
Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles

 ,
, 
 and
and
Abstract
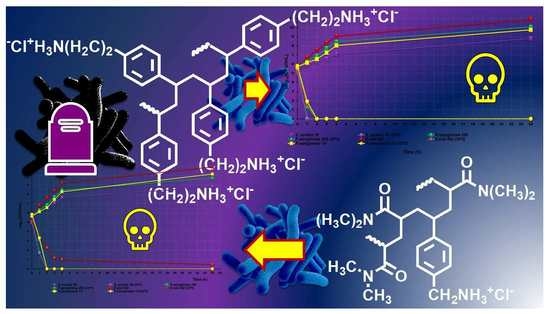
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Instruments
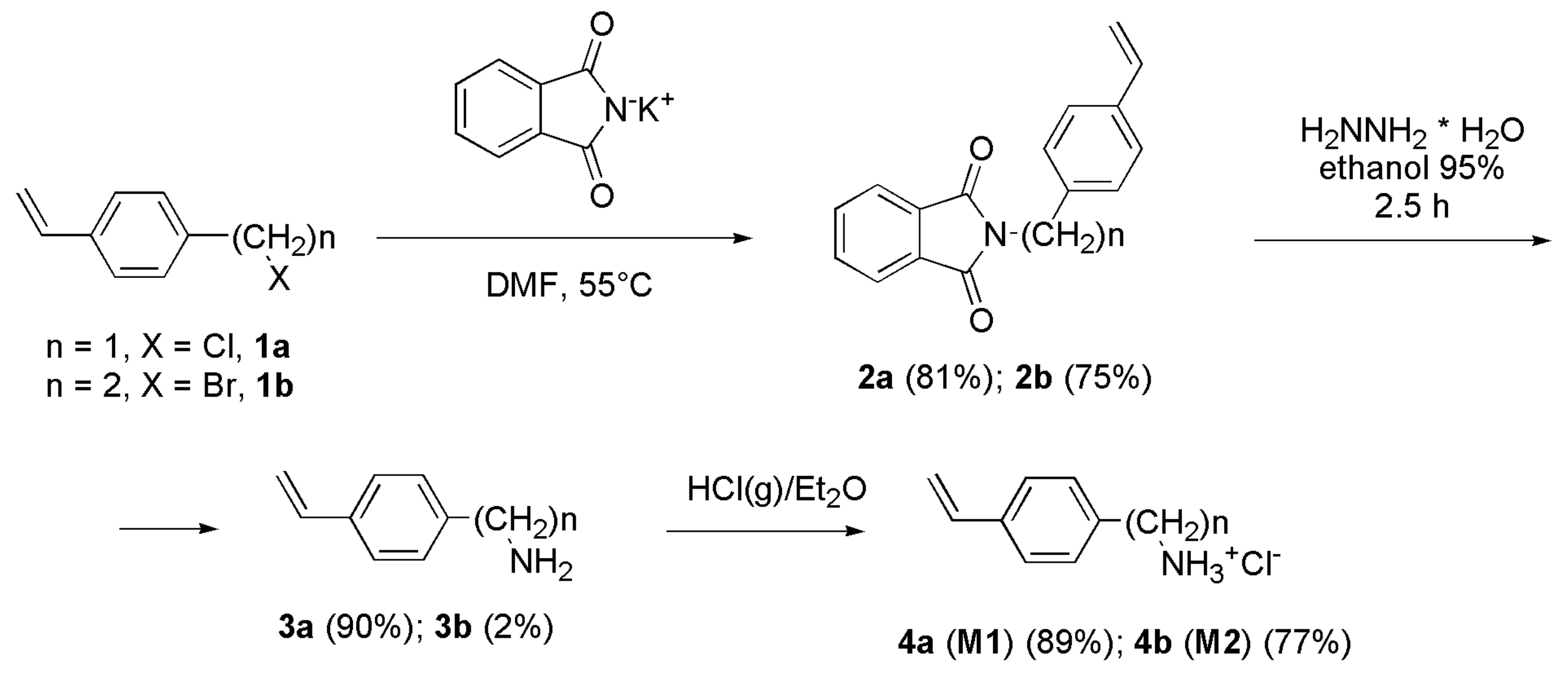
2.2. N-[(4-Vinylphenyl) alkyl] Phthalimides 2a and 2b
2.2.1. N-(4-Vinylbenzyl) Phthalimide (2a)
2.2.2. N-[2-(4-Vinylphenyl) ethyl] Phthalimide (2b)
2.3. Hydrazinolysis of Phthalimides 2a and 2b
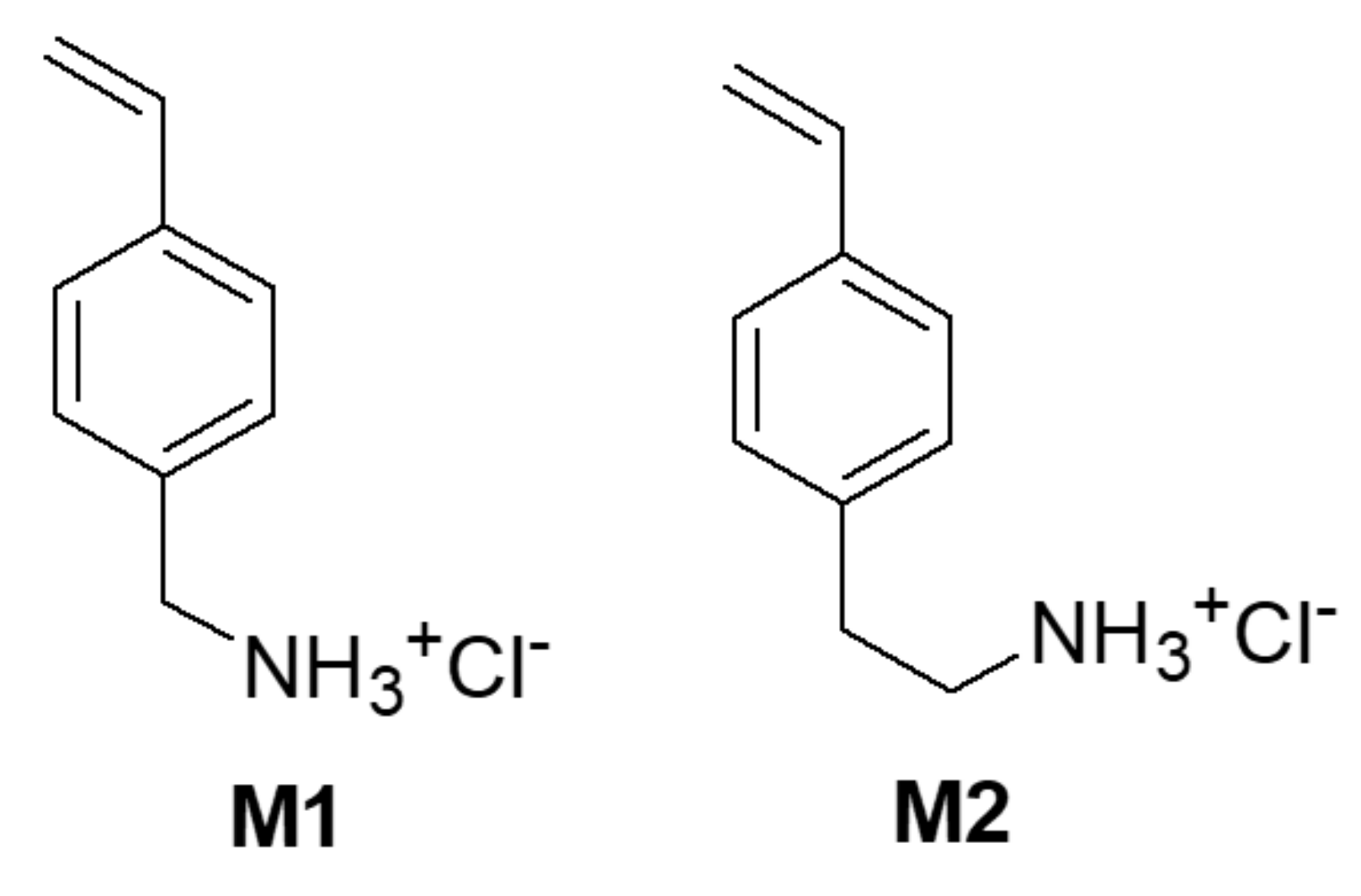
2.4. Synthesis of Hydrochlorides 4a and 4b: 4-AMSTY and 4-AESTY (Monomers M1 and M2)
2.4.1. 4-Aminomethylstyrene Hydrochloride (4-AMSTY) 4a (M1)
2.4.2. 4-Aminoethylstyrene hydrochloride (4-AESTY) 4b (M2)
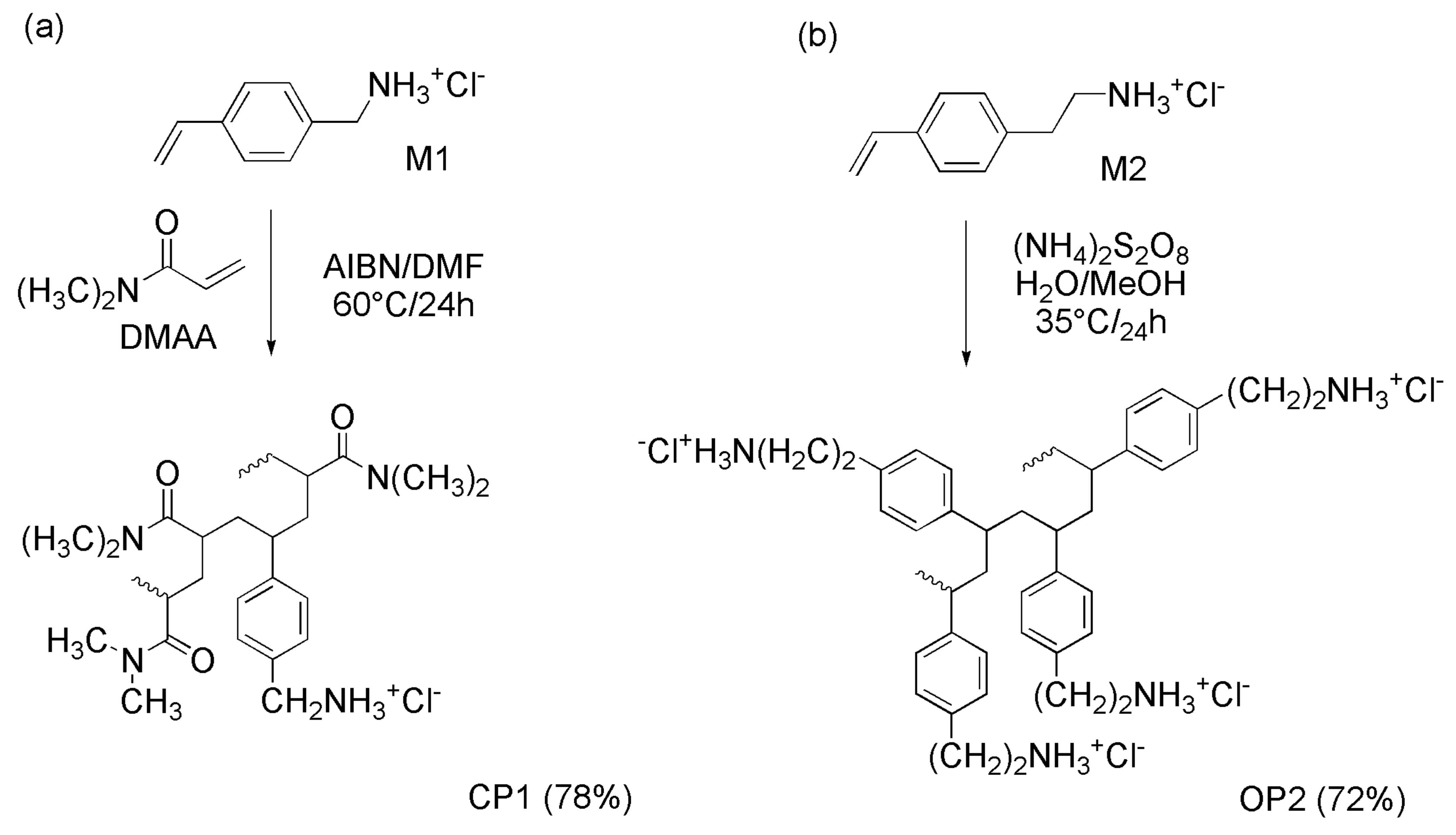
2.5. Radical Solution Copolymerization of M1 with N,N-Dimethyl Acrylamide (DMAA) (CP1)
2.6. Radical Solution Polymerizations of M2 (OP2)
2.7. Determination of the Relative Molecular Mass (Mr) of CP1 and OP2
2.7.1. Determination of Intrinsic Viscosity [η] of CP1 and OP2
2.8. Determination of NH2 Equivalents Contained in CP1 and OP2
2.9. Dynamic Light Scattering (DLS) Analysis
2.10. Scanning Electron Microscopy (SEM)
2.11. Potentiometric Titration of CP1 and OP2 NPs
2.12. Microbiology
2.12.1. Microorganisms
2.12.2. Determination of the Antibacterial Properties of CP1 and OP2
Determination of the Minimal Inhibitory Concentrations (MICs)
Determination of Minimal Bactericidal Concentrations (MBCs)
2.12.3. Time-Kill Experiments
3. Results and Discussion
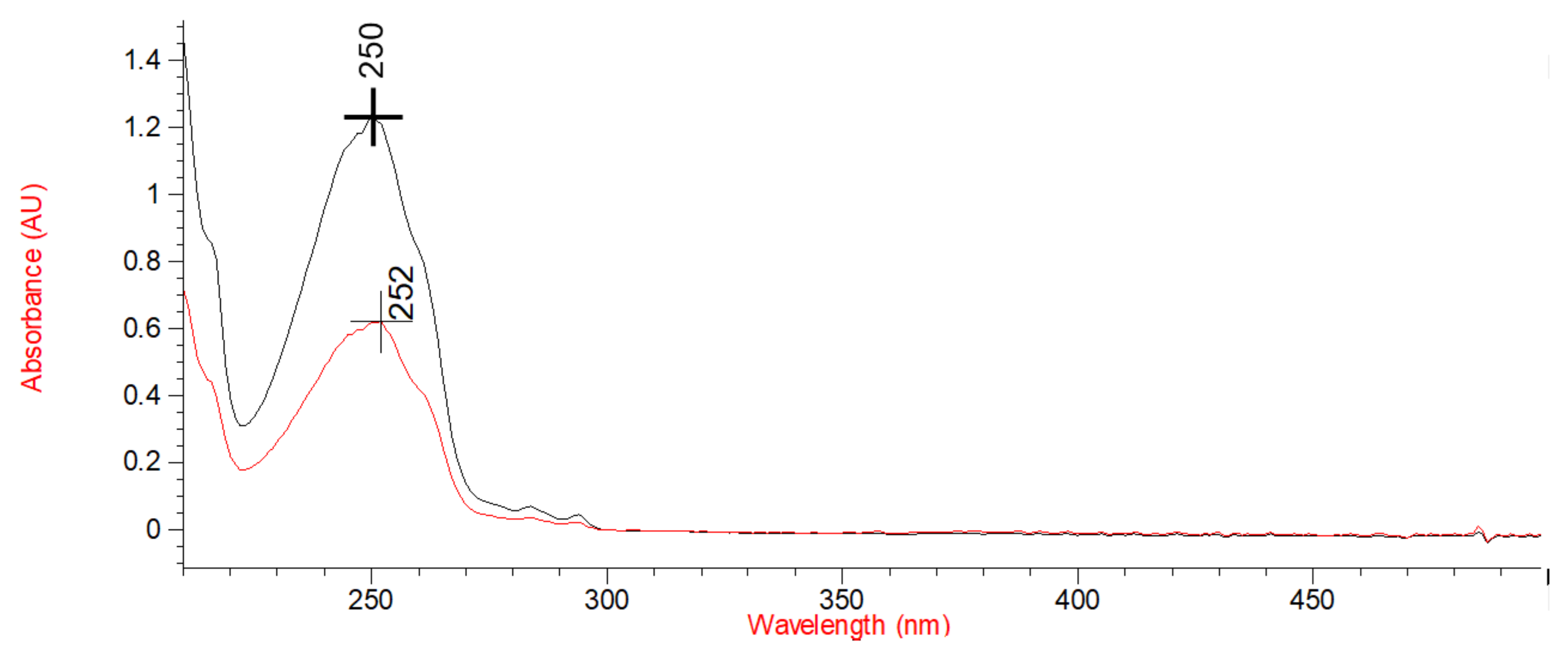
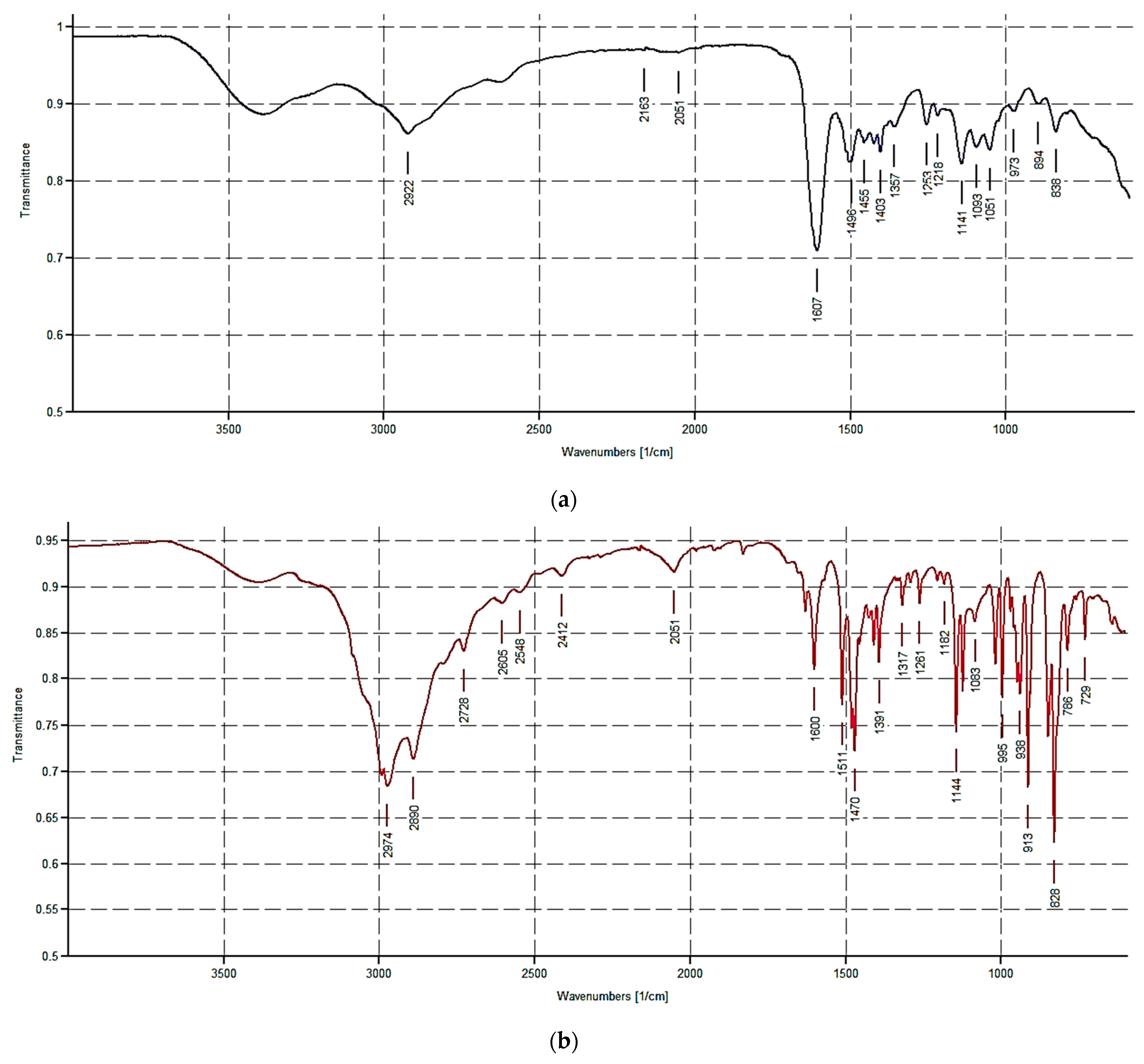
3.1. Synthesis and Spectrophotometric Characterization of Monomers M1 and M2
The Design of M1 and M2 Structure
3.2. Radical Polymerizations in Solution
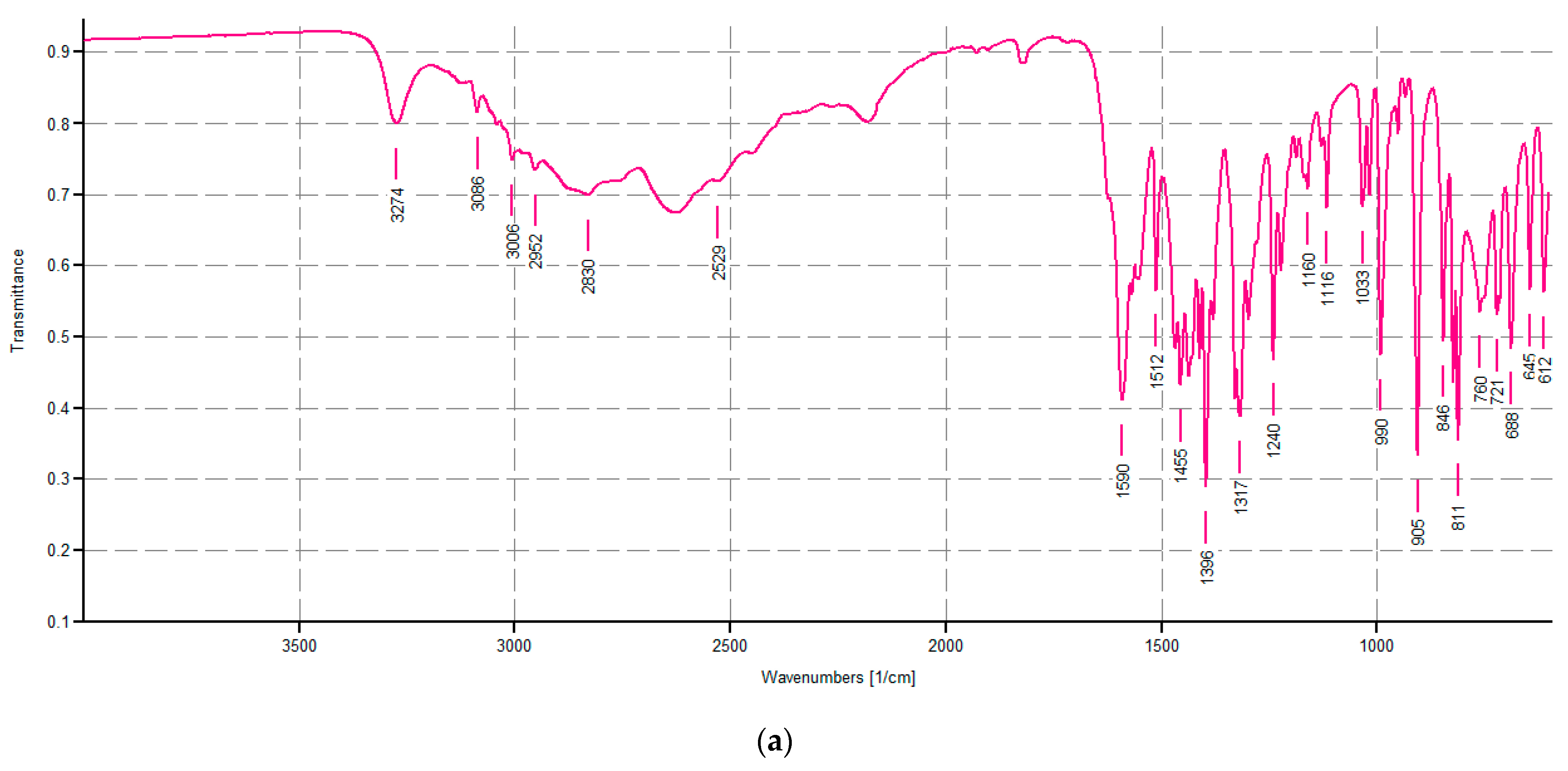
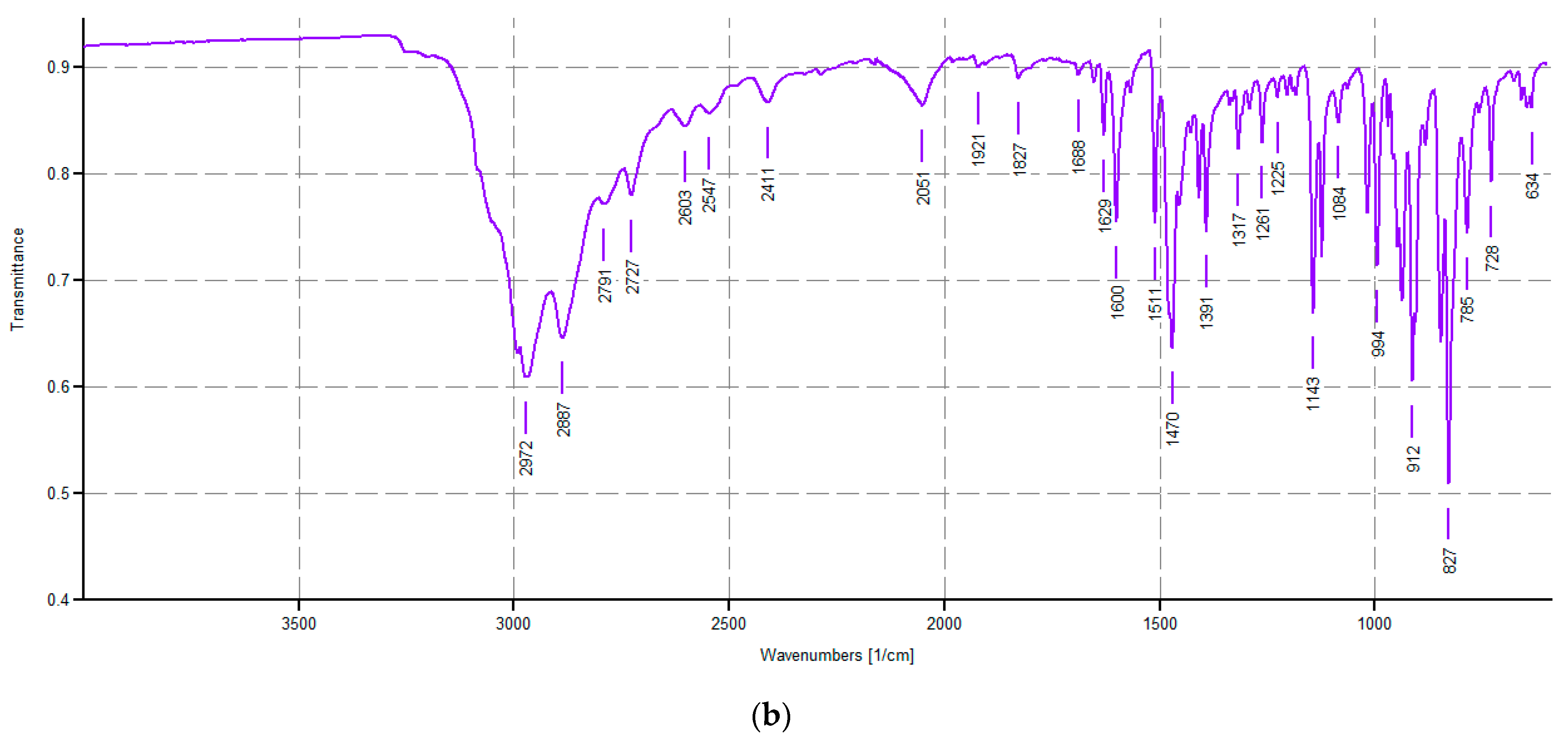
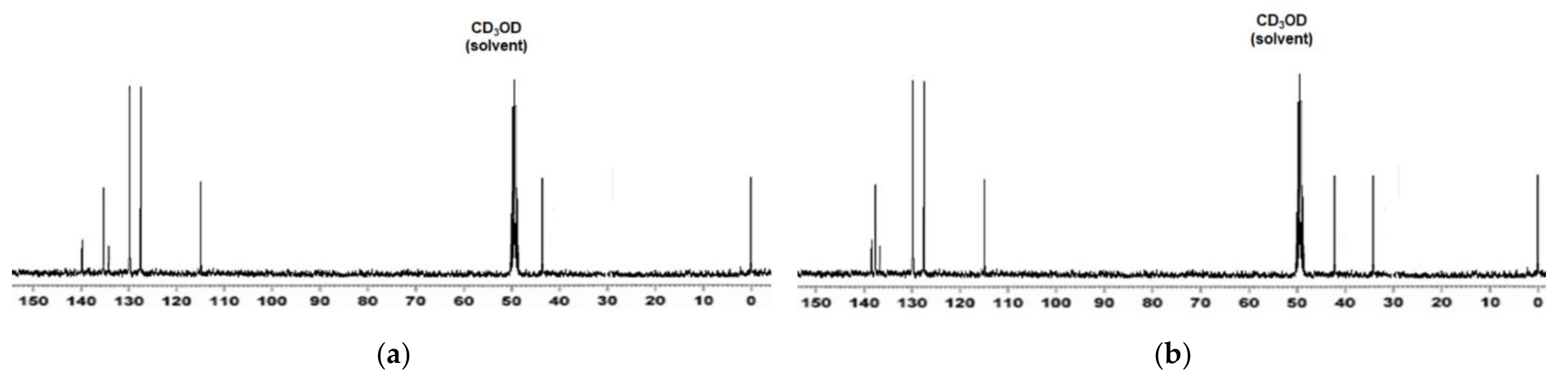
Spectroscopic Characterization of CP1 and OP2
3.3. Determination of the Relative Molecular Mass (Mr) of CP1 and OP2
