
Cost-Effective Calculation of Collective Electronic Excitations in Graphite Intercalated Compounds


Abstract
:1. Introduction
2. Method
3. Results and Discussion
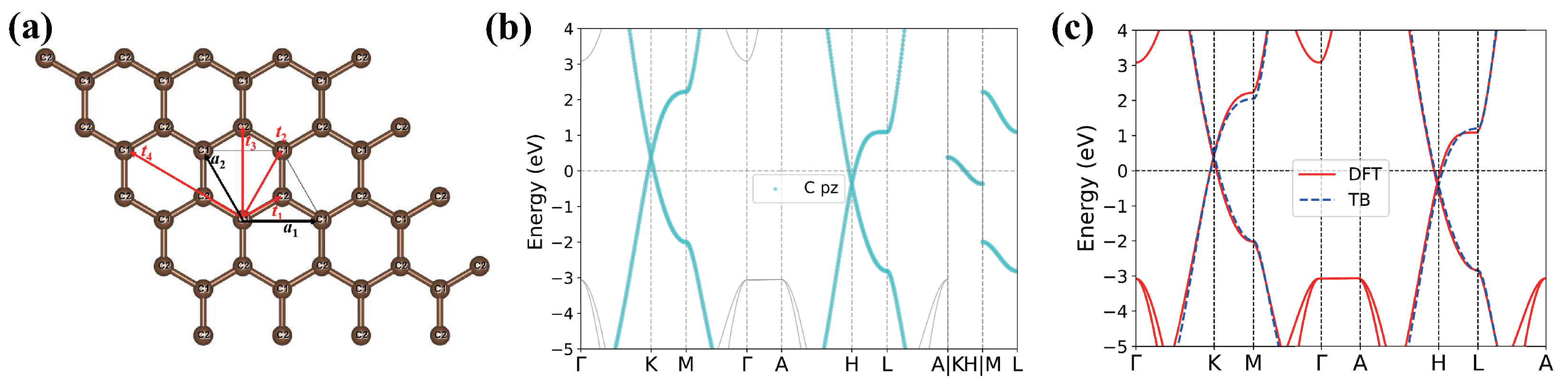
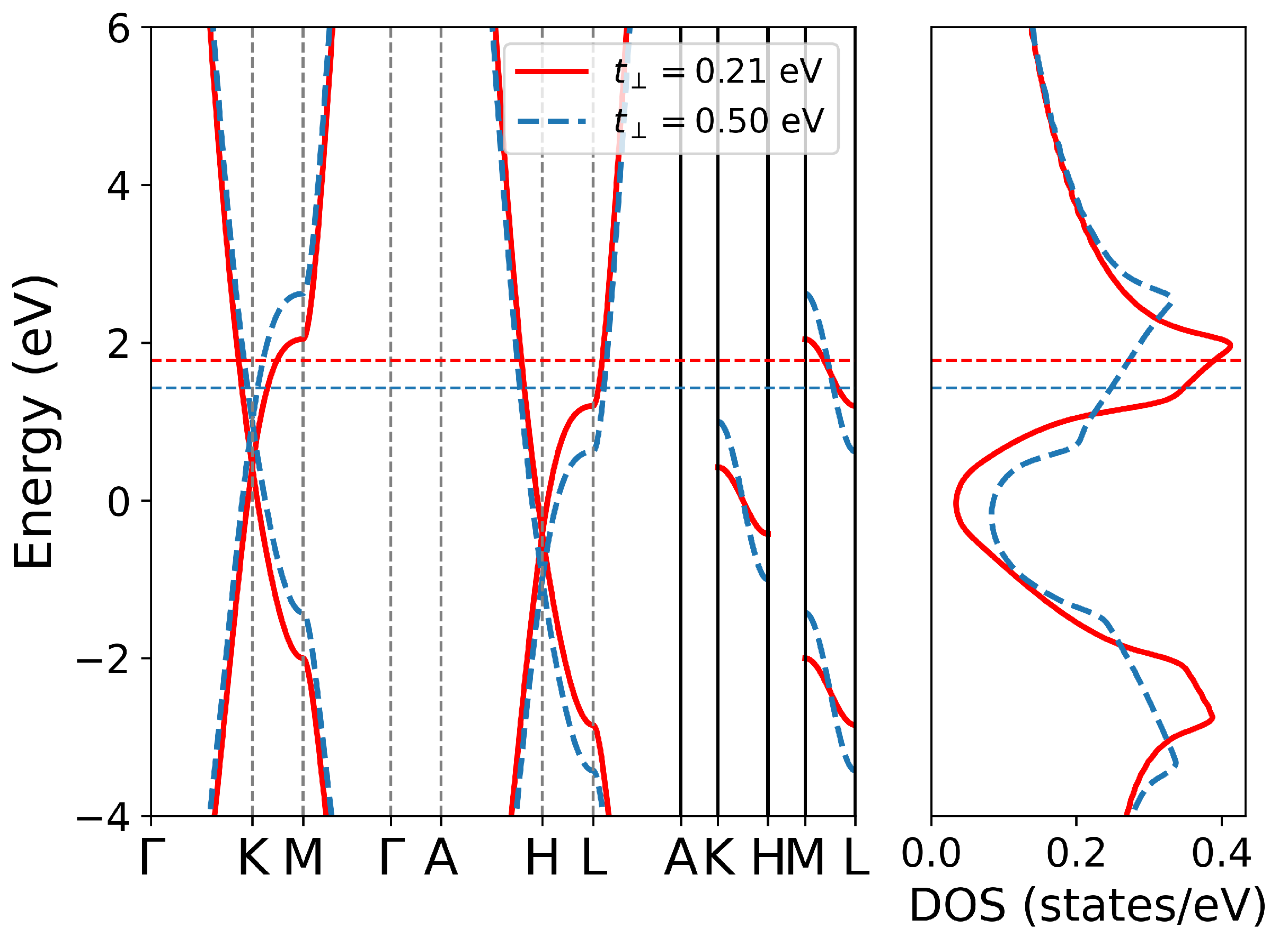
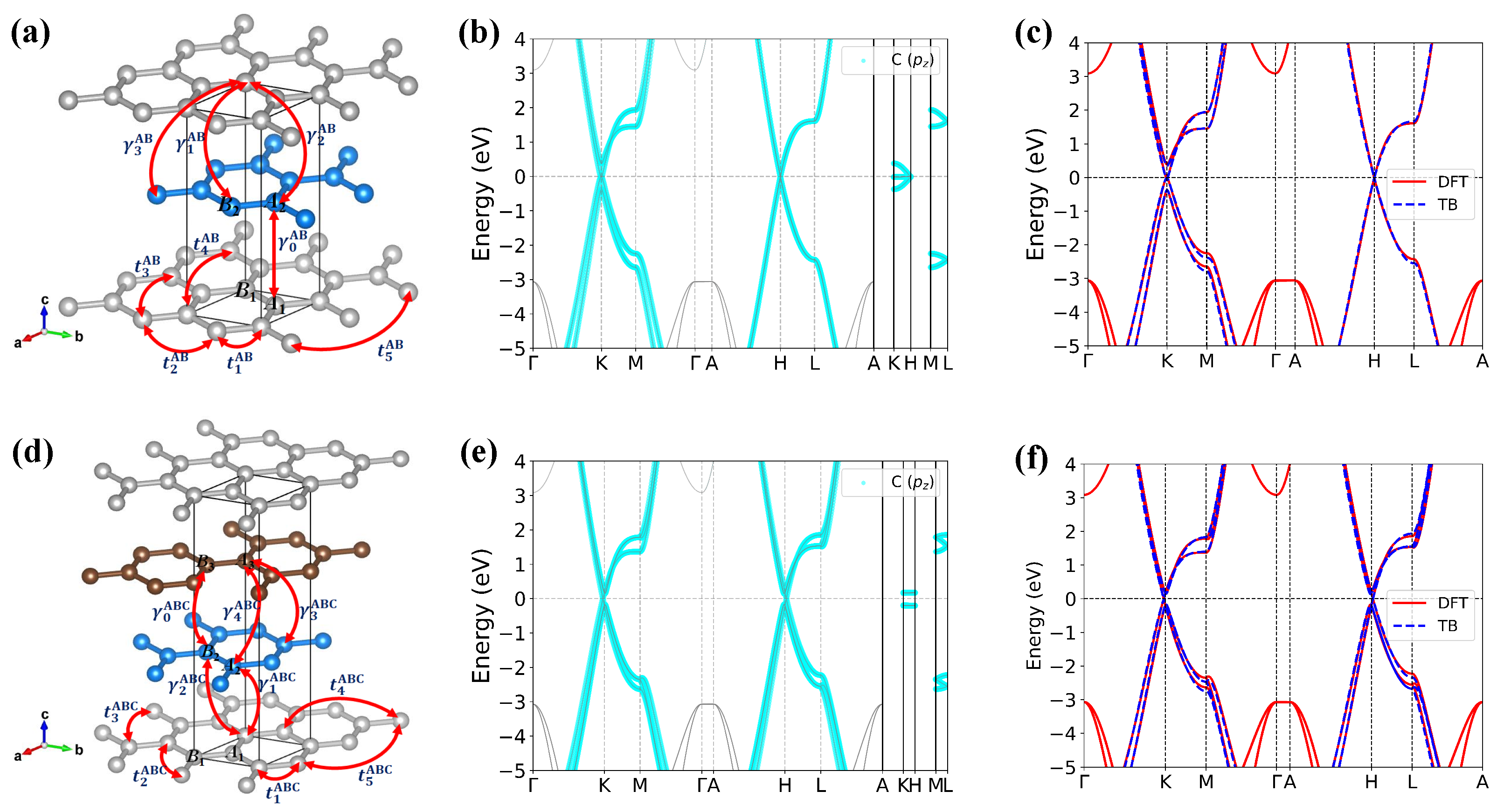
3.1. TB Model of AA-Stacking Graphite
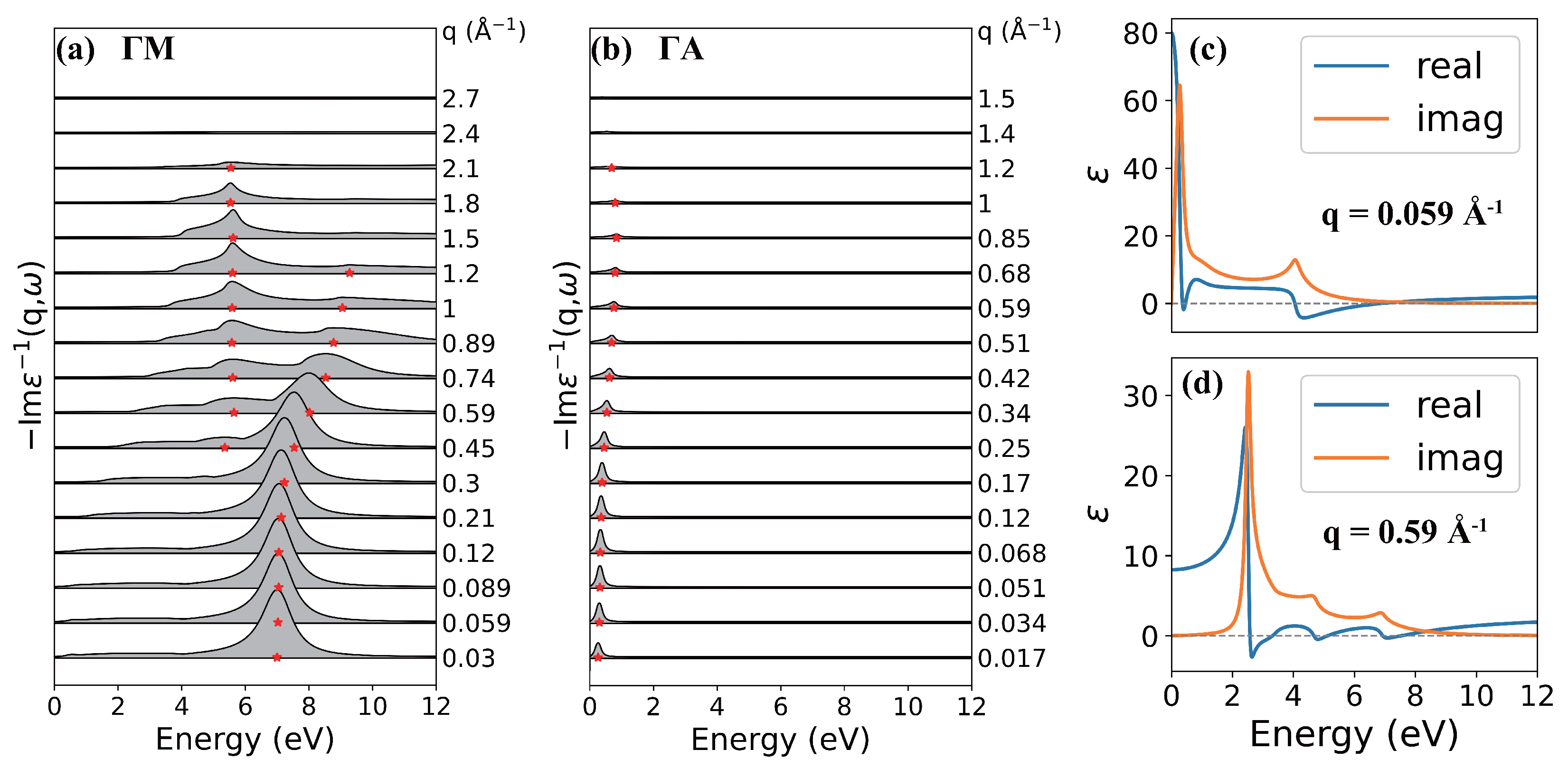
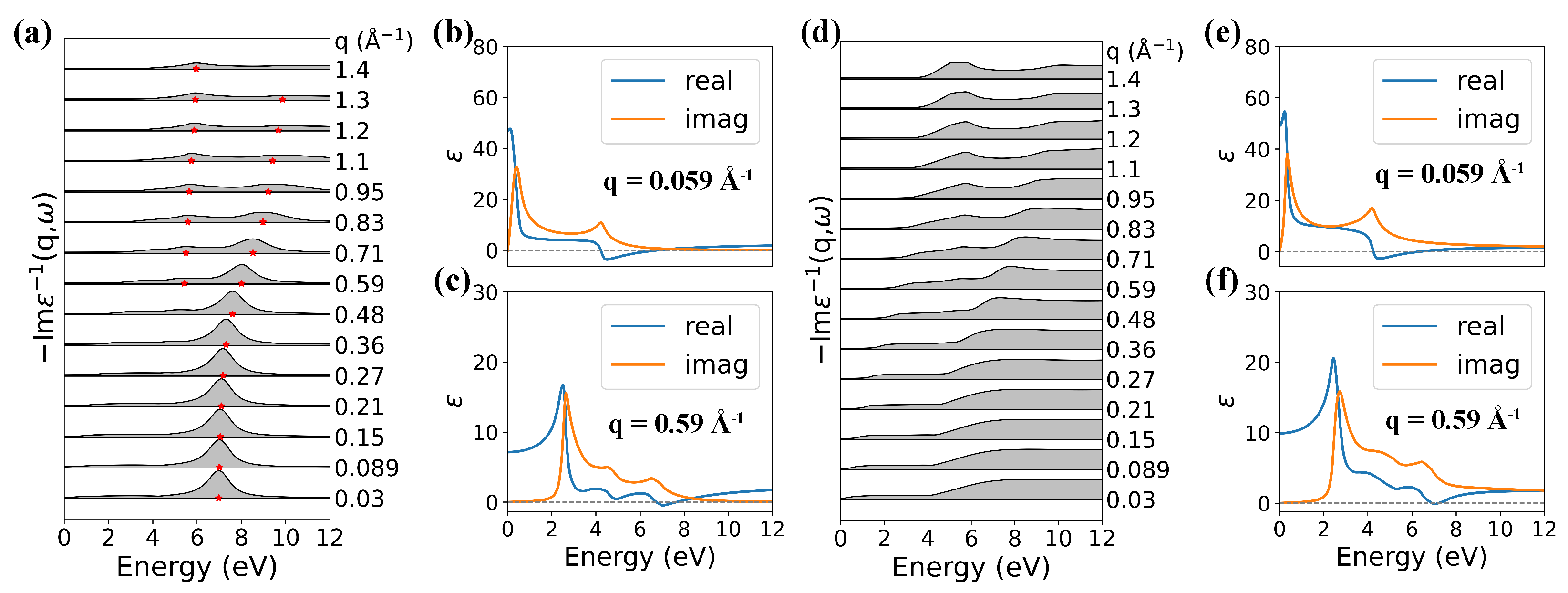
3.2. Plasmon Excitations
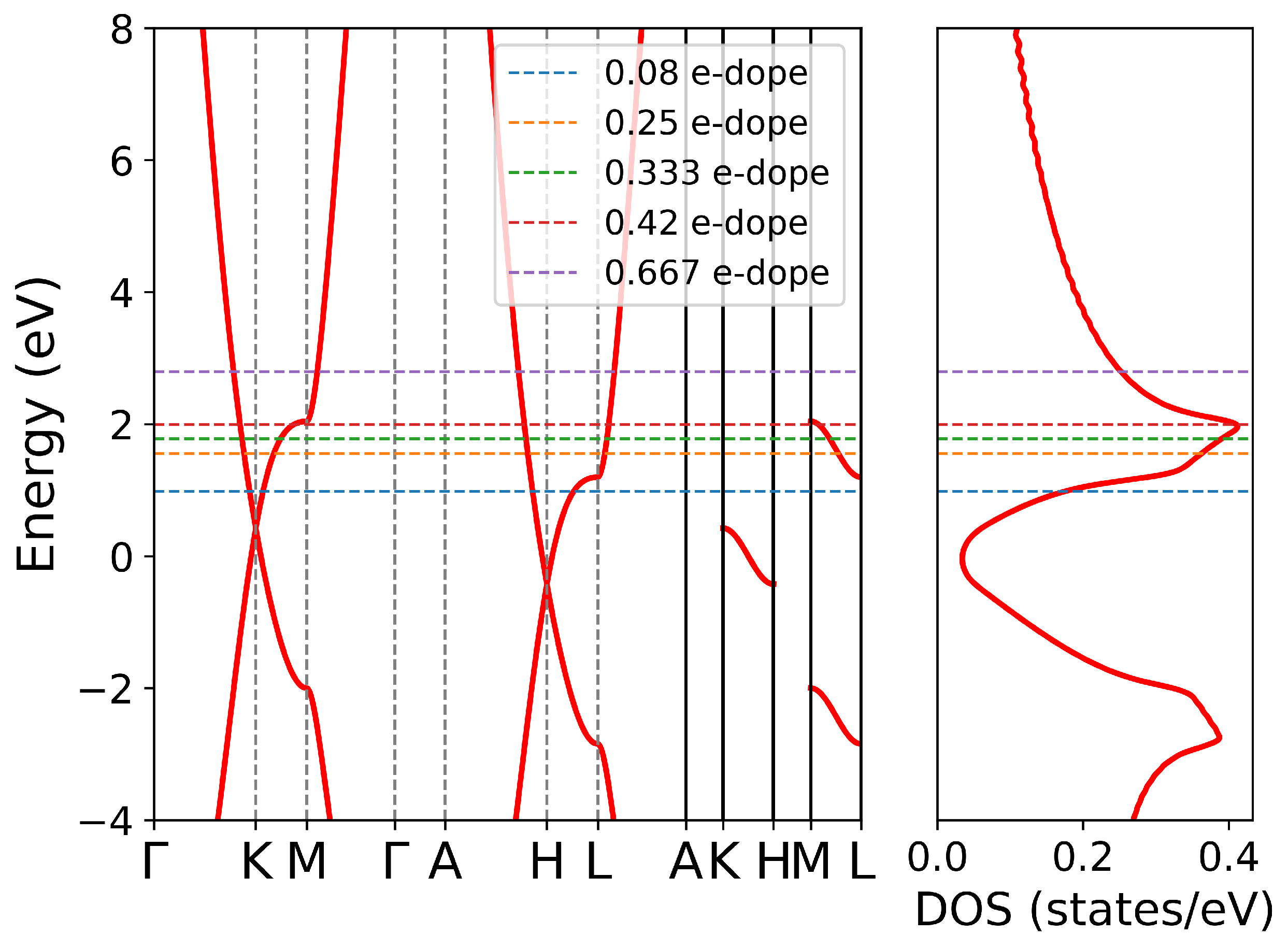
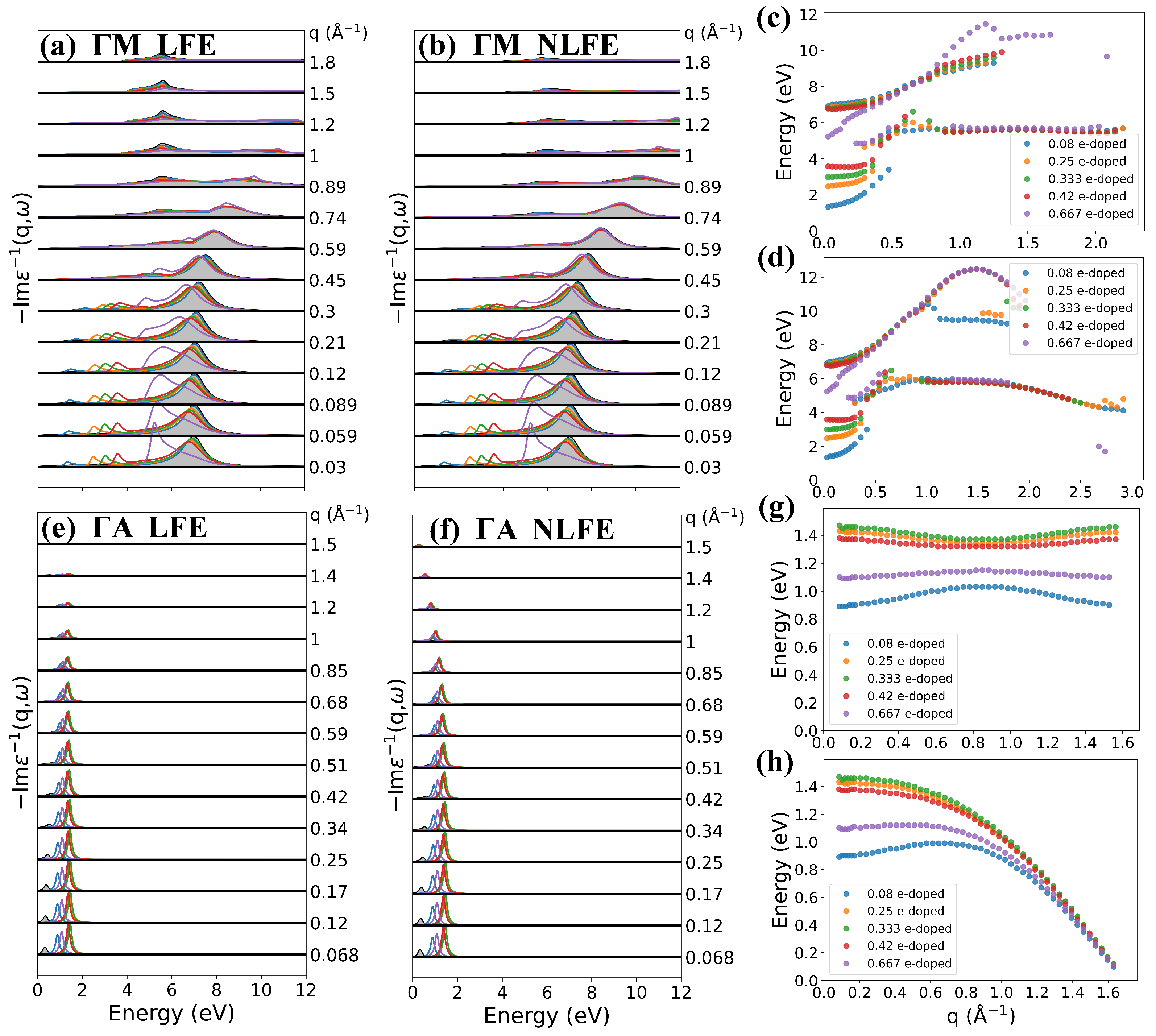
3.3. Effect of Doping Level
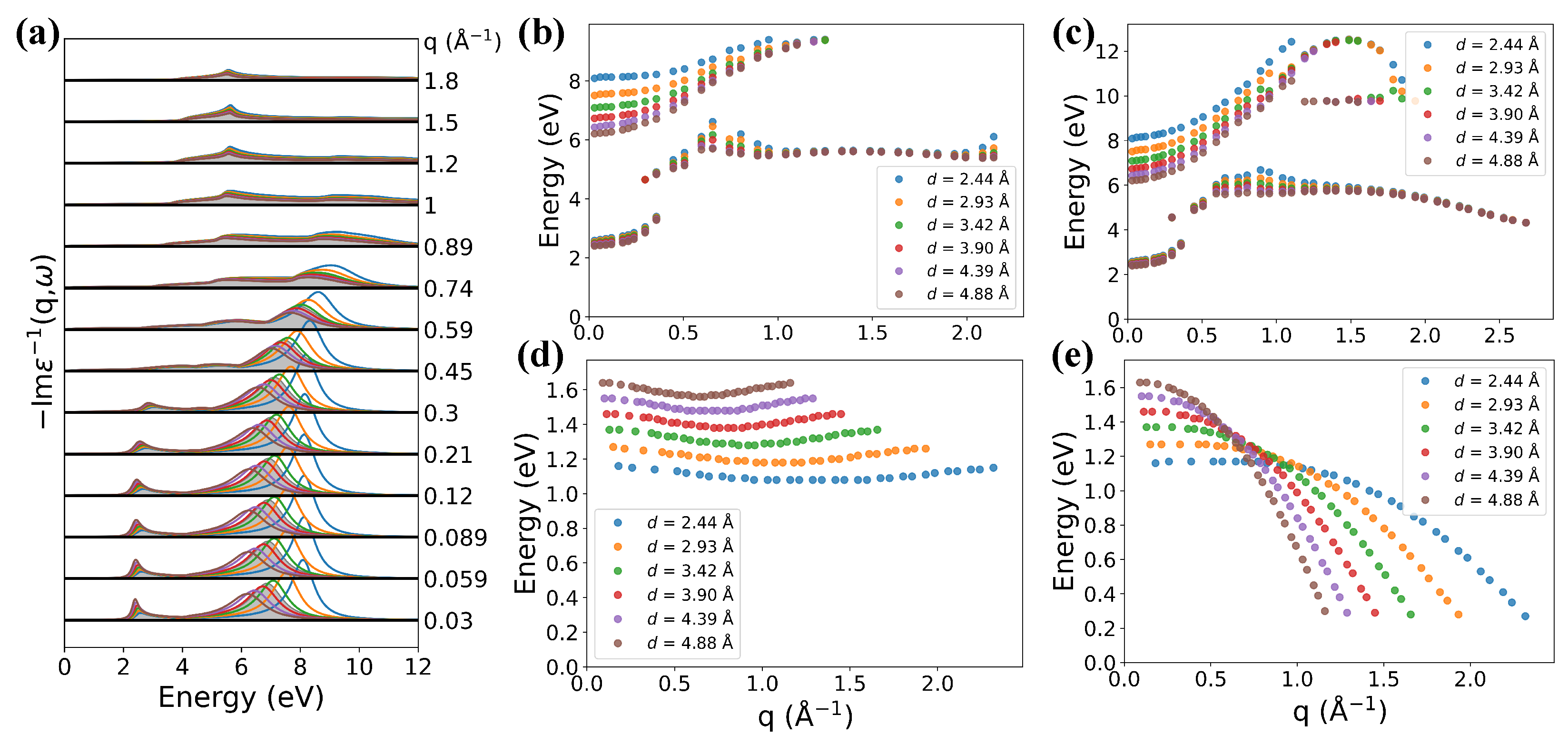
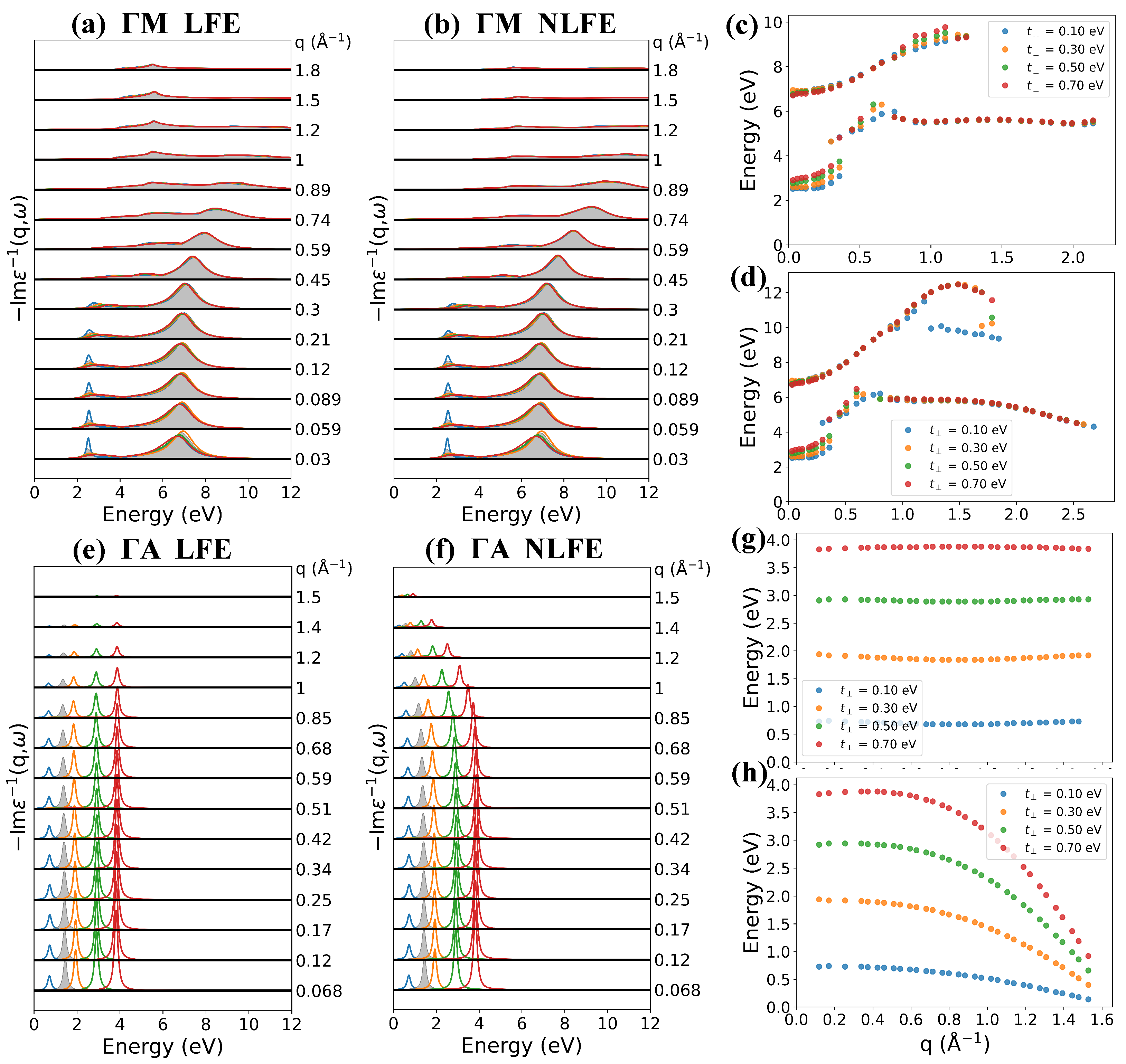
3.4. Effect of Interlayer Distance and Hopping
3.5. Effect of Stacking Order
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Tight-Binding Model
Appendix B. Approximations in Practical
Appendix C. First-Principle Calculation Details
Appendix D. Tight-Binding Model of AB and ABC Stacking Graphite


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stacking Order | AB | ABC |
|---|---|---|
| 0 |
References
- Dresselhaus, M.; Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 1981, 30, 139–326. [Google Scholar] [CrossRef]
- Wan, J.; Lacey, S.D.; Dai, J.; Bao, W.; Fuhrer, M.S.; Hu, L. Tuning two-dimensional nanomaterials by intercalation: Materials, properties and applications. Chem. Soc. Rev. 2016, 45, 6742–6765. [Google Scholar] [CrossRef] [PubMed]
