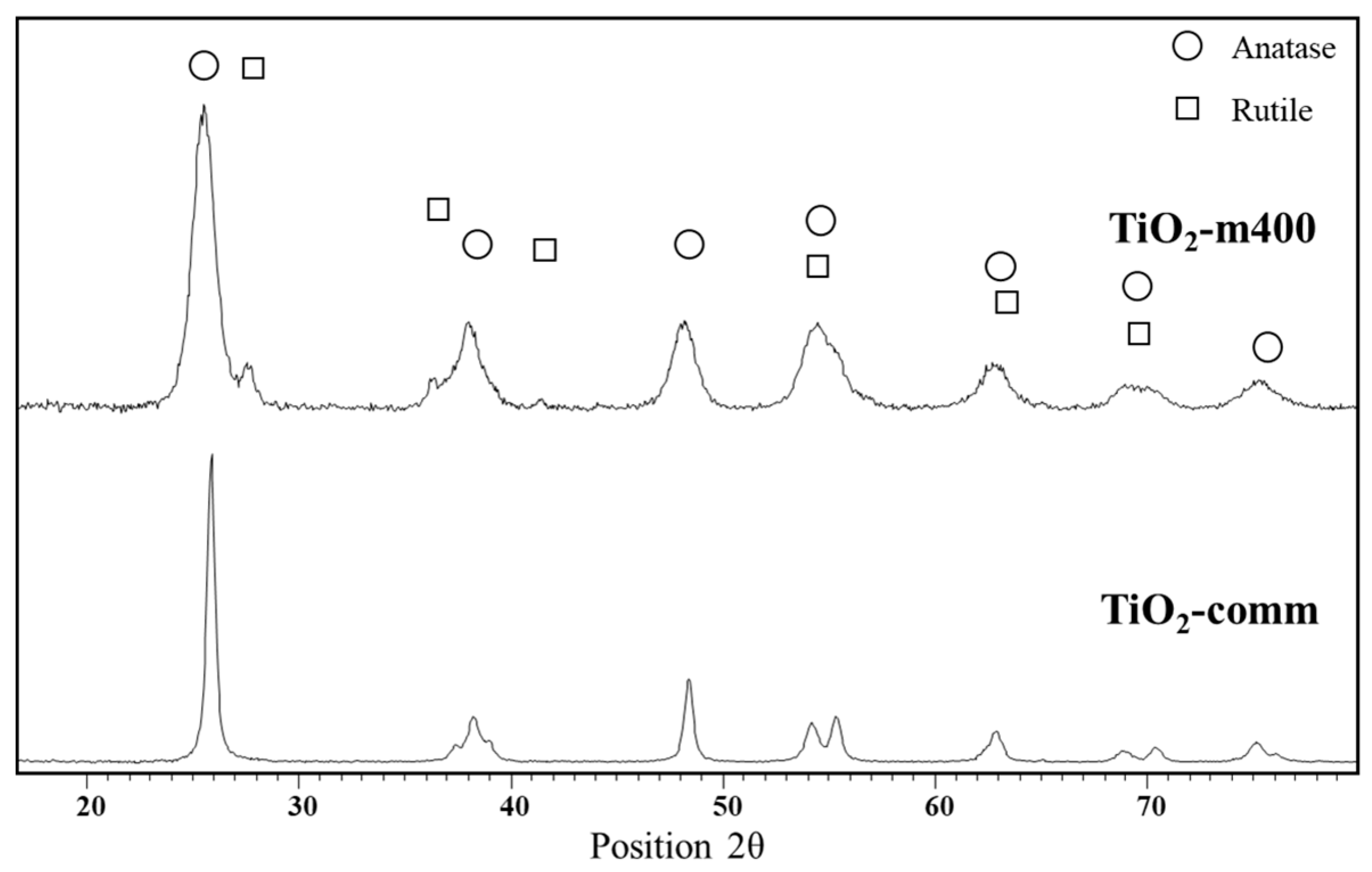{kind=link}
{kind=link}
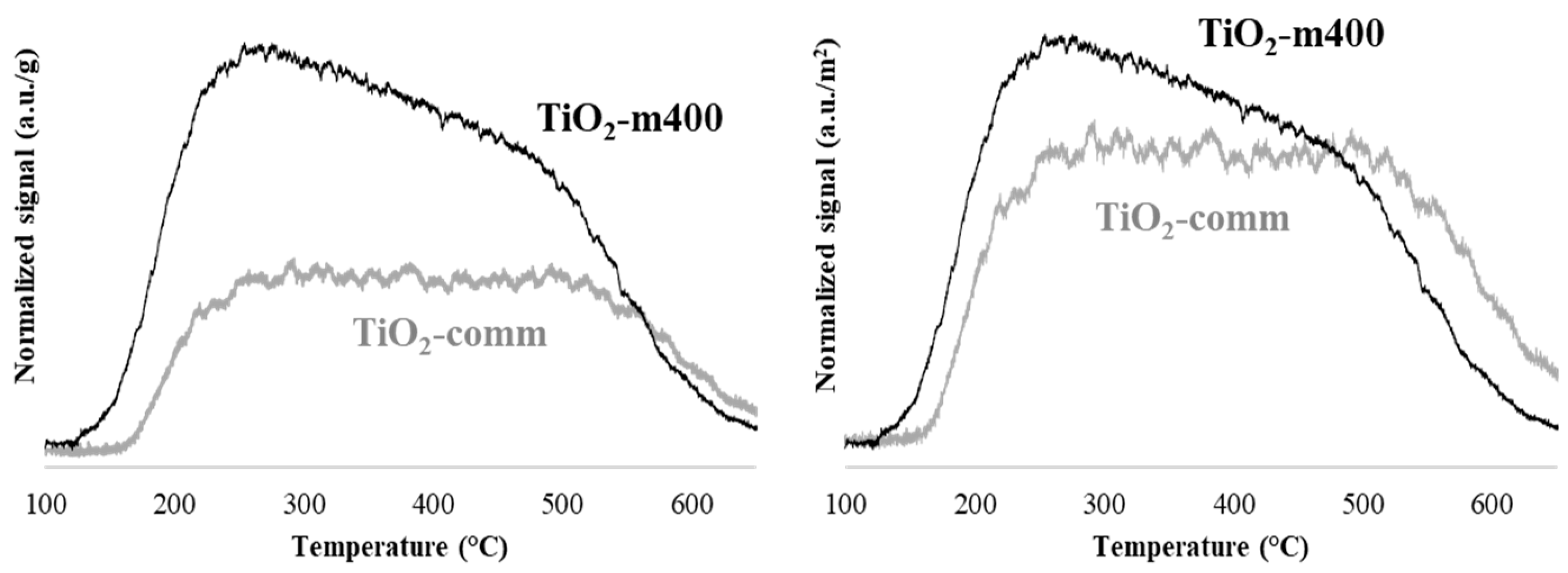{kind=link}
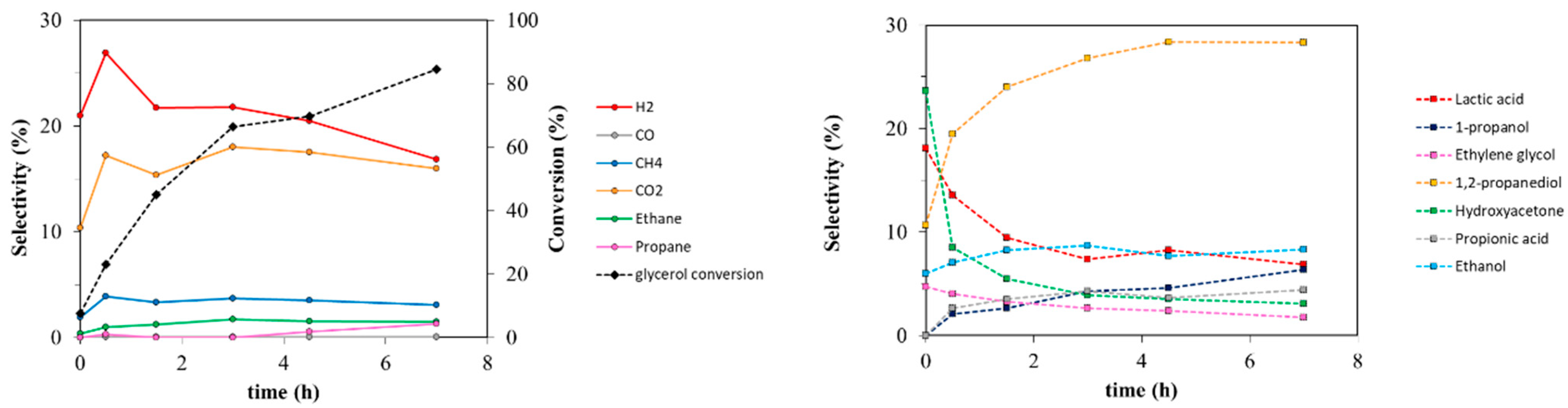{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | ΔH025 °C | |
|---|---|---|
| Glycerol Aqueous Phase Reforming (APR) | C3H5(OH)3 (l) + 3H2O (g) → 3CO2 (g) + 7H2 (g) | 128 kJ/mol |
| Water Gas Shift (WGS) | H2O + CO → H2 + CO2 | −41 kJ/mol |
| CO methanation | 3H2 + CO → CH4 + H2O | −206 kJ/mol |
| CO2 methanation | 4H2 + CO2 → CH4 + 2H2O | −165 kJ/mol |
2. Materials and Methods
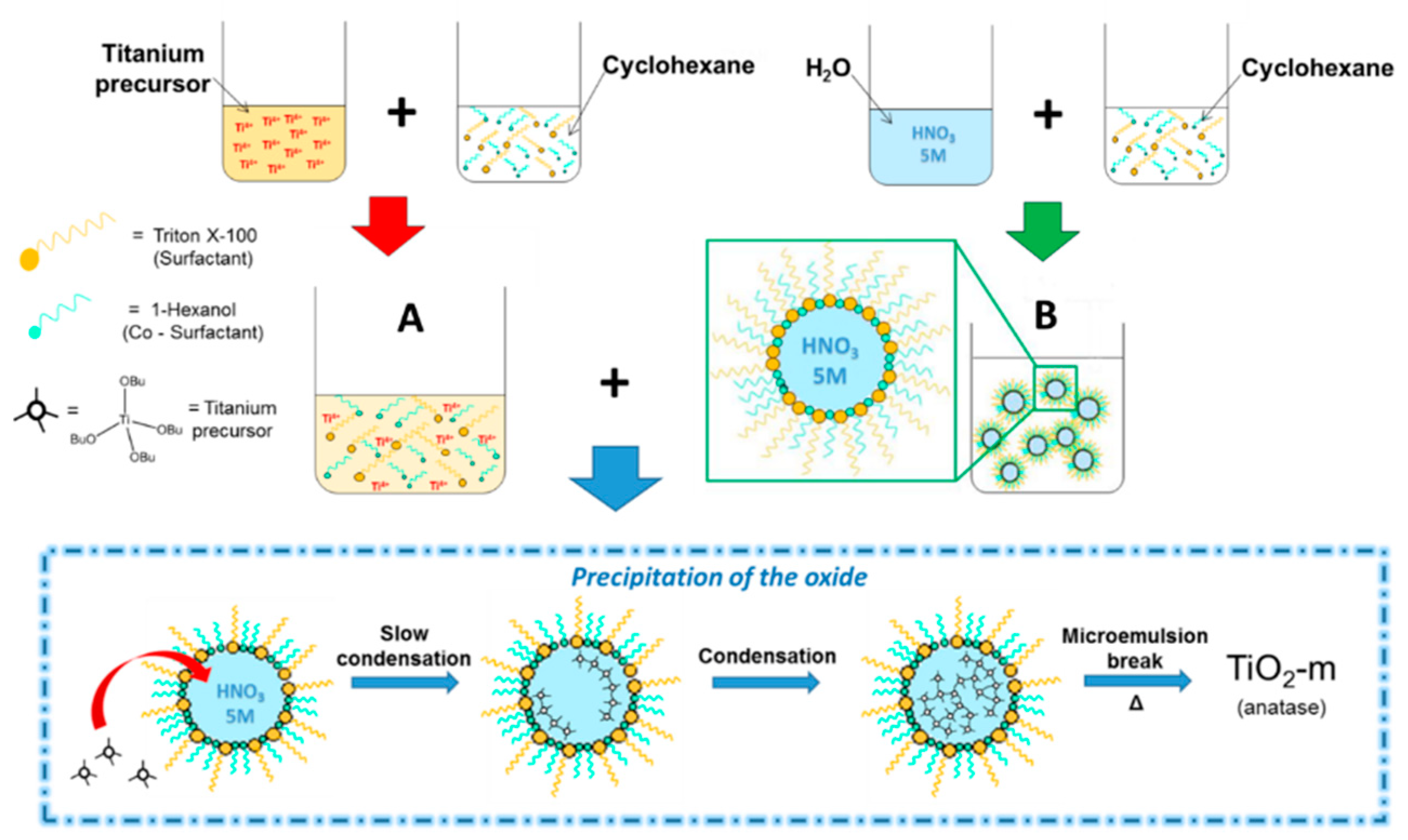
2.1. Synthesis of TiO2 by Microemulsion (TiO2-m)
2.1.1. Synthesis Background and Current Modifications
2.1.2. Synthetic Procedure
2.2. Impregnation of the Active Phase
2.3. Characterization of the Catalyst
2.4. APR Reaction
3. Results
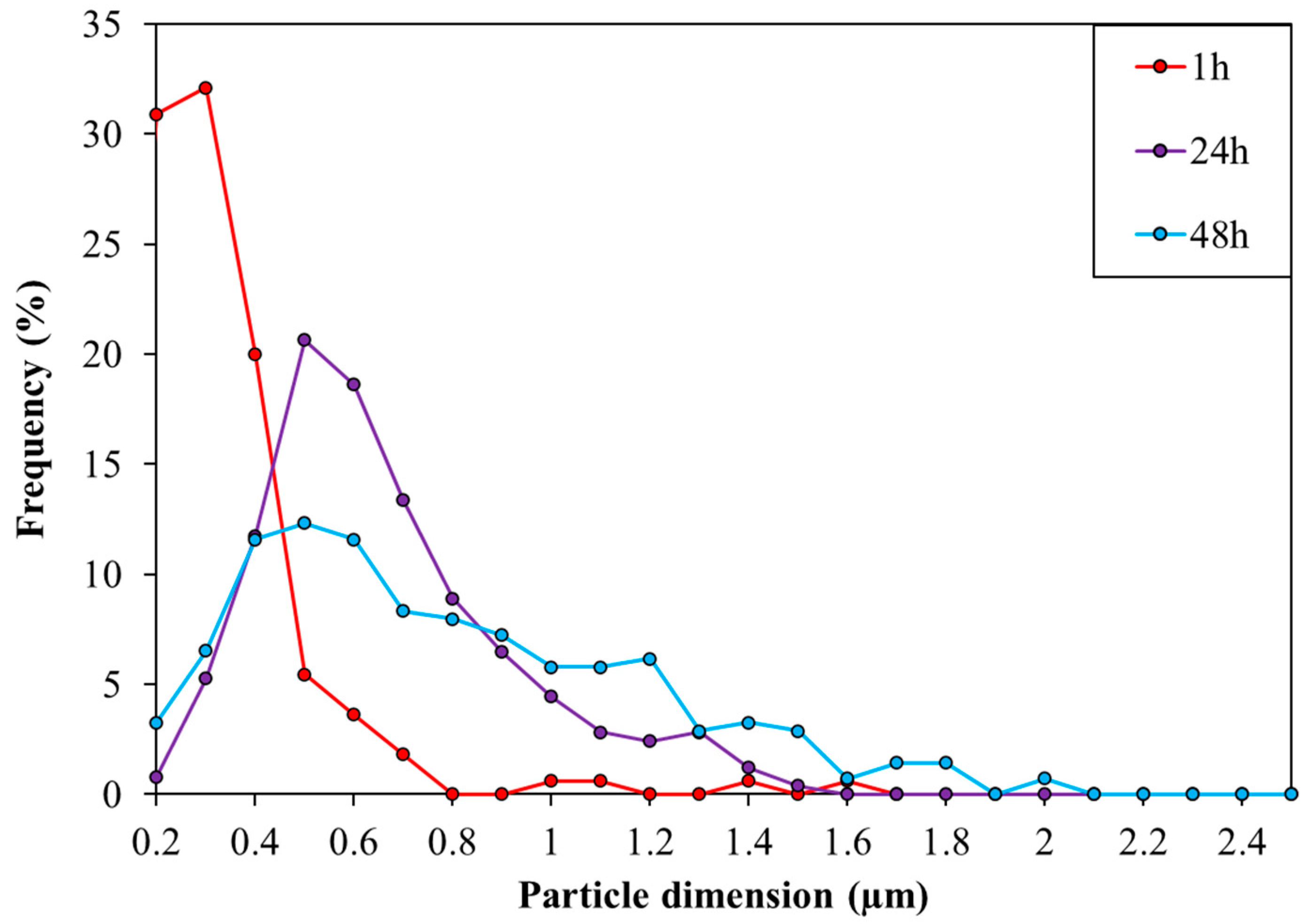
3.1. Optimization of the TiO2 Microemulsion Preparation
3.2. Catalytic Activity in APR Reaction
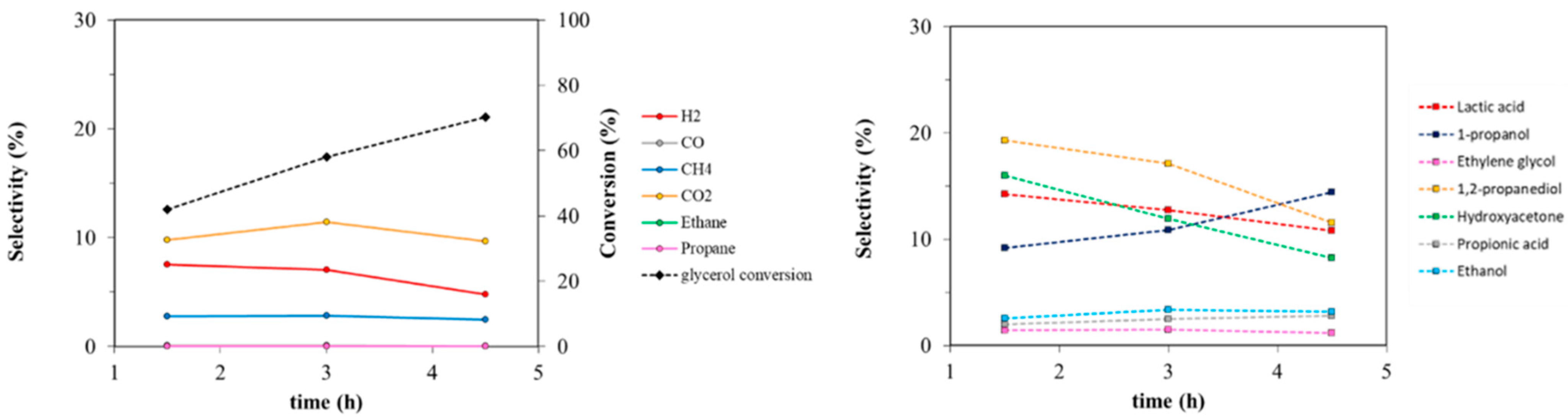
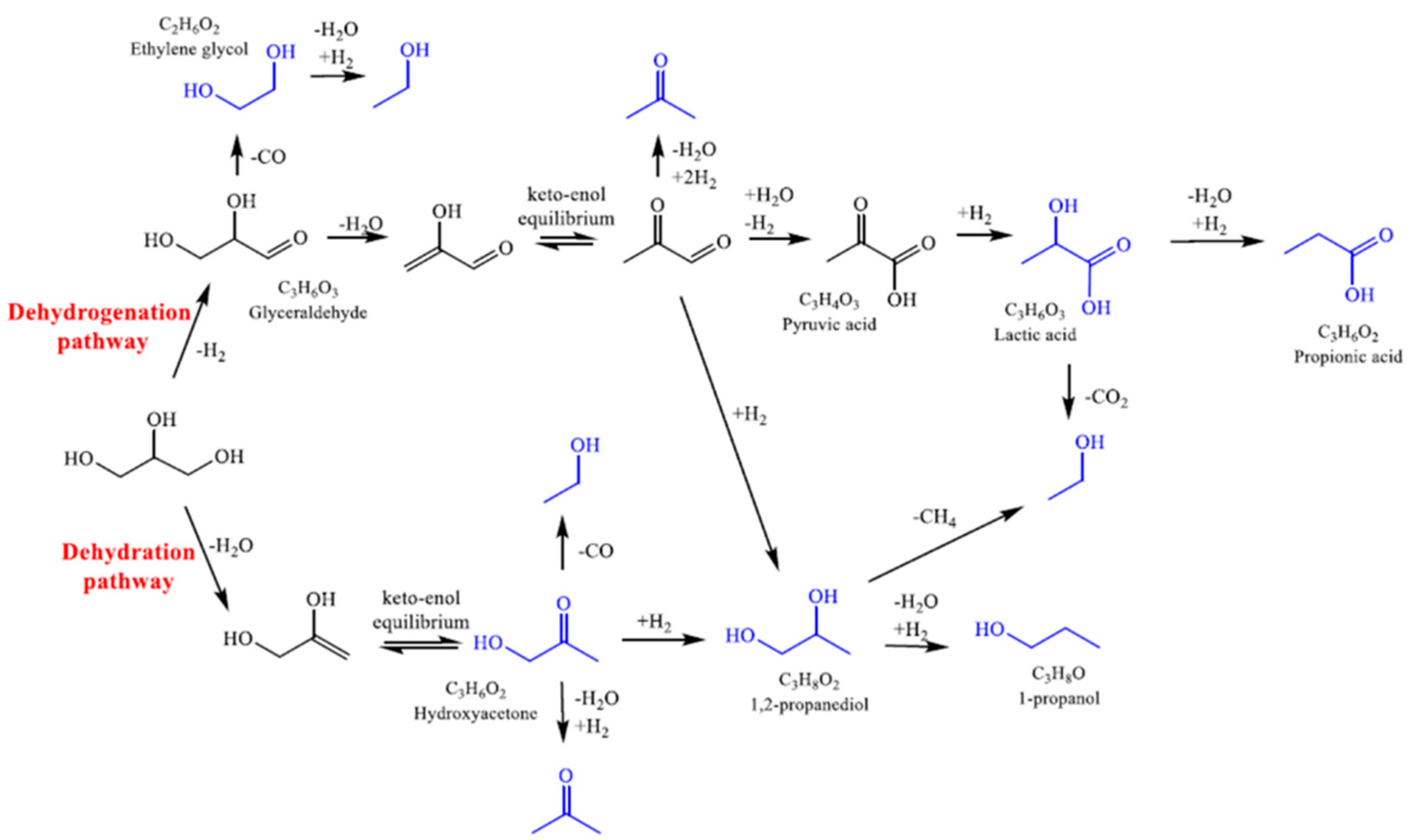
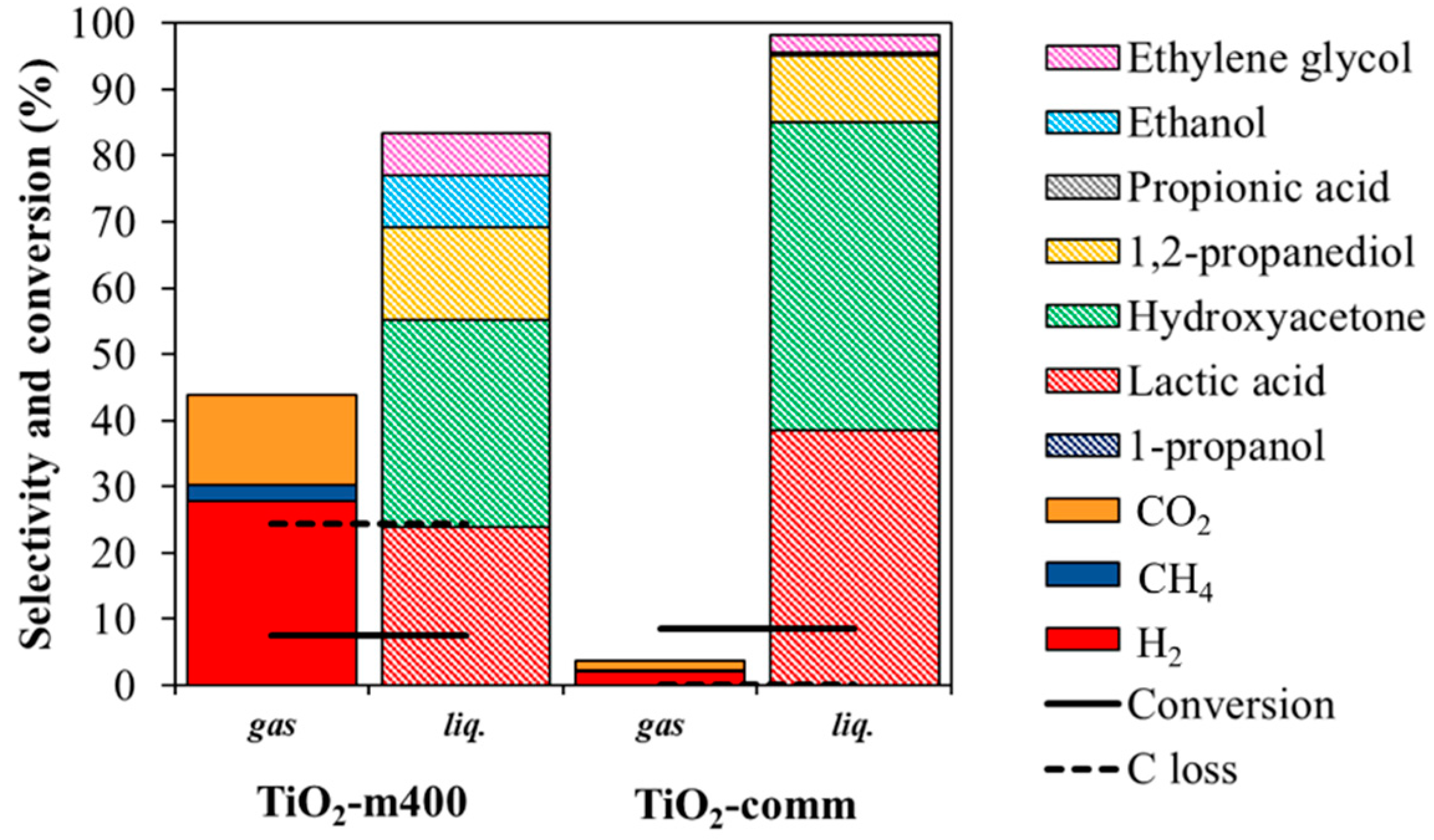
3.2.1. Effect of Reaction Time and Mechanism of Reaction
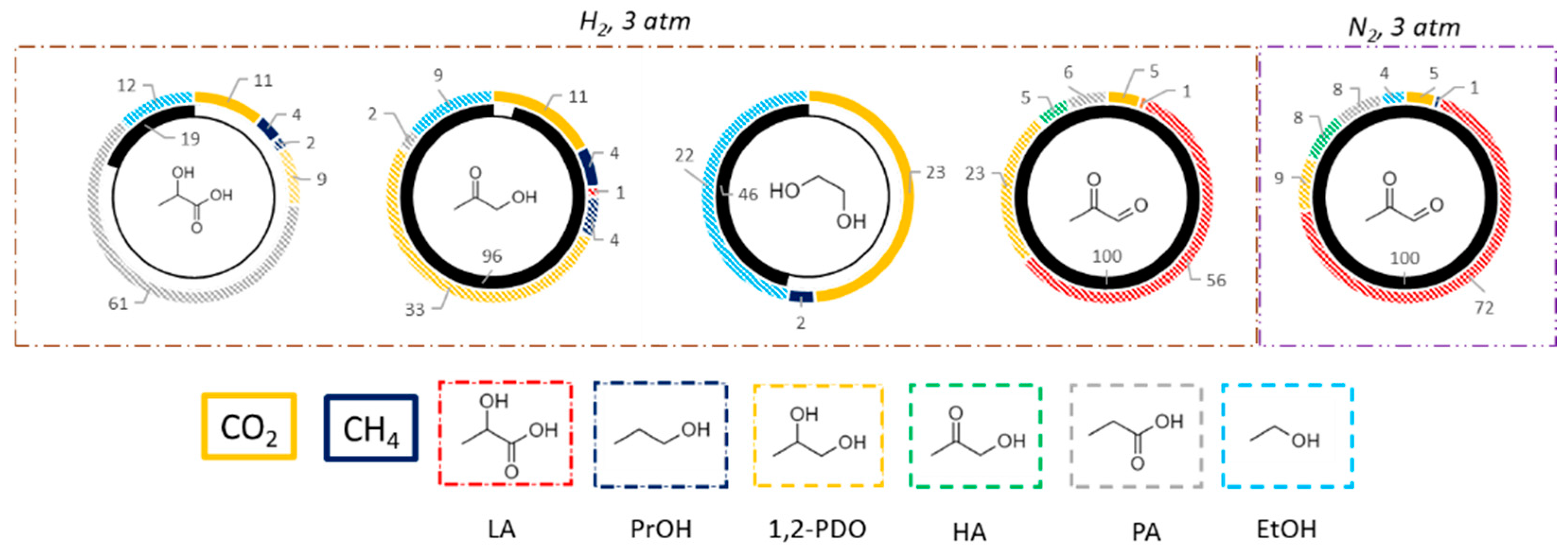
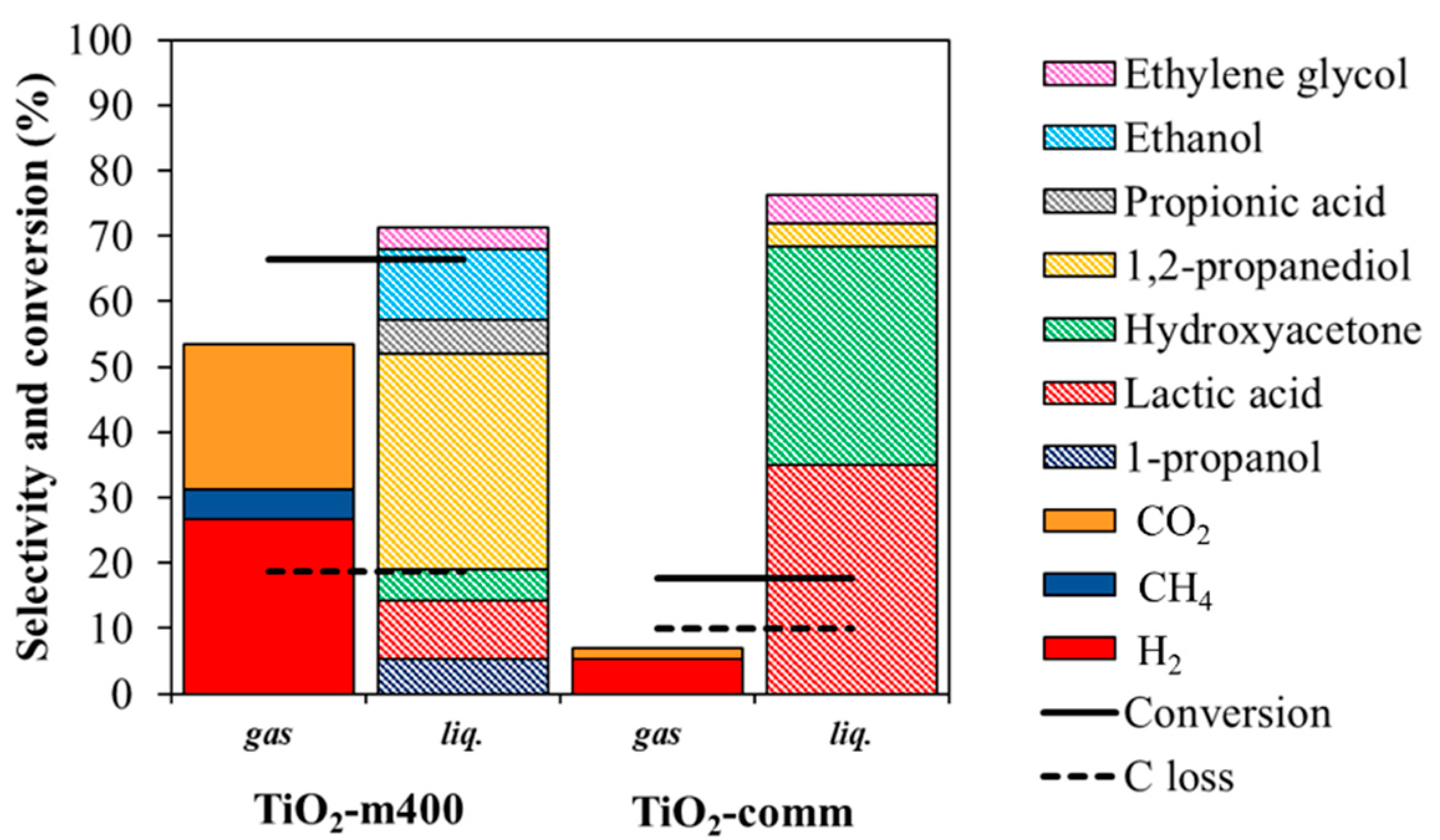
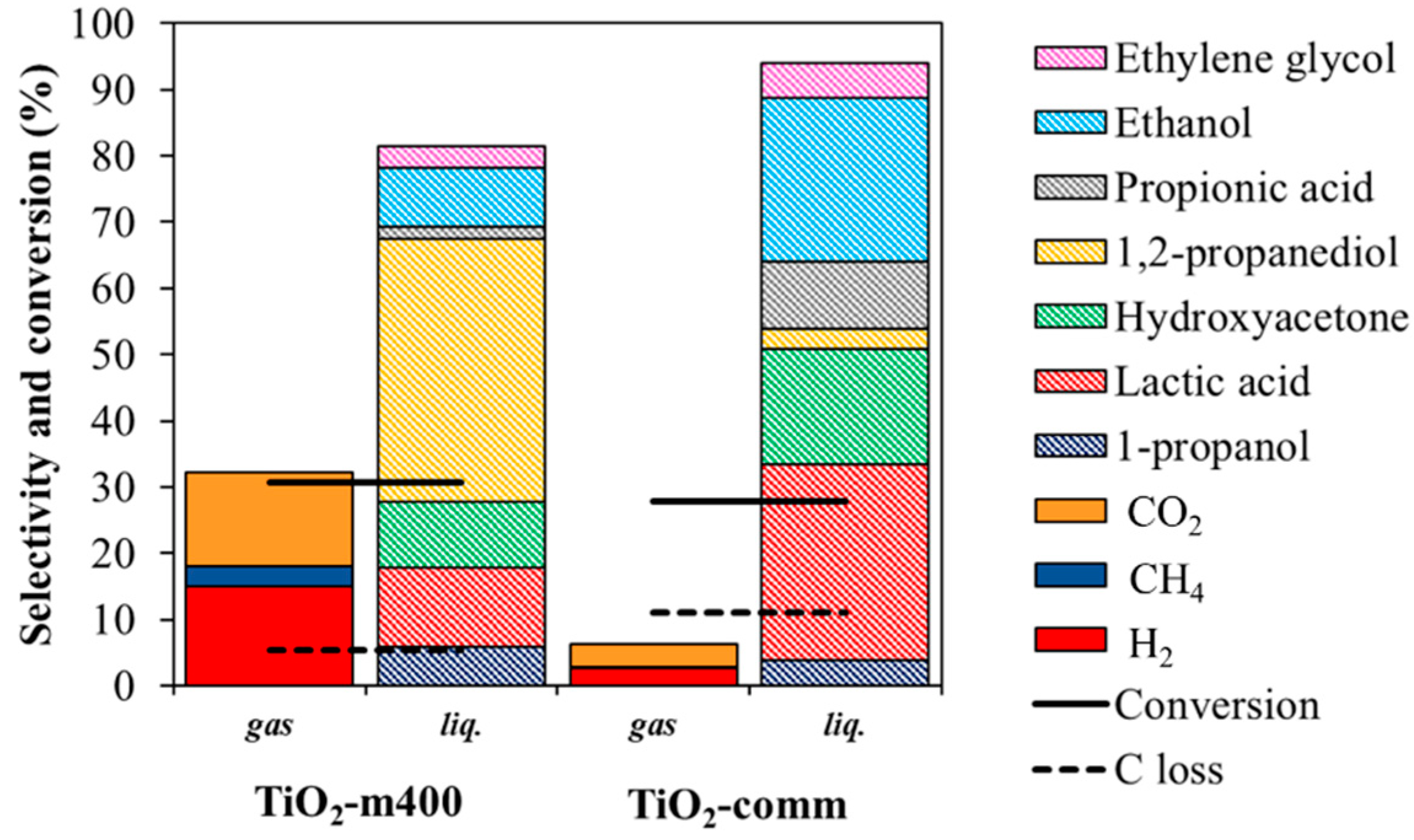
3.2.2. Comparison within Pt over TiO2-m400 and TiO2-comm
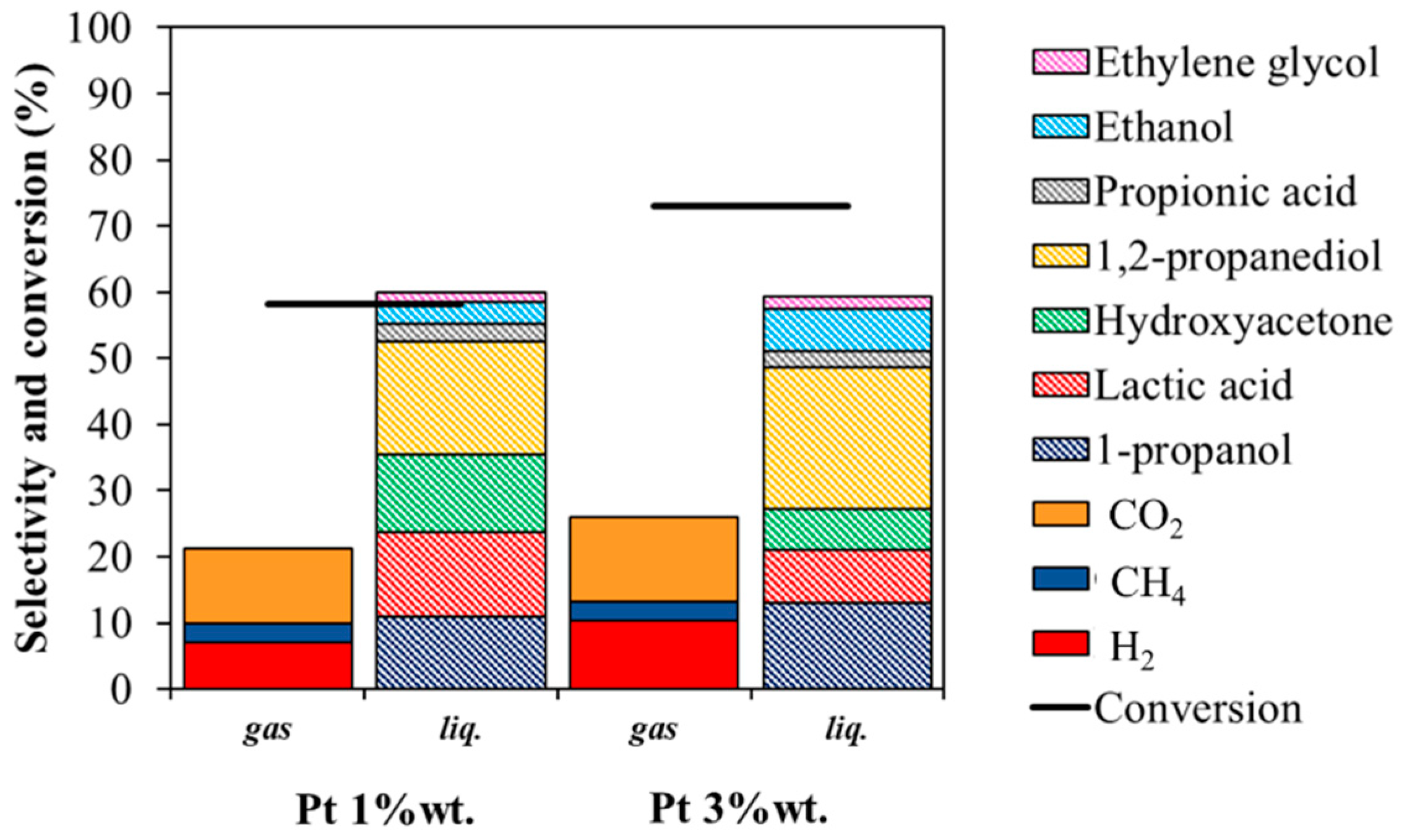
3.2.3. Effect of Metal Loading
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from Catalytic Reforming of Biomass-Derived Hydrocarbons in Liquid Water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef]
- Huber, G.W.; Dumesic, J.A. An Overview of Aqueous-Phase Catalytic Processes for Production of Hydrogen and Alkanes in a Biorefinery. Catal. Today 2006, 111, 119–132. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. A Review of Catalytic Issues and Process Conditions for Renewable Hydrogen and Alkanes by Aqueous-Phase Reforming of Oxygenated Hydrocarbons over Supported Metal Catalysts. Appl. Catal. B Environ. 2005, 56, 171–186. [Google Scholar] [CrossRef]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-Phase Catalytic Processing of Biomass-derived Oxygenated Hydrocarbons to Fuels and Chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Huber, G.W.; Shabaker, J.W.; Evans, S.T.; Dumesic, J.A. Aqueous-Phase Reforming of Ethylene Glycol over Supported Pt and Pd Bimetallic Catalysts. Appl. Catal. B Environ. 2006, 62, 226–235. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-Phase Reforming of Ethylene Glycol on Silica-Supported Metal Catalysts. Appl. Catal. B Environ. 2003, 43, 13–26. [Google Scholar] [CrossRef]
- Shabaker, J.; Davda, R.; Huber, G.; Cortright, R.; Dumesic, J. Aqueous-Phase Reforming of Methanol and Ethylene Glycol over Alumina-Supported Platinum Catalysts. J. Catal. 2003, 215, 344–352. [Google Scholar] [CrossRef]
- Coronado, I.; Stekrova, M.; Reinikainen, M.; Simell, P.; Lefferts, L.; Lehtonen, J. A Review of Catalytic Aqueous-Phase Reforming of Oxygenated Hydrocarbons Derived from Biorefinery Water Fractions. Int. J. Hydrogen Energy 2016, 41, 11003–11032. [Google Scholar] [CrossRef]
