

Ethanol Electro-Oxidation on Catalysts with S-ZrO2-Decorated Graphene as Support in Fuel Cell Applications




Abstract
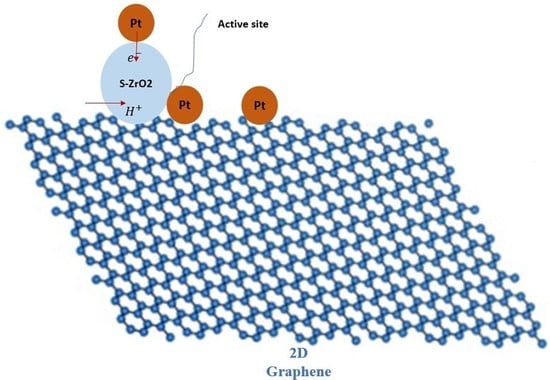
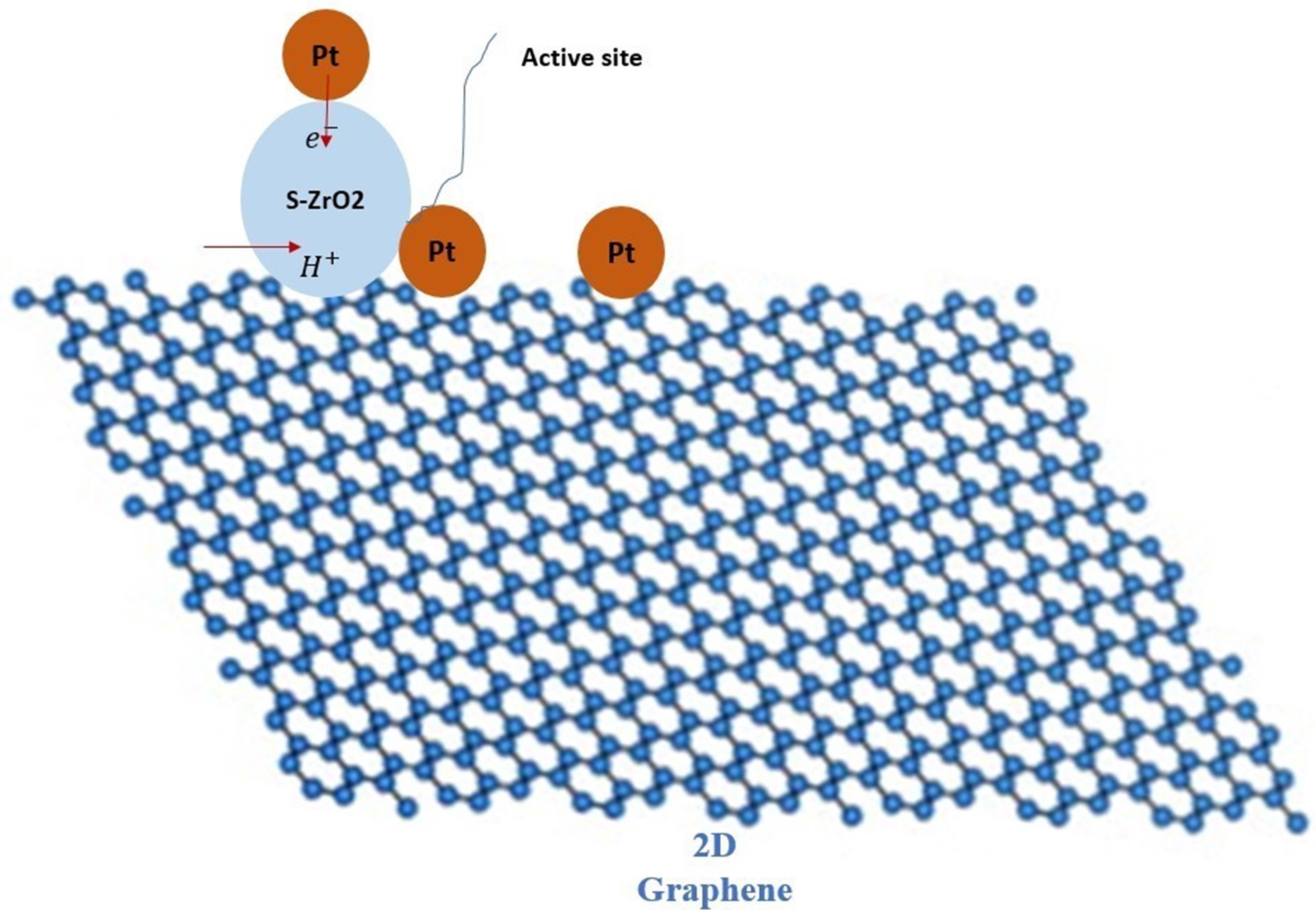
:
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Experimental Methods
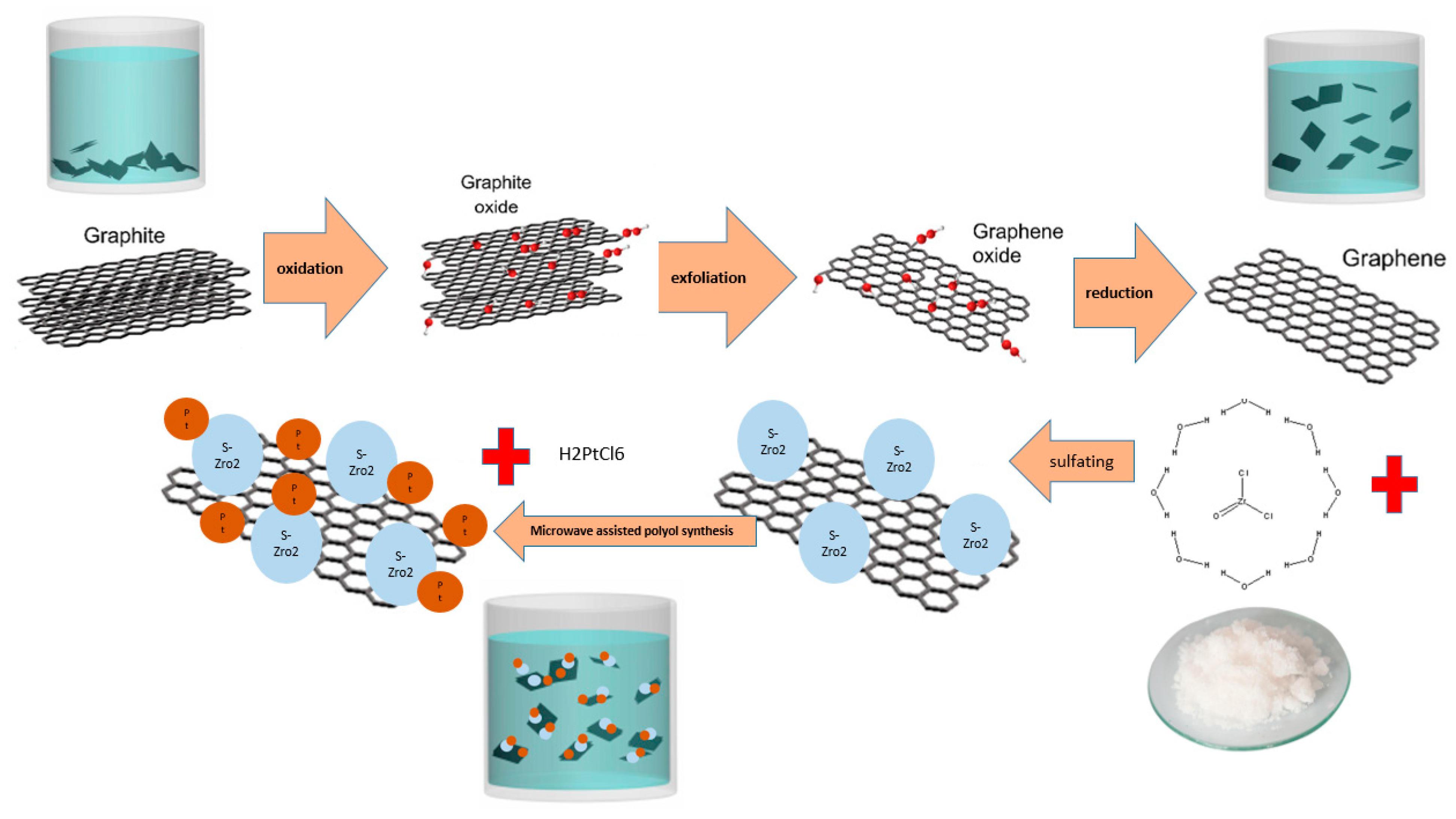
2.2.1. Preparation of GO and GNPs by Chemical Method
2.2.2. Preparation of S-ZrO2-GNP Support
2.2.3. Polyol Synthesis of Pt/S-ZrO2-GNP Nanocomposites by the Aide of Microwave
2.2.4. Characterization
3. Results and Discussion
3.1. Physical Characterization
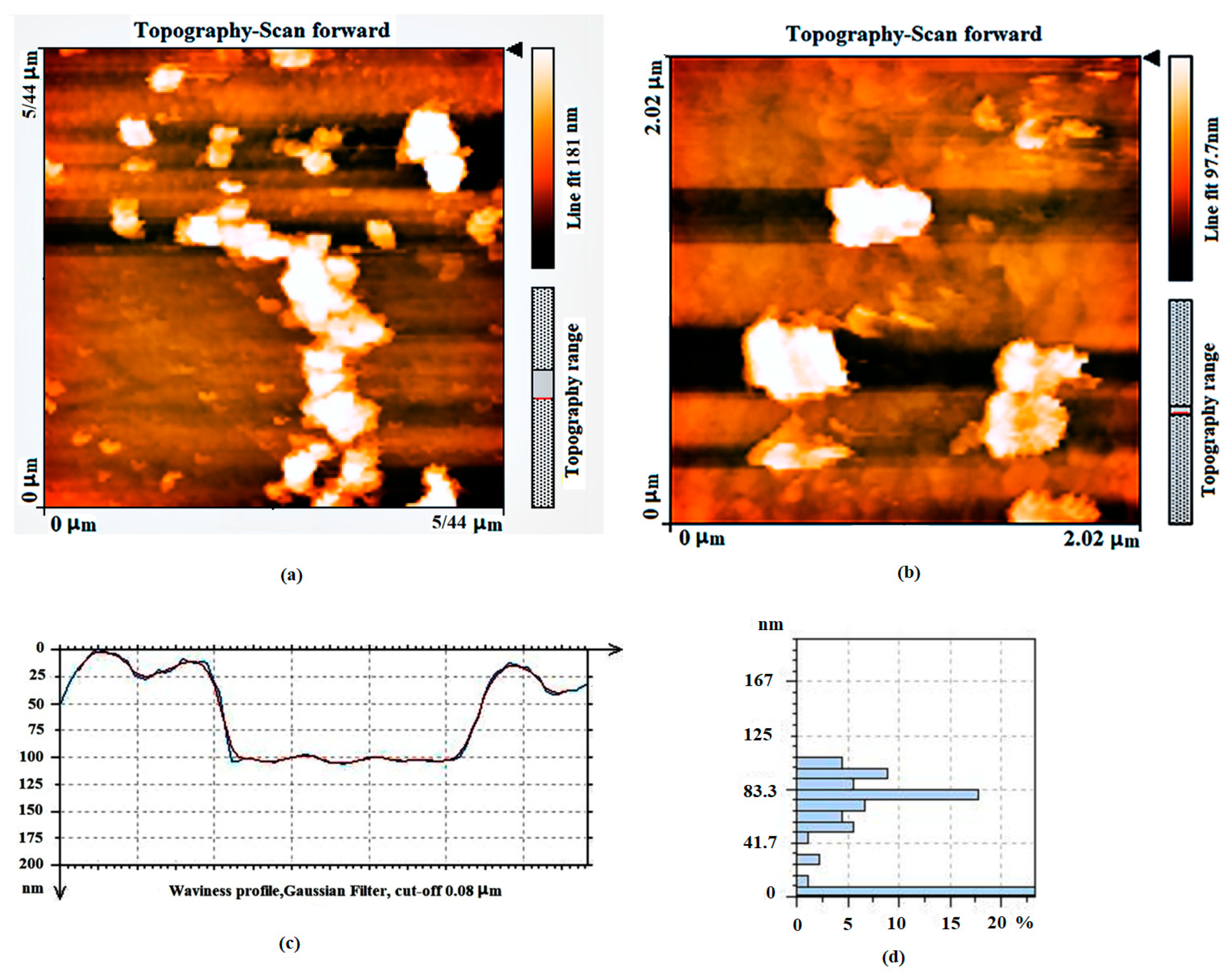
3.1.1. Topographic Study of GNPs
3.1.2. Morphology of ZrO2-GNP Support and Pt Nanoparticles
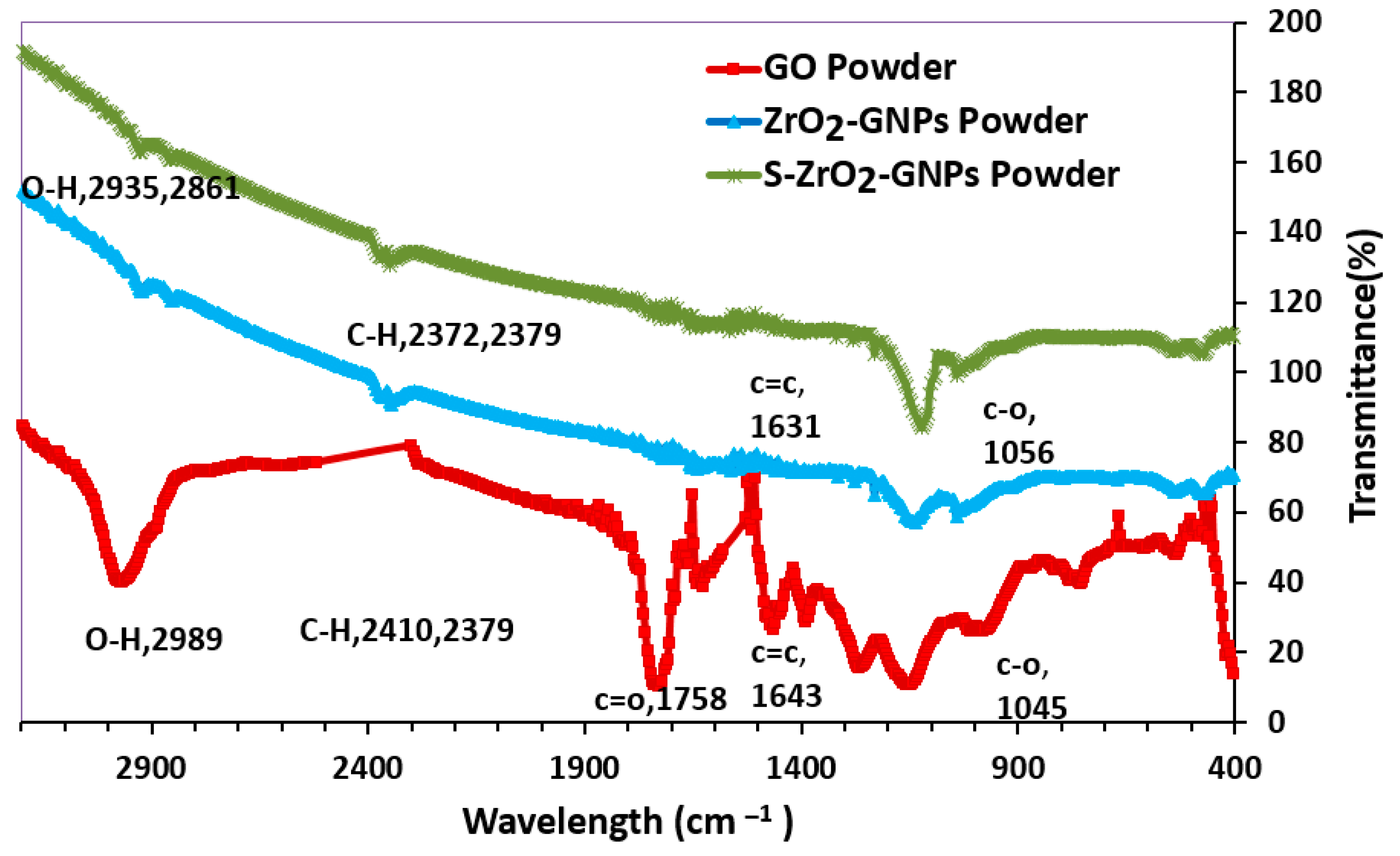
3.1.3. Structural Support Specifications
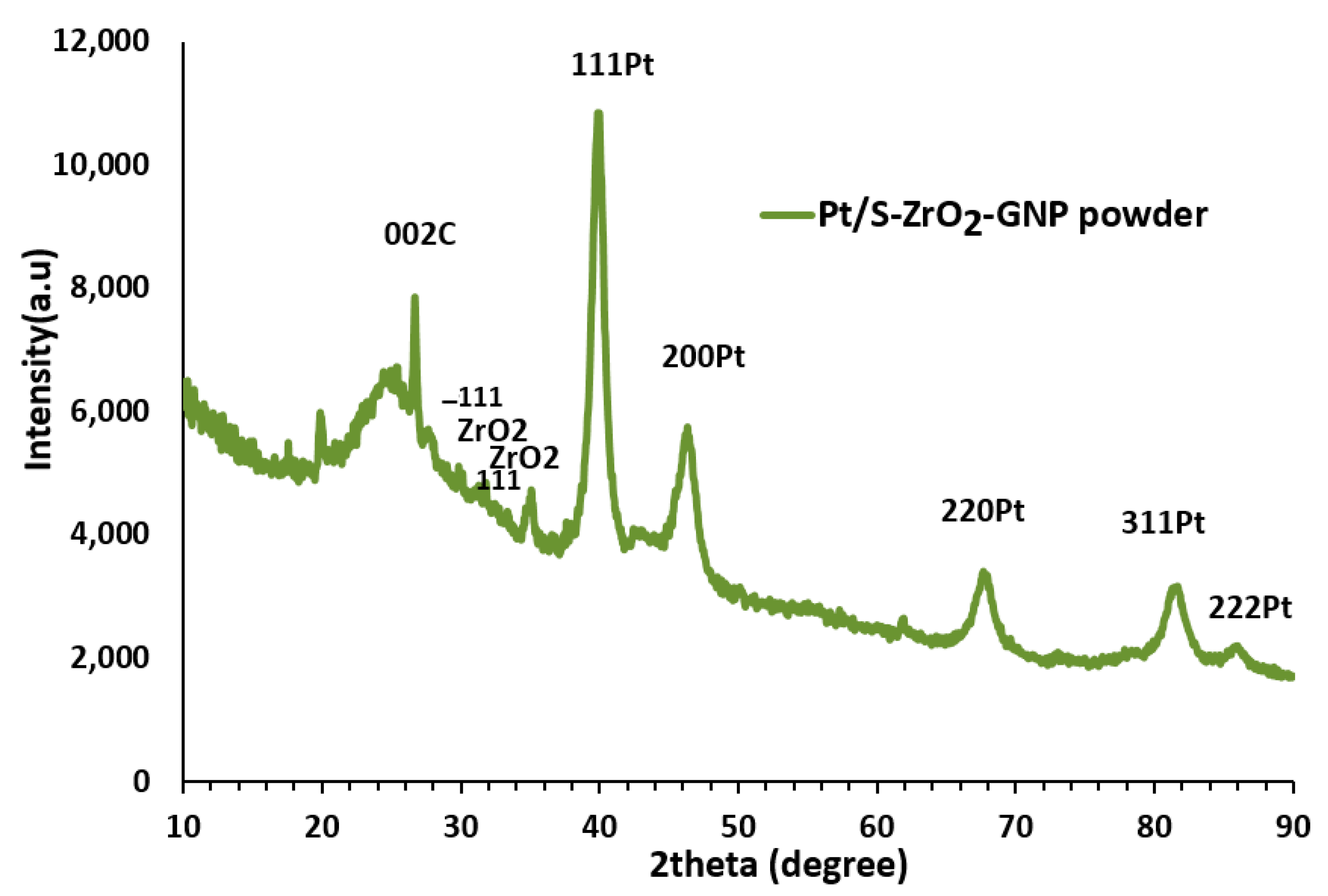
3.1.4. XRD Pattern Characterization
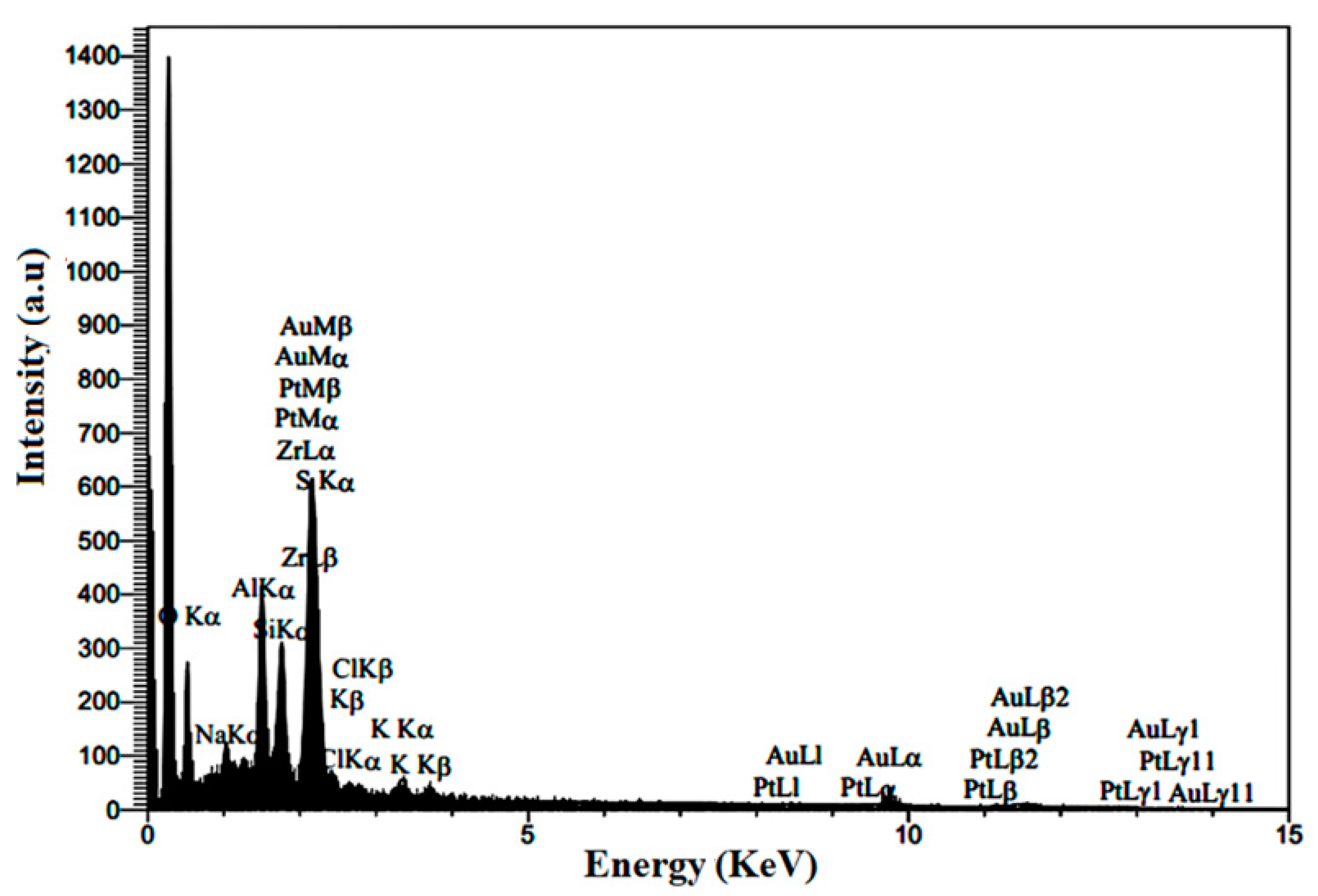
3.1.5. Composition of Prepared Electrocatalyst
3.2. Electrochemical Assessments
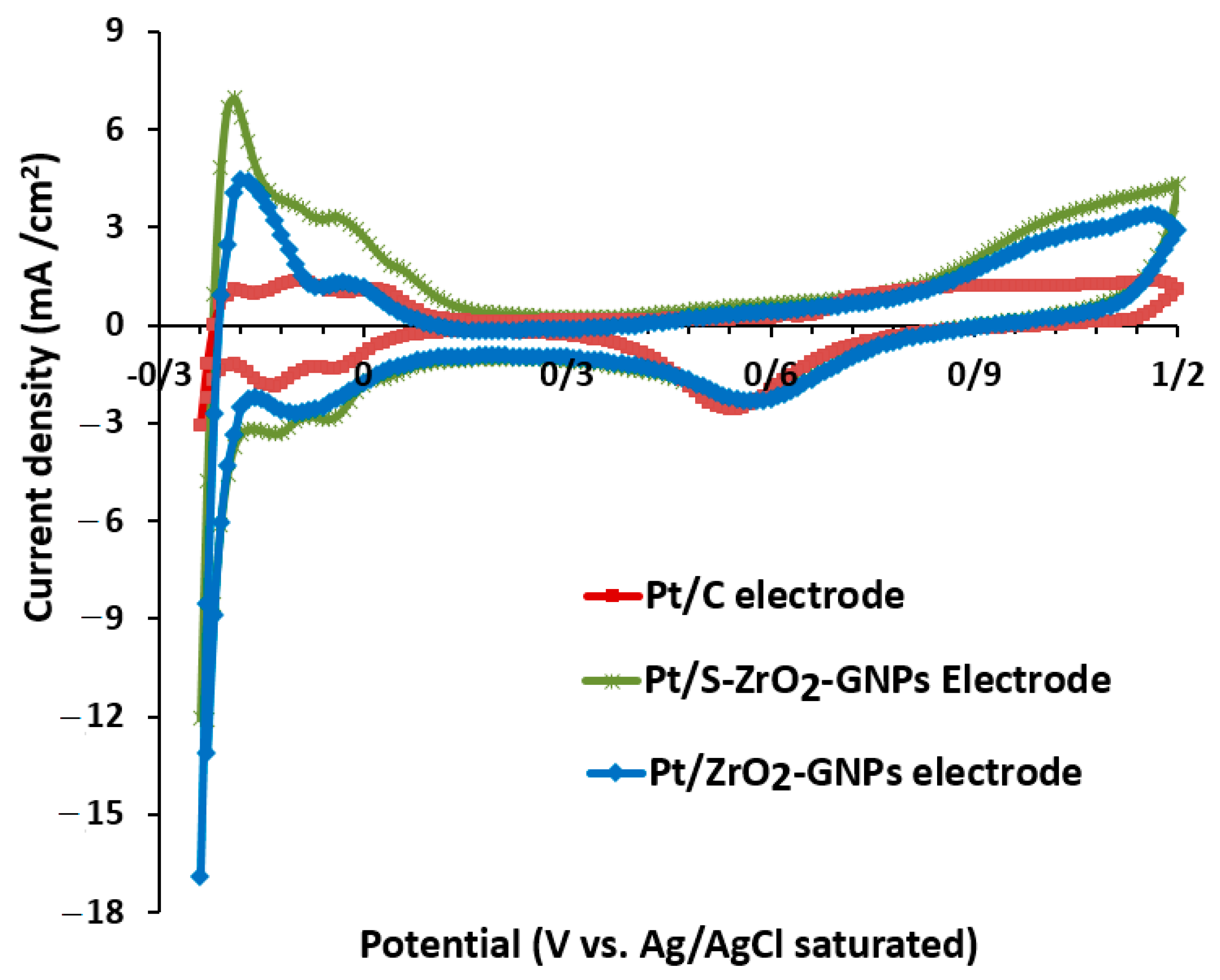
3.2.1. Electrochemical Active Surface Area of Electrocatalysts
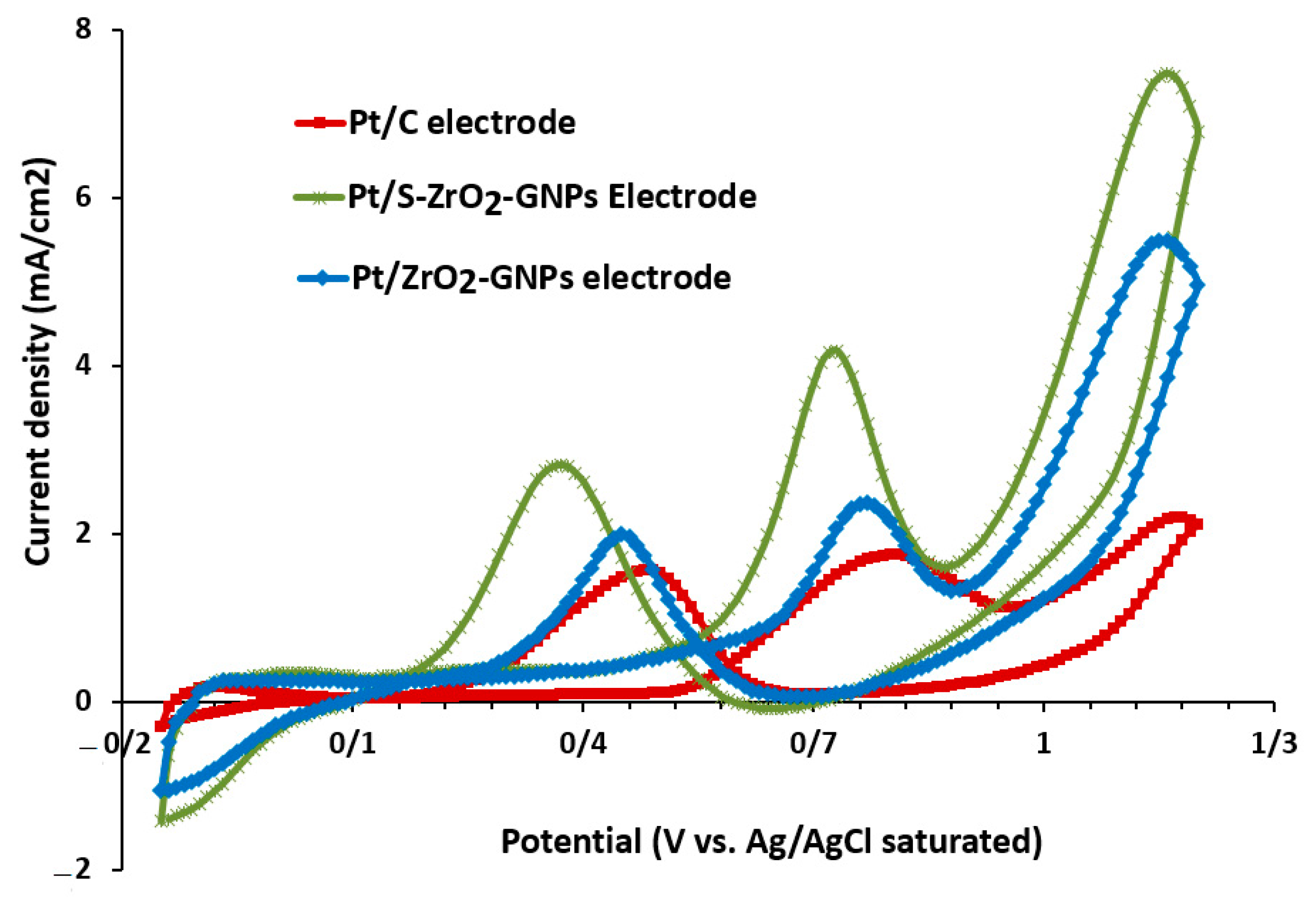
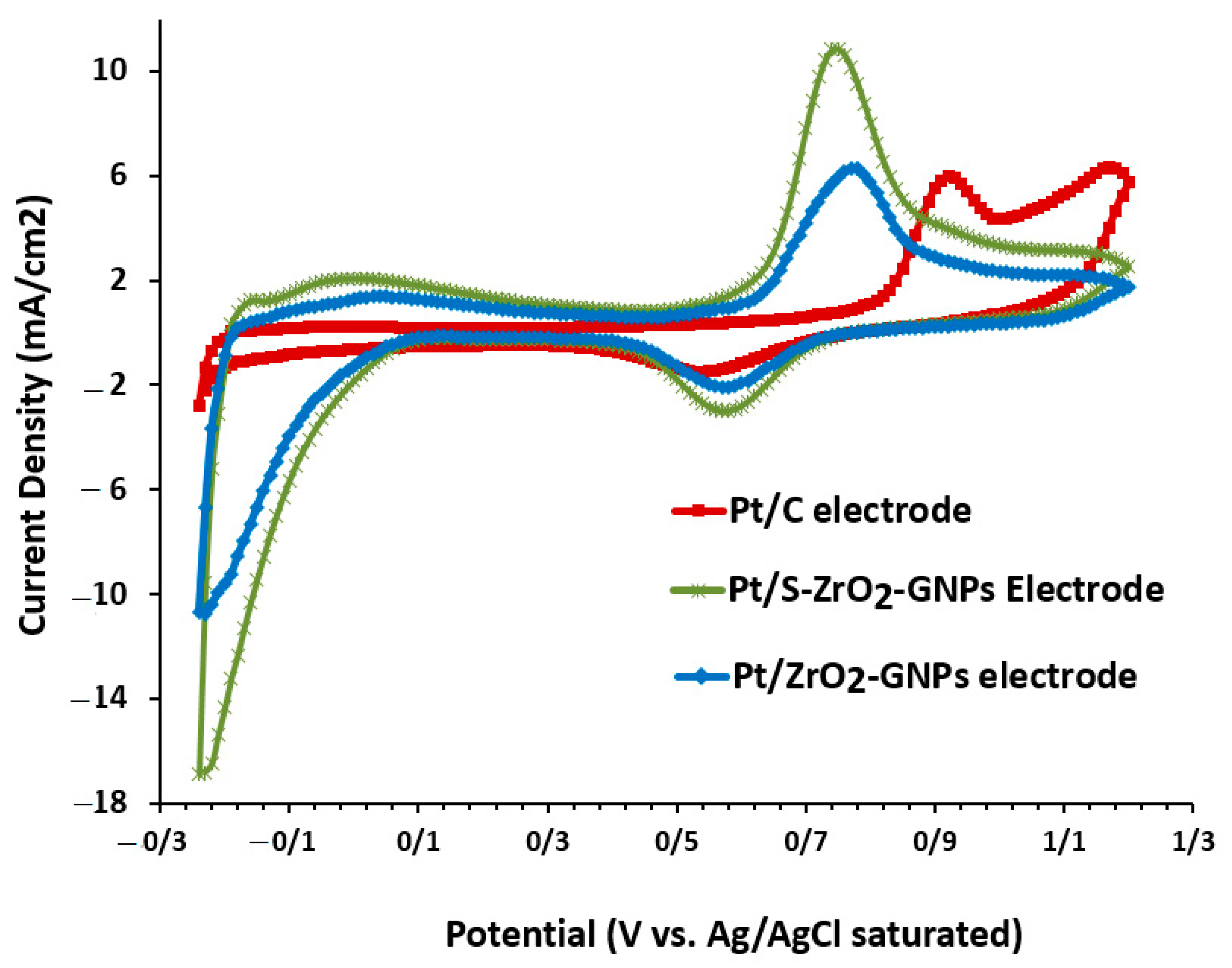
3.2.2. Performance Estimate of Catalyst through Ethanol Oxidation Reaction (EOR) and Carbon Monoxide (CO) Tolerance
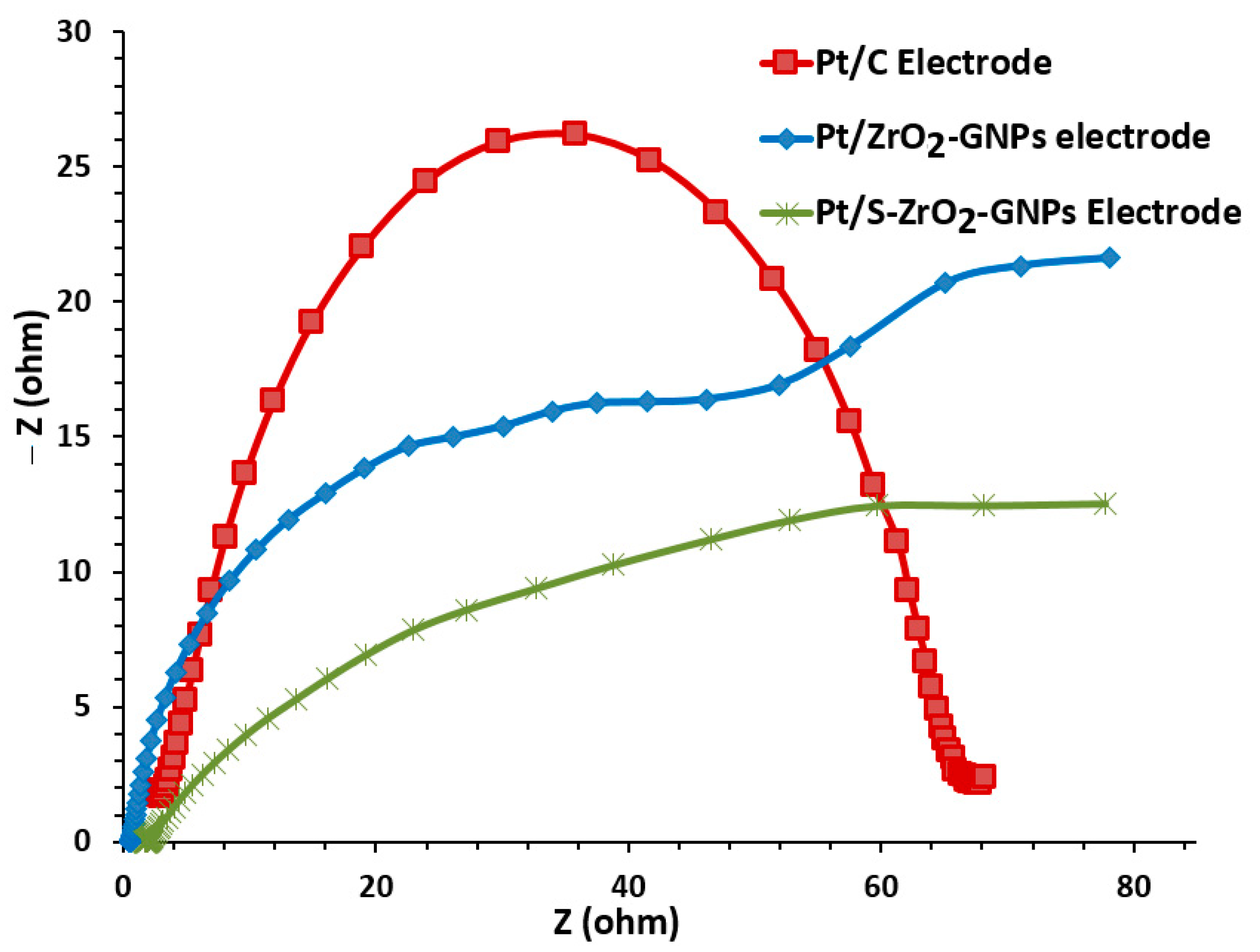
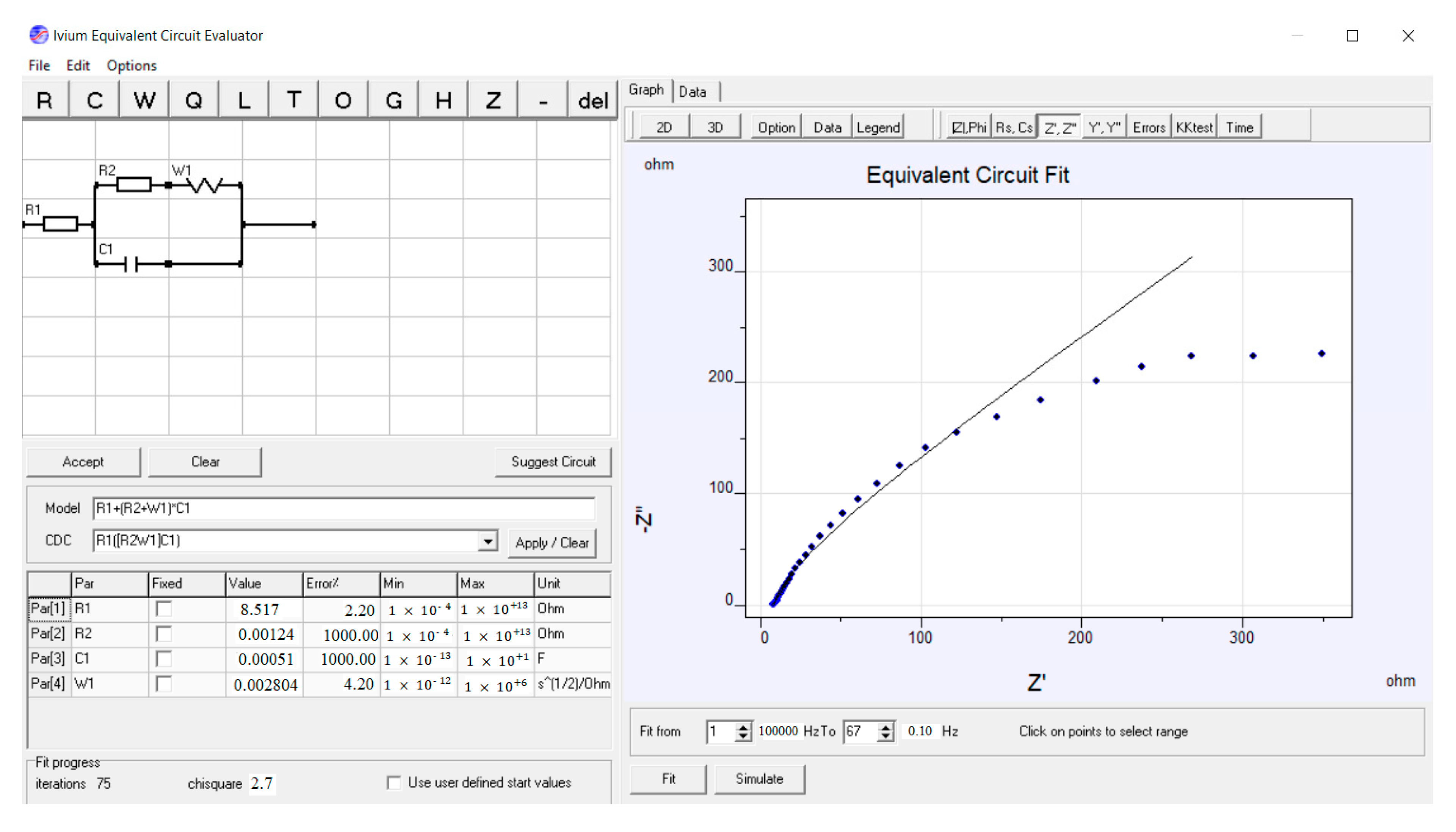
3.2.3. Electrochemical Impedance Spectroscopy (EIS) Investigations of the Electrodes
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Halim, E.M.; Chemchoub, S.; El Attar, A.; Salih, F.E.; Oularbi, L.; El Rhazi, M. Recent advances in anode metallic catalysts supported on conducting polymer-based materials for direct alcohol fuel cells. Front. Energy Res. 2022, 10, 843736. [Google Scholar] [CrossRef]
- Chen, C.; Yang, P. Performance of an air-breathing direct methanol fuel cell. J. Power Sources 2003, 123, 37–42. [Google Scholar] [CrossRef]
- Vigier, F.; Coutanceau, C.; Perrard, A.; Belgsir, E.; Lamy, C. Development of anode catalysts for a direct ethanol fuel cell. J. Appl. Electrochem. 2004, 34, 439–446. [Google Scholar] [CrossRef]