- Khrapach, I.; Withers, F.; Bointon, T.H.; Polyushkin, D.K.; Barnes, W.L.; Russo, S.; Craciun, M.F. Novel highly conductive and transparent graphene-based conductors. Adv. Mater. 2012, 24, 2844–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, W.; Wan, J.; Han, X.; Cai, X.; Zhu, H.; Kim, D.; Ma, D.; Xu, Y.; Munday, J.N.; Drew, H.D.; et al. Approaching the limits of transparency and conductivity in graphitic materials through lithium intercalation. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bointon, T.H.; Khrapach, I.; Yakimova, R.; Shytov, A.V.; Craciun, M.F.; Russo, S. Approaching magnetic ordering in graphene materials by FeCl3 intercalation. Nano Lett. 2014, 14, 1751–1755. [Google Scholar] [CrossRef] [Green Version]
- Hannay, N.; Geballe, T.; Matthias, B.; Andres, K.; Schmidt, P.; MacNair, D. Superconductivity in graphitic compounds. Phys. Rev. Lett. 1965, 14, 225–226. [Google Scholar] [CrossRef]
- Weller, T.E.; Ellerby, M.; Saxena, S.S.; Smith, R.P.; Skipper, N.T. Superconductivity in the intercalated graphite compounds C6Yb and C6Ca. Nat. Phys. 2005, 1, 39–41. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Boeri, L.; O’Brien, J.; Razavi, F.; Kremer, R. Superconductivity in heavy alkaline-earth intercalated graphites. Phys. Rev. Lett. 2007, 99, 027001. [Google Scholar] [CrossRef]