3.4. Determinations of NH2 Content of CP1 and OP2
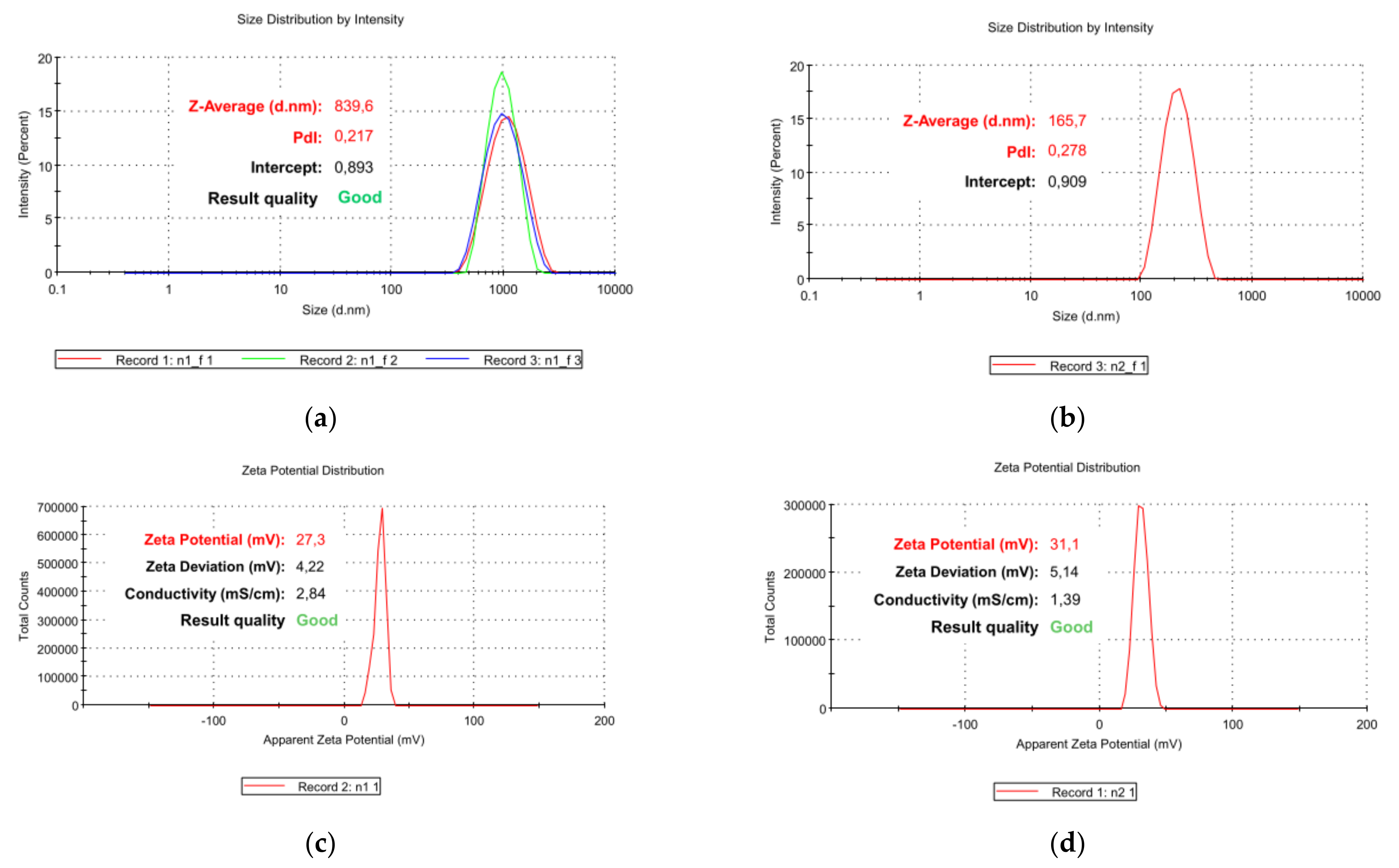
3.5. Particle Size, ζ-p and PDI of CP1 and OP2
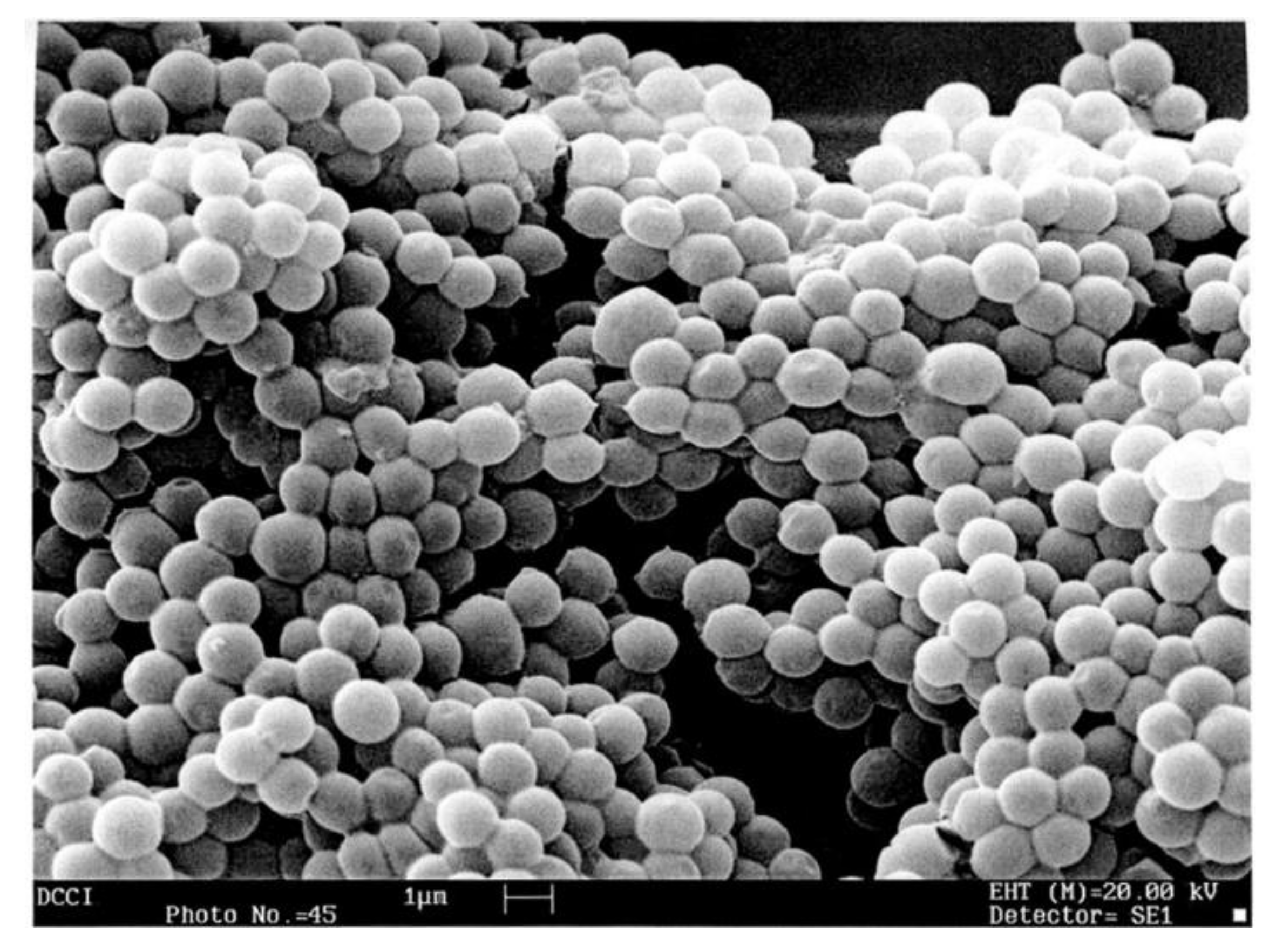
3.6. Scanning Electron Microscopy (SEM)
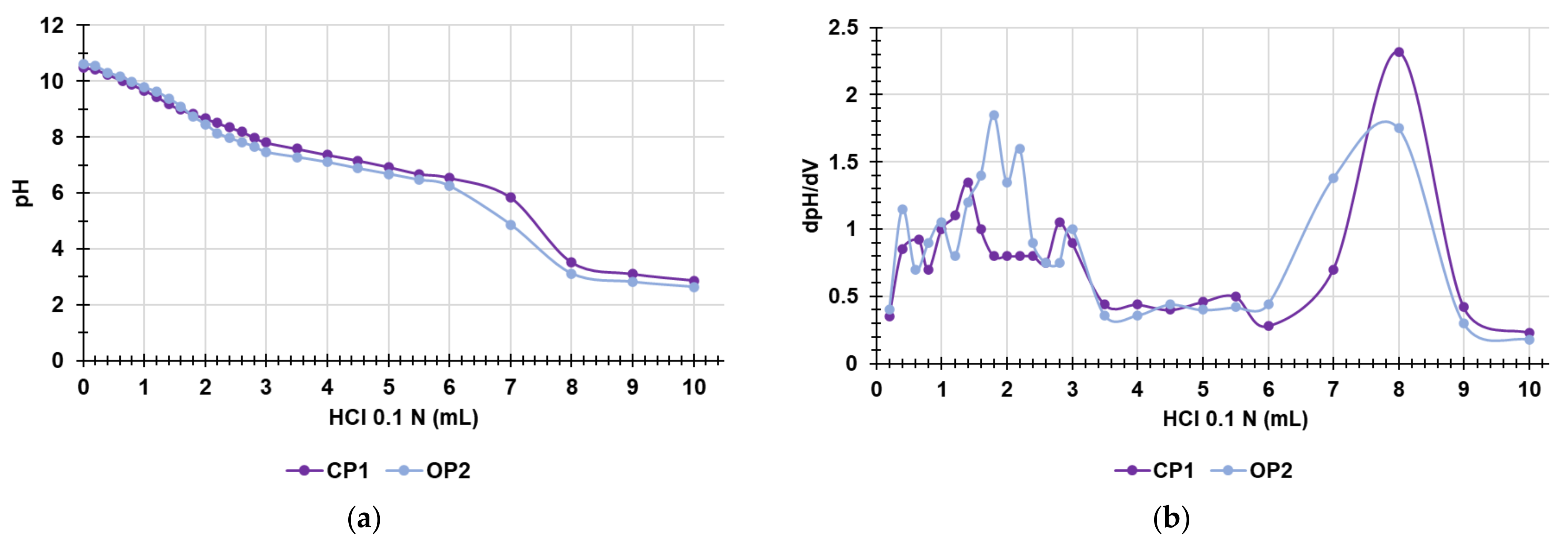
3.7. Potentiometric Titration of CP1 and OP2 NPs
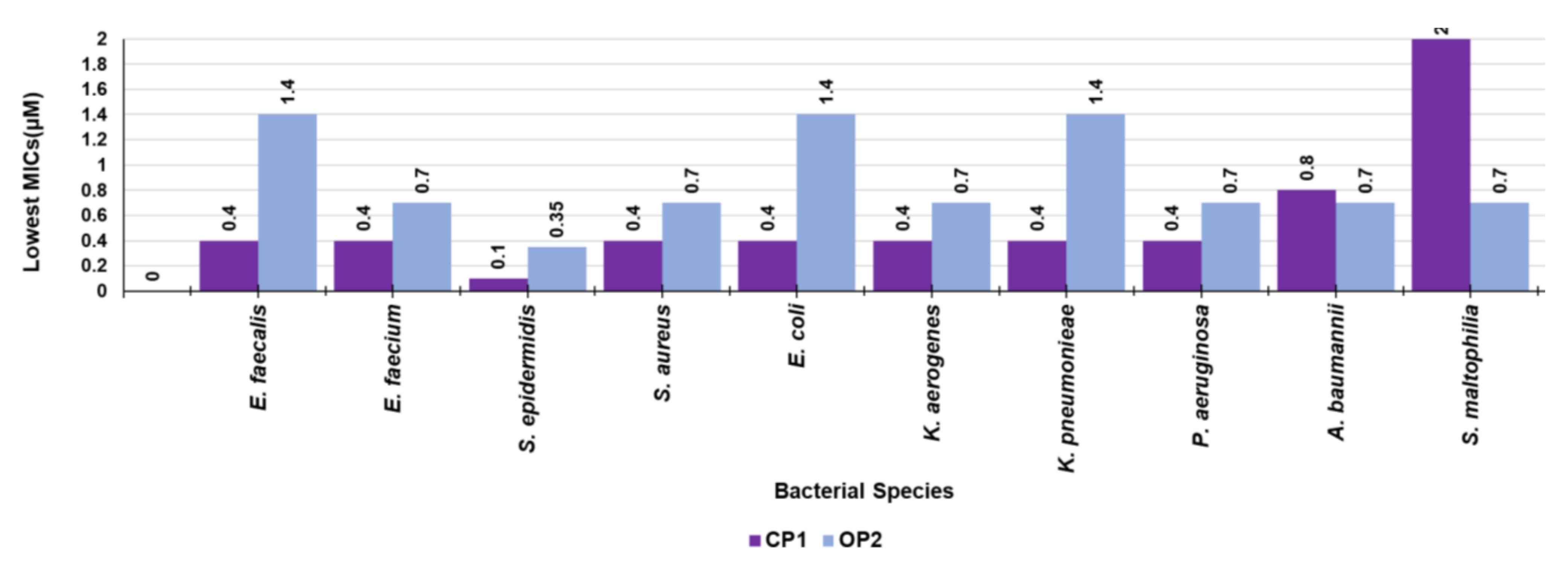
3.8. Antibacterial Properties
3.8.1. Antimicrobial Activity of CP1 and of OP2
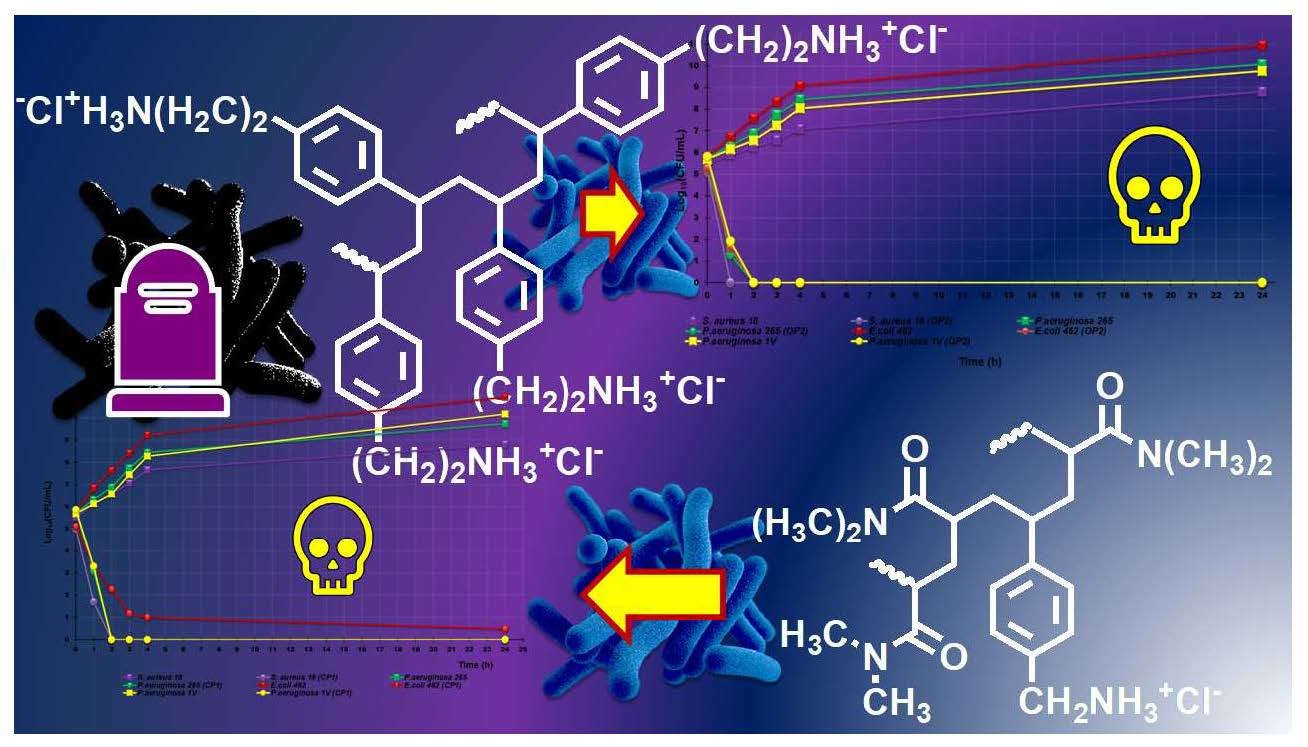
3.8.2. Time-Killing Curves
4. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alfei, S.; Schito, A.M. From Nanobiotechnology, Positively Charged Biomimetic Dendrimers as Novel Antibacterial Agents: A Review. Nanomaterials 2020, 10, 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, X.; Yu, L.; Wu, L.; Hao, X.; Liu, Q.; Lin, L.; Huang, Z.; Ruan, Z.; Weng, S.; et al. Quaternized carbon quantum dots with broad-spectrum antibacterial activity for the treatment of wounds infected with mixed bacteria. Acta Biomater. 2022, 138, 528–544. [Google Scholar] [CrossRef]
- Alfei, S.; Schito, A.M. Positively Charged Polymers as Promising Devices against Multidrug Resistant Gram-Negative Bacteria: A Review. Polymers 2020, 12, 1195. [Google Scholar] [CrossRef] [PubMed]
- Gelman, M.A.; Weisblum, B.; Lynn, D.M.; Gellman, S.H. Biocidal activity of polystyrenes that are cationic by virtue of protonation. Org. Lett. 2004, 6, 557–560. [Google Scholar] [CrossRef]
- Palermo, E.; Kuroda, K. Chemical structure of cationic groups in amphiphilic polymethacrylates modulates the antimicrobial and hemolytic activities. Biomacromolecules 2009, 10, 1416–1428. [Google Scholar] [CrossRef]
- Schito, A.M.; Alfei, S. Antibacterial Activity of Non-Cytotoxic, Amino Acid-Modified Polycationic Dendrimers against Pseudomonas aeruginosa and Other Non-Fermenting Gram-Negative Bacteria. Polymers 2020, 12, 1818. [Google Scholar] [CrossRef] [PubMed]