- Lamy, C.; Rousseau, S.; Belgsir, E.; Coutanceau, C.; Léger, J.-M. Recent progress in the direct ethanol fuel cell: Development of new platinum–tin electrocatalysts. Electrochim. Acta 2004, 49, 3901–3908. [Google Scholar] [CrossRef]
- Spinacé, E.; Neto, A.O.; Linardi, M. Electro-oxidation of methanol and ethanol using PtRu/C electrocatalysts prepared by spontaneous deposition of platinum on carbon-supported ruthenium nanoparticles. J. Power Sources 2004, 129, 121–126. [Google Scholar] [CrossRef]
- Jiang, L.; Zhou, Z.; Li, W.; Zhou, W.; Song, S.; Li, H.; Sun, A.G.; Xin, Q. Effects of treatment in different atmosphere on Pt3Sn/C electrocatalysts for ethanol electro-oxidation. Energy Fuels 2004, 18, 866–871. [Google Scholar] [CrossRef]
- Wang, H.; Jusys, Z.; Behm, R. Ethanol electrooxidation on a carbon-supported Pt catalyst: Reaction kinetics and product yields. J. Phys. Chem. B 2004, 108, 19413–19424. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, H.; Zhang, N.; Kong, F.; Liu, H.; Yin, G. Role of Pt-pyridinic nitrogen sites in methanol oxidation on Pt/polypyrrole-carbon black catalyst. J. Power Sources 2012, 197, 44–49. [Google Scholar] [CrossRef]
- Lamy, C.; Belgsir, E.; Leger, J. Electrocatalytic oxidation of aliphatic alcohols: Application to the direct alcohol fuel cell (DAFC). J. Appl. Electrochem. 2001, 31, 799–809. [Google Scholar] [CrossRef]
- Datta, J.; Singh, S.; Das, S.; Bandyopadhyay, N. A comprehensive study on the effect of Ru addition to Pt electrodes for direct ethanol fuel cell. Bull. Mater. Sci. 2009, 32, 643–652. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Z.; Song, S.; Li, W.; Sun, G.; Tsiakaras, P.; Xin, Q. Pt based anode catalysts for direct ethanol fuel cells. Appl. Catal. B 2003, 46, 273–285. [Google Scholar] [CrossRef]
- Song, S.; Tsiakaras, P. Recent progress in direct ethanol proton exchange membrane fuel cells (DE-PEMFCs). Appl. Catal. B 2006, 63, 187–193. [Google Scholar] [CrossRef]
- Brandalise, M.; Verjulio-Silva, R.; Tusi, M.; Correa, O.; Farias, L.; Linardi, M.; Spinacé, E.V.; Neto, A.O. Electro-oxidation of ethanol using PtRuBi/C electrocatalyst prepared by borohydride reduction. Ionics 2009, 15, 743. [Google Scholar] [CrossRef]