- Davda, R.R.; Dumesic, J.A. Renewable Hydrogen by Aqueous-Phase Reforming of Glucose. Chem. Commun. 2004, 36–37. [Google Scholar] [CrossRef]
- Wen, G.; Xu, Y.; Xu, Z.; Tian, Z. Characterization and Catalytic Properties of the Ni/Al2O3 Catalysts for Aqueous-Phase Reforming of Glucose. Catal. Lett. 2009, 129, 250–257. [Google Scholar] [CrossRef]
- Fasolini, A.; Cucciniello, R.; Paone, E.; Mauriello, F.; Tabanelli, T. A Short Overview on the Hydrogen Production Via Aqueous Phase Reforming (APR) of Cellulose, C6-C5 Sugars and Polyols. Catalysts 2019, 9, 917. [Google Scholar] [CrossRef] [Green Version]
- Taccardi, N.; Assenbaum, D.; Berger, M.E.M.; Bösmann, A.; Enzenberger, F.; Wölfel, R.; Neuendorf, S.; Goeke, V.; Schödel, N.; Maass, H.-J.; et al. Catalytic Production of Hydrogen from Glucose and Other Carbohydrates under Exceptionally Mild Reaction Conditions. Green Chem. 2010, 12, 1150–1156. [Google Scholar] [CrossRef]
- Kirilin, A.V.; Tokarev, A.V.; Kustov, L.M.; Salmi, T.; Mikkola, J.-P.; Murzin, D.Y. Aqueous Phase Reforming of Xylitol and Sorbitol: Comparison and Influence of Substrate Structure. Appl. Catal. A Gen. 2012, 435–436, 172–180. [Google Scholar] [CrossRef]
- Vaidya, P.D.; Lopez-Sanchez, J.A. Review of Hydrogen Production by Catalytic Aqueous-Phase Reforming. ChemistrySelect 2017, 2, 6563–6576. [Google Scholar] [CrossRef]
- Kirilin, A.; Wärnå, J.; Tokarev, A.; Murzin, D.Y. Kinetic Modeling of Sorbitol Aqueous-Phase Reforming over Pt/Al2O3. Ind. Eng. Chem. Res. 2014, 53, 4580–4588. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, T.; Ma, L.; Li, Y.; Zhang, Q.; Zhang, X. Investigation on the Xylitol Aqueous-Phase Reforming Performance for Pentane Production over Pt/HZSM-5 and Ni/HZSM-5 Catalysts. Appl. Energy 2012, 90, 51–57. [Google Scholar] [CrossRef]
- Sladkovskiy, D.A.; Godina, L.I.; Semikin, K.V.; Sladkovskaya, E.V.; Smirnova, D.A.; Murzin, D.Y. Process Design and Techno-Economical Analysis of Hydrogen Production by Aqueous Phase Reforming of Sorbitol. Chem. Eng. Res. Des. 2018, 134, 104–116. [Google Scholar] [CrossRef]
- Murzin, D.Y.; Garcia, S.; Russo, V.; Kilpiö, T.; Godina, L.I.; Tokarev, A.V.; Kirilin, A.V.; Simakova, I.L.; Poulston, S.; Sladkovskiy, D.A.; et al. Kinetics, Modeling, and Process Design of Hydrogen Production by Aqueous Phase Reforming of Xylitol. Ind. Eng. Chem. Res. 2017, 56, 13240–13253. [Google Scholar] [CrossRef]
- Godina, L.I.; Kirilin, A.V.; Tokarev, A.V.; Simakova, I.L.; Murzin, D.Y. Sibunit-Supported Mono-and Bimetallic Catalysts Used in Aqueous-Phase Reforming of Xylitol. Ind. Eng. Chem. Res. 2018, 57, 2050–2067. [Google Scholar] [CrossRef]
- Tabanelli, T.; Giliberti, C.; Mazzoni, R.; Cucciniello, R.; Cavani, F. An Innovative Synthesis Pathway to Benzodioxanes: The Peculiar Reactivity of Glycerol Carbonate and Catechol. Green Chem. 2019, 21, 329–338. [Google Scholar] [CrossRef]
- Soriano, M.D.; Chieregato, A.; Zamora, S.; Basile, F.; Cavani, F.; López Nieto, J.M. Promoted Hexagonal Tungsten Bronzes as Selective Catalysts in the Aerobic Transformation of Alcohols: Glycerol and Methanol. Top. Catal. 2016, 59, 178–185. [Google Scholar] [CrossRef]