- Heguri, S.; Kawade, N.; Fujisawa, T.; Yamaguchi, A.; Sumiyama, A.; Tanigaki, K.; Kobayashi, M. Superconductivity in the Graphite Intercalation Compound BaC6. Phys. Rev. Lett. 2015, 114, 247201. [Google Scholar] [CrossRef]
- Ichinokura, S.; Sugawar, K.; Takayama, A.; Takahashi, T.; Hasegawa, S. Superconducting Calcium-Intercalated Bilayer Graphene. ACS Nano 2016, 10, 2761–2765. [Google Scholar] [CrossRef] [Green Version]
- Csányi, G.; Littlewood, P.; Nevidomskyy, A.H.; Pickard, C.J.; Simons, B. The role of the interlayer state in the electronic structure of superconducting graphite intercalated compounds. Nat. Phys. 2005, 1, 42–45. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.H.; Camacho, J.; Upton, M.; Fedorov, A.; Howard, C.; Ellerby, M.; Valla, T. Electronic structure of superconducting KC8 and nonsuperconducting LiC6 graphite intercalation compounds: Evidence for a graphene-sheet-driven superconducting state. Phys. Rev. Lett. 2011, 106, 187002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazin, I. Intercalant-driven superconductivity in YbC6 and CaC6. Phys. Rev. Lett. 2005, 95, 