- Wu, B.; Cui, R.; Gao, Y.; Jiang, Z. Ethanol electrocatalytic oxidation on highly dispersed Pt-TiO 2/C catalysts. Russ. J. Electrochem. 2009, 45, 731–735. [Google Scholar] [CrossRef]
- Dos Anjos, D.; Hahn, F.; Léger, J.-M.; Kokoh, K.B.; Tremiliosi-Filho, G. In situ FTIRS studies of the electrocatalytic oxidation of ethanol on Pt alloy electrodes. J. Solid State Electrochem. 2007, 11, 1567–1573. [Google Scholar] [CrossRef]
- Zhang, J. PEM Fuel Cell Electrocatalysts and Catalyst Layers; Fundamentals and applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008; pp. 165–287. [Google Scholar] [CrossRef]
- Iwasita, T.; Hoster, H.; John-Anacker, A.; Lin, W.; Vielstich, W. Methanol oxidation on PtRu electrodes. Influence of surface structure and Pt− Ru atom distribution. Langmuir 2000, 16, 522–529. [Google Scholar] [CrossRef]
- Chen, Z.; Qiu, X.; Lu, B.; Zhang, S.; Zhu, W.; Chen, L. Synthesis of hydrous ruthenium oxide supported platinum catalysts for direct methanol fuel cells. Electrochem. Commun. 2005, 7, 593–596. [Google Scholar] [CrossRef]
- Suffredini, H.; Tricoli, V.; Avaca, L.; Vatistas, N. Sol–gel method to prepare active Pt–RuO2 coatings on carbon powder for methanol oxidation. Electrochem. Commun. 2004, 6, 1025–1028. [Google Scholar] [CrossRef]
- Santos, A.L.; Profeti, D.; Olivi, P. Electrooxidation of methanol on Pt microparticles dispersed on SnO2 thin films. Electrochim. Acta 2005, 50, 2615–2621. [Google Scholar] [CrossRef]
- Jiang, L.; Sun, G.; Zhou, Z.; Sun, S.; Wang, Q.; Yan, S.; Li, H.; Tian, J.; Guo, J.; Zhou, B.; et al. Size-controllable synthesis of monodispersed SnO2 nanoparticles and application in electrocatalysts. J. Phys. Chem. B 2005, 109, 8774–8778. [Google Scholar] [CrossRef]
- Raghuveer, V.; Viswanathan, B. Synthesis, characterization and electrochemical studies of Ti-incorporated tungsten trioxides as platinum support for methanol oxidation. J. Power Sources 2005, 144, 1–10. [Google Scholar] [CrossRef]
- Bai, Y.; Wu, J.; Xi, J.; Wang, J.; Zhu, W.; Chen, L.; Qiu, X. Electrochemical oxidation of ethanol on Pt–ZrO2/C catalyst. Electrochem. Commun. 2005, 7, 1087–1090. [Google Scholar] [CrossRef]