- Schito, A.M.; Schito, G.C.; Alfei, S. Synthesis and Antibacterial Activity of Cationic Amino Acid-Conjugated Dendrimers Loaded with a Mixture of Two Triterpenoid Acids. Polymers 2021, 13, 521. [Google Scholar] [CrossRef]
- Ganewatta, M.S.; Tang, C. Controlling macromolecular structures towards effective antimicrobial polymers. Polymer 2015, 63, A1–A29. [Google Scholar] [CrossRef]
- Alfei, S.; Piatti, G.; Caviglia, D.; Schito, A.M. Synthesis, Characterization, and Bactericidal Activity of a 4-Ammoniumbuthylstyrene-Based Random Copolymer. Polymers 2021, 13, 1140. [Google Scholar] [CrossRef]
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Alfei, S. Broad-Spectrum Bactericidal Activity of a Synthetic Random Copolymer Based on 2-Methoxy-6-(4-Vinylbenzyloxy)-Benzylammonium Hydrochloride. Int. J. Mol. Sci. 2021, 22, 5021. [Google Scholar] [CrossRef]
- Ergene, C.; Yasuharab, K.; Palermo, E.F. Biomimetic antimicrobial polymers: Recent advances in molecular design. Polym. Chem. 2018, 9, 2407–2427. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, G.J.; Som, A.; Madkour, A.E.; Eren, T.; Tew, G.N. Infectious disease: Connecting innate immunity to biocidal polymers. Mater. Sci. Eng. R Rep. 2007, 57, 28–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K.; Sugishita, K.; Fujii, N.; Miyajima, K. Molecular Basis for Membrane Selectivity of an Antimicrobial Peptide, Magainin 2. Biochemistry 1995, 34, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Marengo, B.; Valenti, G.E.; Domenicotti, C. Synthesis of Polystyrene-Based Cationic Nanomaterials with Pro-Oxidant Cytotoxic Activity on Etoposide-Resistant Neuroblastoma Cells. Nanomaterials 2021, 11, 977. [Google Scholar] [CrossRef]
- Wulff, G.; Sarhan, A.; Gimpel, J.; Lohmar, E. Über enzymanalog gebaute Polymere, III. Zur Synthese von polymerisierbaren D-Glycerinsäurederivaten. Chem. Ber. 1974, 107, 3364–3376. [Google Scholar] [CrossRef]