- Boga, D.A.; Liu, F.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Aqueous-Phase Reforming of Crude Glycerol: Effect of Impurities on Hydrogen Production. Catal. Sci. Technol. 2016, 6, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Seretis, A.; Tsiakaras, P. Crude Bio-Glycerol Aqueous Phase Reforming and Hydrogenolysis over Commercial SiO2Al2O3 Nickel Catalyst. Renew. Energy 2016, 97, 373–379. [Google Scholar] [CrossRef]
- Remón, J.; Giménez, J.R.; Valiente, A.; García, L.; Arauzo, J. Production of Gaseous and Liquid Chemicals by Aqueous Phase Reforming of Crude Glycerol: Influence of Operating Conditions on the Process. Energy Convers. Manag. 2016, 110, 90–112. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, P.D.; Rodrigues, A.E. Glycerol Reforming for Hydrogen Production: A Review. Chem. Eng. Technol. Ind. Chem. Plant Equip. Process Eng. Biotechnol. 2009, 32, 1463–1469. [Google Scholar] [CrossRef]
- Fasolini, A.; Cespi, D.; Tabanelli, T.; Cucciniello, R.; Cavani, F. Hydrogen from Renewables: A Case Study of Glycerol Reforming. Catalysts 2019, 9, 722. [Google Scholar] [CrossRef] [Green Version]
- Schwengber, C.A.; Alves, H.J.; Schaffner, R.A.; da Silva, F.A.; Sequinel, R.; Bach, V.R.; Ferracin, R.J. Overview of Glycerol Reforming for Hydrogen Production. Renew. Sustain. Energy Rev. 2016, 58, 259–266. [Google Scholar] [CrossRef]
- Morales-Marín, A.; Ayastuy, J.L.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A. Nickel Aluminate Spinel-Derived Catalysts for the Aqueous Phase Reforming of Glycerol: Effect of Reduction Temperature. Appl. Catal. B Environ. 2019, 244, 931–945. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Dumesic, J.A. Kinetics of Aqueous-Phase Reforming of Oxygenated Hydrocarbons: Pt/Al2O3 and Sn-Modified Ni Catalysts. Ind. Eng. Chem. Res. 2004, 43, 3105–3112. [Google Scholar] [CrossRef]
- Wawrzetz, A.; Peng, B.; Hrabar, A.; Jentys, A.; Lemonidou, A.A.; Lercher, J.A. Towards Understanding the Bifunctional Hydrodeoxygenation and Aqueous Phase Reforming of Glycerol. J. Catal. 2010, 269, 411–420. [Google Scholar] [CrossRef]
- Vásquez, P.B.; Tabanelli, T.; Monti, E.; Albonetti, S.; Bonincontro, D.; Dimitratos, N.; Cavani, F. Gas-Phase Catalytic Transfer Hydrogenation of Methyl Levulinate with Ethanol over ZrO2. ACS Sustain. Chem. Eng. 2019, 7, 8317–8330. [Google Scholar] [CrossRef]
- Ciftci, A.; Ligthart, D.A.J.M.; Sen, A.O.; van Hoof, A.J.F.; Friedrich, H.; Hensen, E.J.M. Pt-Re Synergy in Aqueous-Phase Reforming of Glycerol and the Water–Gas Shift Reaction. J. Catal. 2014, 311, 88–101. [Google Scholar] [CrossRef]
- Papageridis, K.N.; Siakavelas, G.; Charisiou, N.D.; Avraam, D.G.; Tzounis, L.; Kousi, K.; Goula, M.A. Comparative Study of Ni, Co, Cu Supported on γ-Alumina Catalysts for Hydrogen Production via the Glycerol Steam Reforming Reaction. Fuel Process. Technol. 2016, 152, 156–175. [Google Scholar] [CrossRef]
- Rahman, M.; Church, T.L.; Minett, A.I.; Harris, A.T. Effect of CeO2 Addition to Al2O3 Supports for Pt Catalysts on the Aqueous-Phase Reforming of Glycerol. ChemSusChem 2013, 6, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.; Xu, Y.; Ma, H.; Xu, Z.; Tian, Z. Production of Hydrogen by Aqueous-Phase Reforming of Glycerol. Int. J. Hydrogen Energy 2008, 33, 6657–6666. [Google Scholar] [CrossRef]