- Song, H.; Qiu, X.; Li, F. Effect of heat treatment on the performance of TiO2-Pt/CNT catalysts for methanol electro-oxidation. Electrochim. Acta 2008, 53, 3708–3713. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kim, D.-Y.; Han, H.; Shul, Y.-G. PtRu/C-Au/TiO2 electrocatalyst for a direct methanol fuel cell. J. Power Sources 2006, 159, 484–490. [Google Scholar] [CrossRef]
- Takahashi, M.; Mori, T.; Ye, F.; Vinu, A.; Kobayashi, H.; Drennan, J. Design of High-Quality Pt–CeO2 Composite Anodes Supported by Carbon Black for Direct Methanol Fuel Cell Application. J. Am. Ceram. Soc. 2007, 90, 1291–1294. [Google Scholar] [CrossRef]
- Wang, J.; Xi, J.; Bai, Y.; Shen, Y.; Sun, J.; Chen, L.; Zhu, W.; Qiu, X. Structural designing of Pt-CeO2/CNTs for methanol electro-oxidation. J. Power Sources 2007, 164, 555–560. [Google Scholar] [CrossRef]
- Guo, D.-J.; Qiu, X.-P.; Chen, L.-Q.; Zhu, W.-T. Multi-walled carbon nanotubes modified by sulfated TiO2–A promising support for Pt catalyst in a direct ethanol fuel cell. Carbon 2009, 47, 1680–1685. [Google Scholar] [CrossRef]
- Song, H.; Qiu, X.; Li, F.; Zhu, W.; Chen, L. Ethanol electro-oxidation on catalysts with TiO2 coated carbon nanotubes as support. Electrochem. Commun. 2007, 9, 1416–1421. [Google Scholar] [CrossRef]
- Sheikhi, S.; Jalali, F. Remarkable electrocatalytic activity of Ni-nanoparticles on MOF-derived ZrO2-porous carbon/reduced graphene oxide towards methanol oxidation. Int. J. Hydrog. Energy 2021, 46, 10723–10738. [Google Scholar] [CrossRef]
- Guo, D.-J.; Qiu, X.-P.; Zhu, W.-T.; Chen, L.-Q. Synthesis of sulfated ZrO2/MWCNT composites as new supports of Pt catalysts for direct methanol fuel cell application. Appl. Catal. B 2009, 89, 597–601. [Google Scholar] [CrossRef]
- Ahmad, K.; Nazir, M.A.; Qureshi, A.K.; Hussain, E.; Najam, T.; Javed, M.S.; Shah, S.S.A.; Tufail, M.K.; Hussain, S.; Khan, N.A.; et al. Engineering of Zirconium based metal-organic frameworks (Zr-MOFs) as efficient adsorbents. Mater. Sci. Eng. B 2020, 262, 114766. [Google Scholar] [CrossRef]
- Barboux, P.; Morineau, R.; Livage, J. Protonic conductivity in hydrates. Solid State Ion. 1988, 27, 221–225. [Google Scholar] [CrossRef]