- Kobayashi, K.; Sumitomo, H.; Ina, Y. A Carbohydrate-Containing Synthetic Polymer Obtained from N-p-Vinylbenzyl-D-gluconamide. Polym. J. 1983, 15, 667–671. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A.; IUPAC. Compendium of Chemical Terminology, 2nd ed.; (the “Gold Book”); Blackwell Scientific Publications: Oxford, UK, 1997; Online version (2019-) created by S. J. Chalk; ISBN 0-9678550-9-8. [Google Scholar] [CrossRef]
- American Polymer Standards Corporation. Available online: http://www.ampolymer.com/Mark-Houwink.html (accessed on 20 July 2022).
- Viscosity of Polymer Solutions. Part I: Intrinsic Viscosity of Dilute Solutions. Available online: https://www.polymerdatabase.com/polymer%20physics/Solution_Viscosity.html (accessed on 21 July 2022).
- Alfei, S.; Castellaro, S. Synthesis and Characterization of Polyester-Based Dendrimers Containing Peripheral Arginine or Mixed Amino Acids as Potential Vectors for Gene and Drug Delivery. Macromol. Res. 2017, 25, 1172–1186. [Google Scholar] [CrossRef]
- EUCAST. European Committee on Antimicrobial Susceptibility Testing. Available online: https://www.eucast.org/ast_of_bacteria/ (accessed on 11 July 2022).
- Alfei, S.; Caviglia, D.; Zorzoli, A.; Marimpietri, D.; Spallarossa, A.; Lusardi, M.; Zuccari, G.; Schito, A.M. Potent and Broad-Spectrum Bactericidal Activity of a Nanotechnologically Manipulated Novel Pyrazole. Biomedicines 2022, 10, 907. [Google Scholar] [CrossRef]
- Pearson, R.D.; Steigbigel, R.T.; Davis, H.T.; Chapman, S.W. Method for reliable determination of minimal lethal anti-biotic concentrations. Antimicrob. Agents Chemother. 1980, 18, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Zorzoli, A.; Marimpietri, D.; Alfei, S. Bactericidal Activity of Non-Cytotoxic Cationic Nanoparticles against Clinically and Environmentally Relevant Pseudomonas spp. Isolates. Pharmaceutics 2021, 13, 1411. [Google Scholar] [CrossRef]