- Ciftci, A.; Peng, B.; Jentys, A.; Lercher, J.A.; Hensen, E.J. Support Effects in the Aqueous Phase Reforming of Glycerol over Supported Platinum Catalysts. Appl. Catal. A Gen. 2012, 431, 113–119. [Google Scholar] [CrossRef]
- Lehnert, K.; Claus, P. Influence of Pt Particle Size and Support Type on the Aqueous-Phase Reforming of Glycerol. Catal. Commun. 2008, 9, 2543–2546. [Google Scholar] [CrossRef]
- El Doukkali, M.; Iriondo, A.; Cambra, J.; Jalowiecki-Duhamel, L.; Mamede, A.; Dumeignil, F.; Arias, P. Pt Monometallic and Bimetallic Catalysts Prepared by Acid Sol–Gel Method for Liquid Phase Reforming of Bioglycerol. J. Mol. Catal. A Chem. 2013, 368, 125–136. [Google Scholar] [CrossRef]
- Manfro, R.L.; da Costa, A.F.; Ribeiro, N.F.P.; Souza, M.M.V.M. Hydrogen Production by Aqueous-Phase Reforming of Glycerol over Nickel Catalysts Supported on CeO2. Fuel Process. Technol. 2011, 92, 330–335. [Google Scholar] [CrossRef]
- Guo, Y.; Azmat, M.U.; Liu, X.; Wang, Y.; Lu, G. Effect of Support’s Basic Properties on Hydrogen Production in Aqueous-Phase Reforming of Glycerol and Correlation between WGS and APR. Appl. Energy 2012, 92, 218–223. [Google Scholar] [CrossRef]
- Barbelli, M.L.; Pompeo, F.; Santori, G.F.; Nichio, N.N. Pt Catalyst Supported on α-Al2O3 Modified with CeO2 and ZrO2 for Aqueous-Phase-Reforming of Glycerol. Catal. Today 2013, 213, 58–64. [Google Scholar] [CrossRef]
- Larimi, A.S.; Kazemeini, M.; Khorasheh, F. Aqueous Phase Reforming of Glycerol Using Highly Active and Stable Pt0.05CexZr0.95-XO2 Ternary Solid Solution Catalysts. Appl. Catal. A Gen. 2016, 523, 230–240. [Google Scholar] [CrossRef]
- Ciftci, A.; Eren, S.; Ligthart, D.M.; Hensen, E.J. Platinum–Rhenium Synergy on Reducible Oxide Supports in Aqueous-Phase Glycerol Reforming. ChemCatChem 2014, 6, 1260–1269. [Google Scholar] [CrossRef]
- Bastan, F.; Kazemeini, M.; Larimi, A.; Maleki, H. Production of Renewable Hydrogen through Aqueous-Phase Reforming of Glycerol over Ni/Al2O3MgO Nano-Catalyst. Int. J. Hydrogen Energy 2018, 43, 614–621. [Google Scholar] [CrossRef]
- Delgado, S.N.; Yap, D.; Vivier, L.; Especel, C. Influence of the Nature of the Support on the Catalytic Properties of Pt-Based Catalysts for Hydrogenolysis of Glycerol. J. Mol. Catal. A Chem. 2013, 367, 89–98. [Google Scholar] [CrossRef]
- Boga, D.A.; Oord, R.; Beale, A.M.; Chung, Y.; Bruijnincx, P.C.; Weckhuysen, B.M. Highly Selective Bimetallic Pt-Cu/Mg(Al)O Catalysts for the Aqueous-Phase Reforming of Glycerol. ChemCatChem 2013, 5, 529–537. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Davda, R.R.; Cortright, R.D.; Dumesic, J.A. Aqueous-Phase Reforming of Ethylene Glycol Over Supported Platinum Catalysts. Catal. Lett. 2003, 88, 1–8. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kim, D.-Y.; Han, H.; Shul, Y.-G. PtRu/C-Au/TiO2 Electrocatalyst for a Direct Methanol Fuel Cell. J. Power Sources 2006, 159, 484–490. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Han, G.; Du, C.; Sun, Y.; Du, L.; An, M.; Yin, G.; Gao, Y.; Song, Y. Superior Catalytic Performance and CO Tolerance of Ru@Pt/C-TiO2 Electrocatalyst toward Methanol Oxidation Reaction. Appl. Surf. Sci. 2019, 473, 943–950. [Google Scholar] [CrossRef]