- Kreuer, K.-D. Proton conductivity: Materials and applications. Chem Mater. 1996, 8, 610–641. [Google Scholar] [CrossRef]
- Norby, T. Solid-state protonic conductors: Principles, properties, progress and prospects. Solid State Ion. 1999, 125, 1–11. [Google Scholar] [CrossRef]
- Alberti, G.; Casciola, M. Solid state protonic conductors, present main applications and future prospects. Solid State Ion. 2001, 145, 3–16. [Google Scholar] [CrossRef]
- Javed, M.S.; Shaheen, N.; Hussain, S.; Li, J.; Shah, S.S.A.; Abbas, Y.; Ahmad, M.A.; Raza, R.; Mai, W. An ultra-high energy density flexible asymmetric supercapacitor based on hierarchical fabric decorated with 2D bimetallic oxide nanosheets and MOF-derived porous carbon polyhedra. J. Mater. Chem. A 2019, 7, 946–957. [Google Scholar] [CrossRef]
- Javed, M.S.; Shah, S.S.A.; Najam, T.; Siyal, S.H.; Hussain, S.; Saleem, M.; Zhao, Z.; Mai, W. Achieving high-energy density and superior cyclic stability in flexible and lightweight pseudocapacitor through synergic effects of binder-free CoGa2O4 2D-hexagonal nanoplates. Nano Energy 2020, 77, 105276. [Google Scholar] [CrossRef]
- Fu, J.; Jiang, X.; Han, W.; Cao, Z. Enhancing the Cycling Stability of Transition-Metal-Oxide-Based Electrochemical Electrode via Pourbaix Diagram Engineering. Energy Storage Mater. 2021, 42, 252–258. [Google Scholar] [CrossRef]
- Wu, Z.; Sun, G.; Jin, W.; Hou, H.; Wang, S.; Xin, Q. Nafion® and nano-size TiO2–SO42− solid superacid composite membrane for direct methanol fuel cell. J. Membr. Sci. 2008, 313, 336–343. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ishihara, A.; Mitsushima, S.; Kamiya, N.; Ota, K.-I. Sulfated-zirconia as a support of Pt catalyst for polymer electrolyte fuel cells. Electrochem. Solid-State Lett. 2007, 10, B105–B107. [Google Scholar] [CrossRef]
- Easton, E.B.; Qi, Z.; Kaufman, A.; Pickup, P.G. Chemical modification of proton exchange membrane fuel cell catalysts with a sulfonated silane. Electrochem. Solid-State Lett. 2001, 4, A59. [Google Scholar] [CrossRef]
- Xu, Z.; Qi, Z.; Kaufman, A. Advanced fuel cell catalysts: Sulfonation of carbon-supported catalysts using 2-aminoethanesulfonic acid. Electrochem. Solid-State Lett. 2003, 6, A171. [Google Scholar] [CrossRef]