- Bertini, V.; Alfei, S.; Pocci, M.; Lucchesini, F.; Picci, N.; Iemma, F. Monomers containing substrate or inhibitor residues for copper amine oxidases and their hydrophilic beaded resins designed for enzyme interaction studies. Tetrahedron 2004, 60, 11407–11414. [Google Scholar] [CrossRef]
- Mackenzie, L.E.; Goode, J.A.; Vakurov, A.; Nampi, P.P.; Saha, S.; Jose, G.; Millner, P.A. The theoretical molecular weight of NaYF4:RE upconversion nanoparticles. Sci. Rep. 2018, 8, 1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, S.; Marengo, B.; Domenicotti, C. Polyester-Based Dendrimer Nanoparticles Combined with Etoposide Have an Improved Cytotoxic and Pro-Oxidant Effect on Human Neuroblastoma Cells. Antioxidants 2020, 9, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, S.; Zuccari, G.; Caviglia, D.; Brullo, C. Synthesis and Characterization of Pyrazole-Enriched Cationic Nanoparticles as New Promising Antibacterial Agent by Mutual Cooperation. Nanomaterials 2022, 12, 1215. [Google Scholar] [CrossRef]
- Alfei, S.; Brullo, C.; Caviglia, D.; Zuccari, G. Preparation and Physicochemical Characterization of Water-Soluble Pyrazole-Based Nanoparticles by Dendrimer Encapsulation of an Insoluble Bioactive Pyrazole Derivative. Nanomaterials 2021, 11, 2662. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Battaglia, G. Exploiting endocytosis for nanomedicines. Cold Spring Harb. Perspect. Biol. 2013, 5, a016980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Mccrate, J.M.; Lee, J.C.-M.; Li, H. The Role of Surface Charge on the Uptake and Biocompatibility of Hydroxyapatite Nanoparticles with Osteoblast Cells. Nanotechnology 2011, 22, 105708. [Google Scholar] [CrossRef] [Green Version]
- Jambhrunkar, S.; Yu, M.; Yang, J.; Zhang, J.; Shrotri, A.; Endo-Munoz, L.; Moreau, J.; Lu, G.; Yu, C. Stepwise Pore Size Reduction of Ordered Nanoporous Silica Materials at Angstrom Precision. J. Am. Chem. Soc. 2013, 135, 8444–8447. [Google Scholar] [CrossRef]