227001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, N.; Hérold, C.; d’Astuto, M.; Garcia, V.; Bellin, C.; Marêché, J.; Lagrange, P.; Loupias, G. Superconductivity of bulk CaC6. Phys. Rev. Lett. 2005, 95, 087003. [Google Scholar] [CrossRef] [Green Version]
- Grüneis, A.; Attaccalite, C.; Rubio, A.; Vyalikh, D.; Molodtsov, S.; Fink, J.; Follath, R.; Eberhardt, W.; Büchner, B.; Pichler, T. Electronic structure and electron-phonon coupling of doped graphene layers in KC8. Phys. Rev. B 2009, 79, 205106. [Google Scholar] [CrossRef] [Green Version]
- Profeta, G.; Calandra, M.; Mauri, F. Phonon-mediated superconductivity in graphene by lithium deposition. Nat. Phys. 2012, 8, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Schülke, W.; Bonse, U.; Nagasawa, H.; Kaprolat, A.; Berthold, A. Interband transitions and core excitation in highly oriented pyrolytic graphite studied by inelastic synchrotron x-ray scattering: Band-structure information. Phys. Rev. B 1988, 38, 2112–2123. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.; Utlaut, M.; Isaacson, M.; Solin, S. Electron-Energy-Loss Study of Stage-1 Potassium-Intercalated Graphite. Phys. Rev. Lett. 1979, 43, 882–886. [Google Scholar] [CrossRef] [Green Version]
- Ritsko, J.J.; Rice, M.J. Plasmon spectra of ferric-chloride-intercalated graphite. Phys. Rev. Lett. 1979, 42, 666–669. [Google Scholar] [CrossRef]