- Xu, Z.; Qi, Z.; Kaufman, A. Superior catalysts for proton exchange membrane fuel cells: Sulfonation of carbon-supported catalysts using sulfate salts. Electrochem. Solid-State Lett. 2005, 8, A313. [Google Scholar] [CrossRef]
- Mizuhata, H.; Nakao, S.-i.; Yamaguchi, T. Morphological control of PEMFC electrode by graft polymerization of polymer electrolyte onto platinum-supported carbon black. J. Power Sources 2004, 138, 25–30. [Google Scholar] [CrossRef]
- Kuroki, H.; Yamaguchi, T. Nanoscale morphological control of anode electrodes by Grafting of Methylsulfonic acid groups onto Platinum–Ruthenium-supported carbon Blacks. J. Electrochem. Soc. 2006, 153, A1417. [Google Scholar] [CrossRef]
- Kim, H.; Lee, W.; Yoo, D. Functionalized carbon support with sulfonated polymer for direct methanol fuel cells. Electrochim. Acta 2007, 52, 2620–2624. [Google Scholar] [CrossRef]
- Huang, H.; Chen, H.; Sun, D.; Wang, X. Graphene nanoplate-Pt composite as a high performance electrocatalyst for direct methanol fuel cells. J. Power Sources 2012, 204, 46–52. [Google Scholar] [CrossRef]
- Bong, S.; Kim, Y.-R.; Kim, I.; Woo, S.; Uhm, S.; Lee, J.; Kim, H. Graphene supported electrocatalysts for methanol oxidation. Electrochem. Commun. 2010, 12, 129–131. [Google Scholar] [CrossRef]
- Dong, L.; Gari, R.R.S.; Li, Z.; Craig, M.M.; Hou, S. Graphene-supported platinum and platinum–ruthenium nanoparticles with high electrocatalytic activity for methanol and ethanol oxidation. Carbon 2010, 48, 781–787. [Google Scholar] [CrossRef]
- Yoo, E.; Okada, T.; Akita, T.; Kohyama, M.; Honma, I.; Nakamura, J. Sub-nano-Pt cluster supported on graphene nanosheets for CO tolerant catalysts in polymer electrolyte fuel cells. J. Power Sources 2011, 196, 110–115. [Google Scholar] [CrossRef]
- Guo, S.; Dong, S.; Wang, E. Three-dimensional Pt-on-Pd bimetallic nanodendrites supported on graphene nanosheet: Facile synthesis and used as an advanced nanoelectrocatalyst for methanol oxidation. ACS Nano 2010, 4, 547–555. [Google Scholar] [CrossRef]
- Wang, H.; Yu, H.; Peng, F.; Lv, P. Methanol electrocatalytic oxidation on highly dispersed Pt/sulfonated-carbon nanotubes catalysts. Electrochem. Commun. 2006, 8, 499–504. [Google Scholar] [CrossRef]