- Petri-Fink, A.; Chastellain, M.; Juillerat-Jeanneret, L.; Ferrari, A.; Hofmann, H. Development of Functionalized Superparamagnetic Iron Oxide Nanoparticles for Interaction with Human Cancer Cells. Biomaterials 2005, 26, 2685–2694. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Spallarossa, A.; Lusardi, M.; Zuccari, G. Successful Dendrimer and Liposome-Based Strategies to Solubilize an Antiproliferative Pyrazole Otherwise Not Clinically Applicable. Nanomaterials 2022, 12, 233. [Google Scholar] [CrossRef]
- Alfei, S.; Catena, S. Synthesis and characterization of fourth generation polyester-based dendrimers with cationic amino acids-modified crown as promising water soluble biomedical devices. Polym. Adv. Technol. 2018, 29, 2735–2749. [Google Scholar] [CrossRef]
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landman, D.; Georgescu, C.; Martin, D.A.; Quale, J. Polymyxins revisited. Clin. Microbiol. Rev. 2008, 21, 449–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, S.; Caviglia, D.; Piatti, G.; Zuccari, G.; Schito, A.M. Bactericidal Activity of a Self-Biodegradable Lysine-Containing Dendrimer against Clinical Isolates of Acinetobacter Genus. Int. J. Mol. Sci. 2021, 22, 7274. [Google Scholar] [CrossRef]
- Sudip, M.; Swagatam, B.; Riya, M.; Jayanta, H. Amphiphilic Cationic Macromolecules Highly Effective Against Multi-Drug Resistant Gram-Positive Bacteria and Fungi With No Detectable Resistance. Front. Bioeng. Biotechnol. 2020, 8, e55. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | Loading (mg, mmol, % 1) | DMAA (mg, mmol) | Solvent (mL) | Initiator (mg, % 2) | Time/Temp. (h/°C) | Polymer (mg, % 3) |
|---|---|---|---|---|---|---|
| M1 | 120.5, 0.71, 41.9 | 167.3, 1.69 | MeOH 1.2 | AIBN 3.4, 1.2 | 24/60 | CP1 223.9, 78 |
| M2 | 197.2, 1.07, 100 | NO | H2O/MeOH 2.5/2.0 | (NH4)2S2O8 3.6, 1.8 | 24/35 | OP2 141.2, 72 |
| Polymer | ηrel (dL/g) | η sp | [η] (dL/g) | a * K (dL/g) * | Mr |
|---|---|---|---|---|---|