- Roth, F.; König, A.; Kramberger, C.; Pichler, T.; Büchner, B.; Knupfer, M. Challenging the nature of low-energy plasmon excitations in CaC6 using electron energy-loss spectroscopy. EPL (Europhys. Lett.) 2013, 102, 17001. [Google Scholar] [CrossRef] [Green Version]
- Shung, K.W.K. Dielectric function and plasmon structure of stage-1 intercalated graphite. Phys. Rev. B 1986, 34, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Huang, C.; Chuu, D. Plasmons in graphite and stage-1 graphite intercalation compounds. Phys. Rev. B 1997, 55, 13961–13971. [Google Scholar] [CrossRef] [Green Version]
- Echeverry, J.; Chulkov, E.V.; Echenique, P.M.; Silkin, V.M. Low-energy plasmonic structure in CaC6. Phys. Rev. B 2012, 85, 205135. [Google Scholar] [CrossRef] [Green Version]
- Echeverry, J.; Chulkov, E.V.; Echenique, P.M.; Silkin, V. Low-energy collective electronic excitations in LiC6, SrC6, and BaC6. Phys. Rev. B 2019, 100, 115137. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Marušić, L. UV-active plasmons in alkali and alkaline-earth intercalated graphene. Phys. Rev. B 2018, 97, 205426. [Google Scholar] [CrossRef] [Green Version]
- Marušić, L.; Despoja, V. Prediction of measurable two-dimensional plasmons in Li-intercalated graphene LiC2. Phys. Rev. B 2016, 95, 201408. [Google Scholar] [CrossRef]