- Liang, R.; Hu, A.; Persic, J.; Zhou, Y.N. Palladium Nanoparticles Loaded on Carbon Modified TiO2 Nanobelts for Enhanced Methanol Electrooxidation. Nano-Micro Lett. 2013, 5, 202–212. [Google Scholar] [CrossRef]
- Al-Mamun, M.R.; Kader, S.; Islam, M.S.; Khan, M.Z.H. Photocatalytic Activity Improvement and Application of UV-TiO2 Photocatalysis in Textile Wastewater Treatment: A Review. J. Environ. Chem. Eng. 2019, 7, 103248. [Google Scholar] [CrossRef]
- Liang, R.; Hu, A.; Li, W.; Zhou, Y.N. Enhanced Degradation of Persistent Pharmaceuticals Found in Wastewater Treatment Effluents Using TiO2 Nanobelt Photocatalysts. J. Nanopart. Res. 2013, 15, 1990. [Google Scholar] [CrossRef]
- Khalid, N.R.; Majid, A.; Tahir, M.B.; Niaz, N.A.; Khalid, S. Carbonaceous-TiO2 Nanomaterials for Photocatalytic Degradation of Pollutants: A Review. Ceram. Int. 2017, 43, 14552–14571. [Google Scholar] [CrossRef]
- Chen, D.; Cheng, Y.; Zhou, N.; Chen, P.; Wang, Y.; Li, K.; Huo, S.; Cheng, P.; Peng, P.; Zhang, R.; et al. Photocatalytic Degradation of Organic Pollutants Using TiO2-Based Photocatalysts: A Review. J. Clean. Prod. 2020, 268, 121725. [Google Scholar] [CrossRef]
- Kumaravel, V.; Mathew, S.; Bartlett, J.; Pillai, S.C. Photocatalytic Hydrogen Production Using Metal Doped TiO2: A Review of Recent Advances. Appl. Catal. B Environ. 2019, 244, 1021–1064. [Google Scholar] [CrossRef]
- Singh, R.; Dutta, S. A Review on H2 Production through Photocatalytic Reactions Using TiO2/TiO2-Assisted Catalysts. Fuel 2018, 220, 607–620. [Google Scholar] [CrossRef]
- Maslova, V.; Fasolini, A.; Offidani, M.; Albonetti, S.; Basile, F. Solar-Driven Valorization of Glycerol towards Production of Chemicals and Hydrogen. Catal. Today 2021. [Google Scholar] [CrossRef]
- Shen, Y.; Mamakhel, A.; Liu, X.; Hansen, T.W.; Tabanelli, T.; Bonincontro, D.; Iversen, B.B.; Prati, L.; Li, Y.; Niemantsverdriet, J.W.H.; et al. Promotion Mechanisms of Au Supported on TiO2 in T and Photocatalytic Glycerol Conversion. J. Phys. Chem. C 2019, 123, 19734–19741. [Google Scholar] [CrossRef]
- Solmi, S.; Rozhko, E.; Malmusi, A.; Tabanelli, T.; Albonetti, S.; Basile, F.; Agnoli, S.; Cavani, F. The Oxidative Cleavage of Trans-1,2-Cyclohexanediol with O2: Catalysis by Supported Au Nanoparticles. Appl. Catal. A Gen. 2018, 557, 89–98. [Google Scholar] [CrossRef]
- Green, I.X.; Tang, W.; McEntee, M.; Neurock, M.; Yates, J.T. Inhibition at Perimeter Sites of Au/TiO2 Oxidation Catalyst by Reactant Oxygen. J. Am. Chem. Soc. 2012, 134, 12717–12723. [Google Scholar] [CrossRef]
- Katal, R.; Masudy-Panah, S.; Tanhaei, M.; Farahani, M.H.D.A.; Jiangyong, H. A Review on the Synthesis of the Various Types of Anatase TiO2 Facets and Their Applications for Photocatalysis. Chem. Eng. J. 2020, 384, 123384. [Google Scholar] [CrossRef]
- Humayun, M.; Raziq, F.; Khan, A.; Luo, W. Modification Strategies of TiO2 for Potential Applications in Photocatalysis: A Critical Review. Green Chem. Lett. Rev. 2018, 11, 86–102. [Google Scholar] [CrossRef] [Green Version]