- Bonet, F.; Delmas, V.; Grugeon, S.; Urbina, R.H.; Silvert, P.; Tekaia-Elhsissen, K. Synthesis of monodisperse Au, Pt, Pd, Ru and Ir nanoparticles in ethylene glycol. Nanostructured Mater. 1999, 11, 1277–1284. [Google Scholar] [CrossRef]
- Wang, G.; Shen, X.; Wang, B.; Yao, J.; Park, J. Synthesis and characterisation of hydrophilic and organophilic graphene nanosheets. Carbon 2009, 47, 1359–1364. [Google Scholar] [CrossRef]
- Noda, L.K.; de Almeida, R.M.; Probst, L.F.D.; Gonçalves, N.S. Characterization of sulfated TiO2 prepared by the sol–gel method and its catalytic activity in the n-hexane isomerization reaction. J. Mol. Catal. A Chem. 2005, 225, 39–46. [Google Scholar] [CrossRef]
- Mangla, O.; Roy, S. Monoclinic zirconium oxide nanostructures having tunable band gap synthesized under extremely non-equilibrium plasma conditions. Multidisciplinary Digital Publishing Institute. Proceedings 2018, 3, 10. [Google Scholar] [CrossRef]
- Ding, S.; Zhao, J.; Yu, Q. Effect of zirconia polymorph on vapor-phase ketonization of propionic acid. Catalysts 2019, 9, 768. [Google Scholar] [CrossRef]
- Manoharan, D.; Loganathan, A.; Kurapati, V.; Nesamony, V.J. Unique sharp photoluminescence of size-controlled sonochemically synthesized zirconia nanoparticles. Ultrason. Sonochem. 2015, 23, 174–184. [Google Scholar] [CrossRef]
- Gregory, N. Elements of X-Ray Diffraction. J. Am. Chem. Soc. 1957, 79, 1773–1774. [Google Scholar] [CrossRef]
- Pozio, A.; De Francesco, M.; Cemmi, A.; Cardellini, F.; Giorgi, L. Comparison of high surface Pt/C catalysts by cyclic voltammetry. J. Power Sources 2002, 105, 13–19. [Google Scholar] [CrossRef]
- Ciureanu, M.; Wang, H. Electrochemical Impedance Study of Electrode-Membrane Assemblies in PEM Fuel Cells: I. Electro-oxidation of H2 and H2/CO Mixtures on Pt-Based Gas-Diffusion Electrodes. J. Electrochem. Soc. 1999, 146, 4031–4040. [Google Scholar] [CrossRef]