- Novko, D. Dopant-induced plasmon decay in graphene. Nano Lett. 2017, 17, 6991–6996. [Google Scholar] [CrossRef]
- Hanke, W.R. Microscopic Theory of Dielectric Screening and Lattice Dynamics in the Wannier Representation. I. Theory. Phys. Rev. B 1973, 8, 4585–4590. [Google Scholar] [CrossRef]
- Hanke, W.; Sham, L. Local-field and excitonic effects in the optical spectrum of a covalent crystal. Phys. Rev. B 1975, 12, 4501–4511. [Google Scholar] [CrossRef]
- Wiser, N. Dielectric constant with local field effects included. Phys. Rev. 1963, 129, 62–69. [Google Scholar] [CrossRef]
- Charlier, J.C.; Michenaud, J.P.; Gonze, X. First-principles study of the electronic properties of simple hexagonal graphite. Phys. Rev. B 1992, 46, 4531–4539. [Google Scholar] [CrossRef] [PubMed]
- Ando, T. Screening effect and impurity scattering in monolayer graphene. J. Phys. Soc. Jpn. 2006, 75, 074716. [Google Scholar] [CrossRef] [Green Version]
- Marinopoulos, A.; Reining, L.; Olevano, V.; Rubio, A.; Pichler, T.; Liu, X.; Knupfer, M.; Fink, J. Anisotropy and interplane interactions in the dielectric response of graphite. Phys. Rev. Lett. 2002, 89, 076402. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yuan, Z. Anisotropic low-energy plasmon excitations in doped graphene: An ab initio study. Solid State Commun. 2011, 151, 1009–1013. [Google Scholar] [CrossRef]