- Solanki, J.N.; Murthy, Z.V.P. Controlled Size Silver Nanoparticles Synthesis with Water-in-Oil Microemulsion Method: A Topical Review. Ind. Eng. Chem. Res. 2011, 50, 12311–12323. [Google Scholar] [CrossRef]
- Ganguli, A.K.; Ganguly, A.; Vaidya, S. Microemulsion-Based Synthesis of Nanocrystalline Materials. Chem. Soc. Rev. 2010, 39, 474–485. [Google Scholar] [CrossRef]
- Buceta, D.; Piñeiro, Y.; Vázquez-Vázquez, C.; Rivas, J.; López-Quintela, M.A. Metallic Clusters: Theoretical Background, Properties and Synthesis in Microemulsions. Catalysts 2014, 4, 356–374. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.; Pedersen, J.S.; Palmqvist, A.E.C. Silver Nanoparticle Formation in Microemulsions Acting Both as Template and Reducing Agent. Langmuir 2005, 21, 11387–11396. [Google Scholar] [CrossRef]
- Wolf, S.; Feldmann, C. Microemulsions: Options to Expand the Synthesis of Inorganic Nanoparticles. Angew. Chem. Int. Ed. 2016, 55, 15728–15752. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Dominguez, M.; Pemartin, K.; Boutonnet, M. Preparation of Inorganic Nanoparticles in Oil-in-Water Microemulsions: A Soft and Versatile Approach. Curr. Opin. Colloid Interface Sci. 2012, 17, 297–305. [Google Scholar] [CrossRef]
- Boutonnet, M.; Sanchez-Dominguez, M. Microemulsion Droplets to Catalytically Active Nanoparticles. How the Application of Colloidal Tools in Catalysis Aims to Well Designed and Efficient Catalysts. Catal. Today 2017, 285, 89–103. [Google Scholar] [CrossRef]
- Basile, F.; Mafessanti, R.; Fasolini, A.; Fornasari, G.; Lombardi, E.; Vaccari, A. Effect of Synthetic Method on CeZr Support and Catalytic Activity of Related Rh Catalyst in the Oxidative Reforming Reaction. J. Eur. Ceram. Soc. 2019, 39, 41–52. [Google Scholar] [CrossRef]
- Fasolini, A.; Ruggieri, S.; Femoni, C.; Basile, F. Highly Active Catalysts Based on the Rh4(CO)12 Cluster Supported on Ce0.5Zr0.5 and Zr Oxides for Low-Temperature Methane Steam Reforming. Catalysts 2019, 9, 800. [Google Scholar] [CrossRef] [Green Version]
- Lolli, A.; Maslova, V.; Bonincontro, D.; Basile, F.; Ortelli, S.; Albonetti, S. Selective Oxidation of HMF via Catalytic and Photocatalytic Processes Using Metal-Supported Catalysts. Molecules 2018, 23, 2792. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.; Kiselev, A.; Österlund, L.; Palmqvist, A.E.C. Microemulsion-Mediated Room-Temperature Synthesis of High-Surface-Area Rutile and Its Photocatalytic Performance. J. Phys. Chem. C 2007, 111, 6789–6797. [Google Scholar] [CrossRef]
- Andersson, M.; Österlund, L.; Ljungström, S.; Palmqvist, A. Preparation of Nanosize Anatase and Rutile TiO2 by Hydrothermal Treatment of Microemulsions and Their Activity for Photocatalytic Wet Oxidation of Phenol. J. Phys. Chem. B 2002, 106, 10674–10679. [Google Scholar] [CrossRef]
- Maslova, V.; Quadrelli, E.A.; Gaval, P.; Fasolini, A.; Albonetti, S.; Basile, F. Highly-Dispersed Ultrafine Pt Nanoparticles on Microemulsion-Mediated TiO2 for Production of Hydrogen and Valuable Chemicals via Oxidative Photo-Dehydrogenation of Glycerol. J. Environ. Chem. Eng. 2021, 9, 105070. [Google Scholar] [CrossRef]
- Wu, M.; Long, J.; Huang, A.; Luo, Y.; Feng, S.; Xu, R. Microemulsion-Mediated Hydrothermal Synthesis and Characterization of Nanosize Rutile and Anatase Particles. Langmuir 1999, 15, 8822–8825. [Google Scholar] [CrossRef]