- Weaver, M.; Chang, S.-C.; Leung, L.-W.; Jiang, X.; Rubel, M.; Szklarczyk, M.; Zurawski, D.; Wieckowski, A. Evaluation of absolute saturation coverages of carbon monoxide on ordered low-index platinum and rhodium electrodes. J. Electroanal. Chem. 1992, 327, 247–260. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrocatalyst and Support Material | Synthesis Technique | Fuel Type | Electrochemical Assessment Methods | Results | Reference |
|---|---|---|---|---|---|
| Pt/S-ZrO2/MWCNT | NaBH4 reduction procedure | Methanol |
|
| [31] |
| Pt/S-TiO2/MWCNT | NaBH4 reduction procedure | Ethanol |
|
| [28] |
| Pt/ZrO2/C | NaBH4 reduction procedure | Ethanol |
|
| [23] |
| Pt/TiO2/CNT | Sol–gel and ethylene glycol reduction method | Ethanol |
|
| [29] |
| Pt/S-CNT | Ethylene glycol reduction method by refluxing | Methanol |
|
| [53] |
| Pt/S-ZrO2/GNPS | Microwave-assisted polyol synthesis method | Ethanol |
|
| Current work |
| Element | Line | Int | W% | A% |
|---|---|---|---|---|
| C | Ka | 452.7 | 60.14 | 83.42 |
| O | Ka | 73.2 | 10.29 | 10.71 |
| Na | Ka | 47.3 | 0.95 | 0.69 |
| Al | Ka | 183.9 | 2.43 | 1.50 |
| Si | Ka | 140.5 | 1.77 | 1.05 |
| S | Ka | 17.8 | 0.3 | 0.15 |
| Cl | Ka | 14.7 | 0.28 | 0.13 |
| K | Ka | 25.5 | 0.57 | 0.24 |
| Zr | Ka | 52.8 | 1.35 | 0.25 |
| Pt | Ka | 9.5 | 19.13 | 1.62 |
| Au | Ka | 1.8 | 2.8 | 0.24 |
| 100 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yaldagard, M.; Shahbaz, M.; Kim, H.W.; Kim, S.S. Ethanol Electro-Oxidation on Catalysts with S-ZrO2-Decorated Graphene as Support in Fuel Cell Applications. Nanomaterials 2022, 12, 3327. https://doi.org/10.3390/nano12193327
Yaldagard M, Shahbaz M, Kim HW, Kim SS. Ethanol Electro-Oxidation on Catalysts with S-ZrO2-Decorated Graphene as Support in Fuel Cell Applications. Nanomaterials. 2022; 12(19):3327. https://doi.org/10.3390/nano12193327
Chicago/Turabian StyleYaldagard, Maryam, Mehrdard Shahbaz, Hyoun Woo Kim, and Sang Sub Kim. 2022. "Ethanol Electro-Oxidation on Catalysts with S-ZrO2-Decorated Graphene as Support in Fuel Cell Applications" Nanomaterials 12, no. 19: 3327. https://doi.org/10.3390/nano12193327
APA StyleYaldagard, M., Shahbaz, M., Kim, H. W., & Kim, S. S. (2022). Ethanol Electro-Oxidation on Catalysts with S-ZrO2-Decorated Graphene as Support in Fuel Cell Applications. Nanomaterials, 12(19), 3327. https://doi.org/10.3390/nano12193327