- Cao, Y.; Fatemi, V.; Demir, A.; Fang, S.; Tomarken, S.L.; Luo, J.Y.; Sanchez-Yamagishi, J.D.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; et al. Correlated insulator behaviour at half-filling in magic-angle graphene superlattices. Nature 2018, 556, 80–84. [Google Scholar] [CrossRef]
- Cao, Y.; Fatemi, V.; Fang, S.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; Jarillo-Herrero, P. Unconventional superconductivity in magic-angle graphene superlattices. Nature 2018, 556, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Berry Phases in Electronic Structure Theory: Electric Polarization, Orbital Magnetization and Topological Insulators; Cambridge University Press: Cambridge, UK, 2018. [Google Scholar]
- Harrison, W.A. Coulomb interactions in semiconductors and insulators. Phys. Rev. B 1985, 31, 2121–2132. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Hamann, D. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 2013, 88, 085117. [Google Scholar] [CrossRef] [Green Version]
- Schlipf, M.; Gygi, F. Optimization algorithm for the generation of ONCV pseudopotentials. Comput. Phys. Commun. 2015, 196, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M.; Alder, B.J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef] [Green Version]
- Methfessel, M.; Paxton, A. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]








| i | (eV) | d (Å) |
|---|---|---|
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| ⊥ |
| Points | Energy Eigenvalues |
|---|---|
| K | |
| M | |
| A | |
| H | |
| L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suo, P.; Mao, L.; Shi, J.; Xu, H. Cost-Effective Calculation of Collective Electronic Excitations in Graphite Intercalated Compounds. Nanomaterials 2022, 12, 1746. https://doi.org/10.3390/nano12101746
Suo P, Mao L, Shi J, Xu H. Cost-Effective Calculation of Collective Electronic Excitations in Graphite Intercalated Compounds. Nanomaterials. 2022; 12(10):1746. https://doi.org/10.3390/nano12101746
Chicago/Turabian StyleSuo, Pengfei, Li Mao, Jing Shi, and Hongxing Xu. 2022. "Cost-Effective Calculation of Collective Electronic Excitations in Graphite Intercalated Compounds" Nanomaterials 12, no. 10: 1746. https://doi.org/10.3390/nano12101746
APA StyleSuo, P., Mao, L., Shi, J., & Xu, H. (2022). Cost-Effective Calculation of Collective Electronic Excitations in Graphite Intercalated Compounds. Nanomaterials, 12(10), 1746. https://doi.org/10.3390/nano12101746