- Liu, Z.; Jian, Z.; Fang, J.; Xu, X.; Zhu, X.; Wu, S. Low-Temperature Reverse Microemulsion Synthesis, Characterization, and Photocatalytic Performance of Nanocrystalline Titanium Dioxide. Int. J. Photoenergy 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zheng, C.; Ma, L.; Huang, X. Microemulsion-Assisted Hydrothermal Preparation and Infrared Radiation Property of TiO2 Nanomaterials with Tunable Morphologies and Crystal Form. Mater. Sci. Semicond. Process. 2015, 31, 295–301. [Google Scholar] [CrossRef]
- Li, X.; Zheng, W.; He, G.; Zhao, R.; Liu, D. Morphology Control of TiO2 Nanoparticle in Microemulsion and Its Photocatalytic Property. ACS Sustain. Chem. Eng. 2014, 2, 288–295. [Google Scholar] [CrossRef]
- González-Verjan, V.A.; Trujillo-Navarrete, B.; Félix-Navarro, R.M.; de León, J.N.D.; Romo-Herrera, J.M.; Calva-Yáñez, J.C.; Hernández-Lizalde, J.M.; Reynoso-Soto, E.A. Effect of TiO2 Particle and Pore Size on DSSC Efficiency. Mater. Renew. Sustain. Energy 2020, 9, 13. [Google Scholar] [CrossRef]
- Lu, C.-H.; Wen, M.-C. Synthesis of Nanosized TiO2 Powders via a Hydrothermal Microemulsion Process. J. Alloys Compd. 2008, 448, 153–158. [Google Scholar] [CrossRef]
- Lu, C.-H.; Wu, W.-H.; Kale, R.B. Microemulsion-Mediated Hydrothermal Synthesis of Photocatalytic TiO2 Powders. J. Hazard. Mater. 2008, 154, 649–654. [Google Scholar] [CrossRef]
- Baamran, K.S.; Tahir, M.; Mohamed, M.; Hussain Khoja, A. Effect of Support Size for Stimulating Hydrogen Production in Phenol Steam Reforming Using Ni-Embedded TiO2 Nanocatalyst. J. Environ. Chem. Eng. 2020, 8, 103604. [Google Scholar] [CrossRef]
- Gopal, M.; Moberly Chan, W.J.; De Jonghe, L.C. Room Temperature Synthesis of Crystalline Metal Oxides. J. Mater. Sci. 1997, 32, 6001–6008. [Google Scholar] [CrossRef]
- Yan, M.; Chen, F.; Zhang, J.; Anpo, M. Preparation of Controllable Crystalline Titania and Study on the Photocatalytic Properties. J. Phys. Chem. B 2005, 109, 8673–8678. [Google Scholar] [CrossRef]
- Satoh, N.; Nakashima, T.; Yamamoto, K. Metastability of Anatase: Size Dependent and Irreversible Anatase-Rutile Phase Transition in Atomic-Level Precise Titania. Sci. Rep. 2013, 3, 1959. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, T.; Do, D.; Nicholson, D. Capillary Condensation of Adsorbates in Porous Materials. Adv. Colloid Interface Sci. 2011, 169, 40–58. [Google Scholar] [CrossRef]
- Sing, K.S. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Das, D.; Shivhare, A.; Saha, S.; Ganguli, A.K. Room Temperature Synthesis of Mesoporous TiO2 Nanostructures with High Photocatalytic Efficiency. Mater. Res. Bull. 2012, 47, 3780–3785. [Google Scholar] [CrossRef]
- Busca, G.; Saussey, H.; Saur, O.; Lavalley, J.C.; Lorenzelli, V. FT-IR Characterization of the Surface Acidity of Different Titanium Dioxide Anatase Preparations. Appl. Catal. 1985, 14, 245–260. [Google Scholar] [CrossRef]
- Bolado, S.; Treviño, R.E.; García-Cubero, M.T.; González-Benito, G. Glycerol Hydrogenolysis to 1, 2 Propanediol over Ru/C Catalyst. Catal. Commun. 2010, 12, 122–126. [Google Scholar] [CrossRef]