| CP1 | 1.84 ± 0.02 | 0.84 ± 0.02 | 1.68 ± 0.02 | 0.93 | 157,306 |
| OP2 | 1.26 ± 0.02 | 0.26 ± 0.02 | 0.52 ± 0.02 | 0.0000250 | 44,514 |
| Polymer | Weight (mg) | HClO4 0.1612 N (mL) | NH2 (mmol) | mequiv.NH2/g | µequiv.NH2/µmol |
|---|---|---|---|---|---|
| CP1 | 24.2 | 4.44 ± 0.020 | 0.7497 ± 0.0032 | 30.98 ± 0.13 | 4917.5 ± 20.6 |
| OP2 | 31.4 | 13.85 ± 0.016 | 2.2328 ± 0.0026 | 71.11 ± 0.24 | 3174.6 ± 10.7 |
| Measure | CP1 NPs | OP2 NPs |
|---|---|---|
| Z-Ave 1 (nm) | 833.4 ± 10.1 | 163.4 ± 5.5 |
| PDI | 0.235 ± 0.022 | 0.301 ± 0.016 |
| ζ-p (mV) | +27.3 ± 4.2 | +31.1 ± 5.1 |
| Polymer | Weight (mg) | HCl 0.1 N (mL) | NH2 (mmol) | mequiv.NH2/g | µequiv.NH2/µmol | Error * (%) |
|---|---|---|---|---|---|---|
| CP1 | 25.5 | 8.0 ± 0.020 | 0.8 ± 0.0020 | 31.37 ± 0.08 | 4979.4 ± 12.7 | 1.3 |
| OP2 | 11.0 | 7.8 ± 0.016 | 0.78 ± 0.0016 | 70.91 ± 0.14 | 3165.6 ± 6.3 | 0.3 |
| Strains | CP1 (157306) 1 | OP2 (44514) 1 | Reference Antibiotic |
|---|---|---|---|
| MIC µM (µg/mL) | MIC µM (µg/mL) | MIC µM (µg/mL) | |
| Gram-positive species of genus Enterococcus | |||
| E. faecalis 365 * | 0.4 (64) | 1.4 (64) | 366.3 (128) 2 |
| E. faecalis 450 * | 0.8 (128) | 1.4 (64) | 366.3 (128) 2 |
| E. faecalis 451 * | 0.4 (64) | 1.4 (64) | 366.3 (128) 2 |
| E. faecium 300 * | 0.4 (64) | 0.7 (32) | 366.3 (128) 2 |
| E. faecium 364 * | 0.4 (64) | 0.7 (32) | 366.3 (128) 2 |
| E. faecium 503 *,TR | 0.4 (64) | 0.7 (32) | 366.3 (128) 2 |
| Gram-positive species of genus Staphylococcus | |||
| S. aureus 18 ** | 0.4 (64) | 0.7 (32) | 386.4 (128) 3, 1275.5 (512) 4 |
| S. aureus 187 ** | 0.8 (128) | 0.7 (32) | 386.4 (128) 3, 1275.5 (512) 4 |
| S. aureus 195 ** | 0.4 (64) | 0.7 (32) | 386.4 (128) 3, 1275.5 (512) 4 |
| S. epidermidis 180 *** | 0.1 (16) | 0.35 (16) | 193.2 (64) 3, 637.8 (256) 4 |
| S. epidermidis 181 *** | 0.1 (16) | 0.35 (16) | 193.2 (64) 3, 637.8 (256) 4 |
| S. epidermidis 363 ** | 0.2 (32) | 0.7 (32) | 193.2 (64) 3, 637.8 (256) 4 |