- Dietrich, P.J.; Lobo-Lapidus, R.J.; Wu, T.; Sumer, A.; Akatay, M.C.; Fingland, B.R.; Guo, N.; Dumesic, J.A.; Marshall, C.L.; Stach, E. Aqueous Phase Glycerol Reforming by PtMo Bimetallic Nano-Particle Catalyst: Product Selectivity and Structural Characterization. Top. Catal. 2012, 55, 53–69. [Google Scholar] [CrossRef]
- Maris, E.P.; Davis, R.J. Hydrogenolysis of Glycerol over Carbon-Supported Ru and Pt Catalysts. J. Catal. 2007, 249, 328–337. [Google Scholar] [CrossRef]
- King, D.L.; Zhang, L.; Xia, G.; Karim, A.M.; Heldebrant, D.J.; Wang, X.; Peterson, T.; Wang, Y. Aqueous Phase Reforming of Glycerol for Hydrogen Production over Pt–Re Supported on Carbon. Appl. Catal. B Environ. 2010, 99, 206–213. [Google Scholar] [CrossRef]
- Lin, Y.-C. Catalytic Valorization of Glycerol to Hydrogen and Syngas. Int. J. Hydrogen Energy 2013, 38, 2678–2700. [Google Scholar] [CrossRef]
- Hara, M. Heterogeneous Lewis Acid Catalysts Workable in Water. Bull. Chem. Soc. Jpn. 2014, 87, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Sinfelt, J.H.; Yates, D.J.C. Catalytic Hydrogenolysis of Ethane over the Noble Metals of Group VIII. J. Catal. 1967, 8, 82–90. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Brookes, C.; Davies, P.R.; Wawata, I. The Adsorption and Reaction of Alcohols on TiO2 and Pd/TiO2 Catalysts. Appl. Catal. A Gen. 2013, 454, 66–73. [Google Scholar] [CrossRef]















| Sample | Surf/Co-Surf. | Stirring Time at r.t. | Heating Type | Heating Time (h) | Rutile vs. Anatase Phase (%) | Particle Dimension Anatase (nm) | SSA * (m2/g) |
|---|---|---|---|---|---|---|---|
| TiO2-m | Yes | 1 h | Reflux | 5 | 7:93 | 4 | 319 |
| TiO2-e | No | 1 h | Reflux | 5 | 64:36 | 5 | 199 |
| TiO2-5 days | Yes | 5 days | - | - | 91:9 | 9 | 123 |
| TiO2-120 °C | Yes | 1 h | Autoclave (120 °C) | 4 | 1:99 | 5 | 249 |
| Crystal Phase | Particle Size (nm) | SBET (m2/g) 1 | Pore Avg. Diameter (nm) | Vpore (cm3/g) | NH3 Uptake (mmol/g) | NH3 Uptake (mmol/m2) | |
|---|---|---|---|---|---|---|---|
| TiO2-m400 | Anatase | 8 | 147 | 4.2 | 0.15 | 18.94 | 0.13 |
| TiO2-comm | Anatase | 24 | 90 | 14.6 | 0.32 | 10.35 | 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fasolini, A.; Lombardi, E.; Tabanelli, T.; Basile, F. Microemulsion Derived Titania Nanospheres: An Improved Pt Supported Catalyst for Glycerol Aqueous Phase Reforming. Nanomaterials 2021, 11, 1175. https://doi.org/10.3390/nano11051175
Fasolini A, Lombardi E, Tabanelli T, Basile F. Microemulsion Derived Titania Nanospheres: An Improved Pt Supported Catalyst for Glycerol Aqueous Phase Reforming. Nanomaterials. 2021; 11(5):1175. https://doi.org/10.3390/nano11051175
Chicago/Turabian StyleFasolini, Andrea, Erica Lombardi, Tommaso Tabanelli, and Francesco Basile. 2021. "Microemulsion Derived Titania Nanospheres: An Improved Pt Supported Catalyst for Glycerol Aqueous Phase Reforming" Nanomaterials 11, no. 5: 1175. https://doi.org/10.3390/nano11051175
APA StyleFasolini, A., Lombardi, E., Tabanelli, T., & Basile, F. (2021). Microemulsion Derived Titania Nanospheres: An Improved Pt Supported Catalyst for Glycerol Aqueous Phase Reforming. Nanomaterials, 11(5), 1175. https://doi.org/10.3390/nano11051175