| Gram-negative species of Enterobacteriaceae family | |||
| E. coli 461 | 0.8 (128) | 1.4 (64) | 96.6 (32) 3 |
| E. coli 462 § | 0.8 (128) | 1.4 (64) | 96.6 (32) 3 |
| E. coli 477 # | >0.8 (128) | 1.4 (64) | 96.6 (32) 3 |
| K. aerogenes 484CAR | 0.8 (128) | 1.4 (64) | 96.6 (32) 3 |
| K. aerogenes 500 # | 0.8 (128) | 0.7 (32) | 96.6 (32) 3 |
| K. aerogenes 501CAR | 0.4 (64) | 1.4 (64) | 96.6 (32) 3 |
| K. pneumoniae 502 # | >0.8 (128) | 1.4 (64) | 96.6 (32) 3 |
| K. pneumoniae 509 # | 0.8 (128) | 2.8 (128) | 96.6 (32) 3 |
| K. pneumoniae 520 # | 0.4 (64) | 1.4 (64) | 96.6 (32) 3 |
| Non-fermenting Gram-negative species | |||
| P. aeruginosa 1V | 0.8 (128) | 0.7 (32) | 76.2 (64) 5 |
| P. aeruginosa CR | 0.8 (128) | 1.4 (64) | 18.5 (16) 6 |
| P. aeruginosa PY | 0.4 (64) | 0.7 (32) | 76.2 (64) 7 |
| A. baumannii 245 | >0.8 (128) | 0.7 (32) | 193.2 (64) 3 |
| A. baumannii 257 | 0.8 (128) | 0.7 (32) | 193.2 (64) 3 |
| S. maltophylia 255 | >0.8 (128) | 0.7 (32) | 117.7 (64) 7 |
| S. maltophylia 280 | >0.8 (128) | 0.7 (32) | 117.7 (64) 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfei, S.; Caviglia, D.; Piatti, G.; Zuccari, G.; Schito, A.M. Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles. Nanomaterials 2022, 12, 2743. https://doi.org/10.3390/nano12162743
Alfei S, Caviglia D, Piatti G, Zuccari G, Schito AM. Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles. Nanomaterials. 2022; 12(16):2743. https://doi.org/10.3390/nano12162743
Chicago/Turabian StyleAlfei, Silvana, Debora Caviglia, Gabriella Piatti, Guendalina Zuccari, and Anna Maria Schito. 2022. "Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles" Nanomaterials 12, no. 16: 2743. https://doi.org/10.3390/nano12162743
APA StyleAlfei, S., Caviglia, D., Piatti, G., Zuccari, G., & Schito, A. M. (2022). Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles. Nanomaterials, 12(16), 2743. https://doi.org/10.3390/nano12162743