
Electronic Structure of Monolayer FeSe on Si(001) from First Principles


Abstract
:1. Introduction
2. Methods
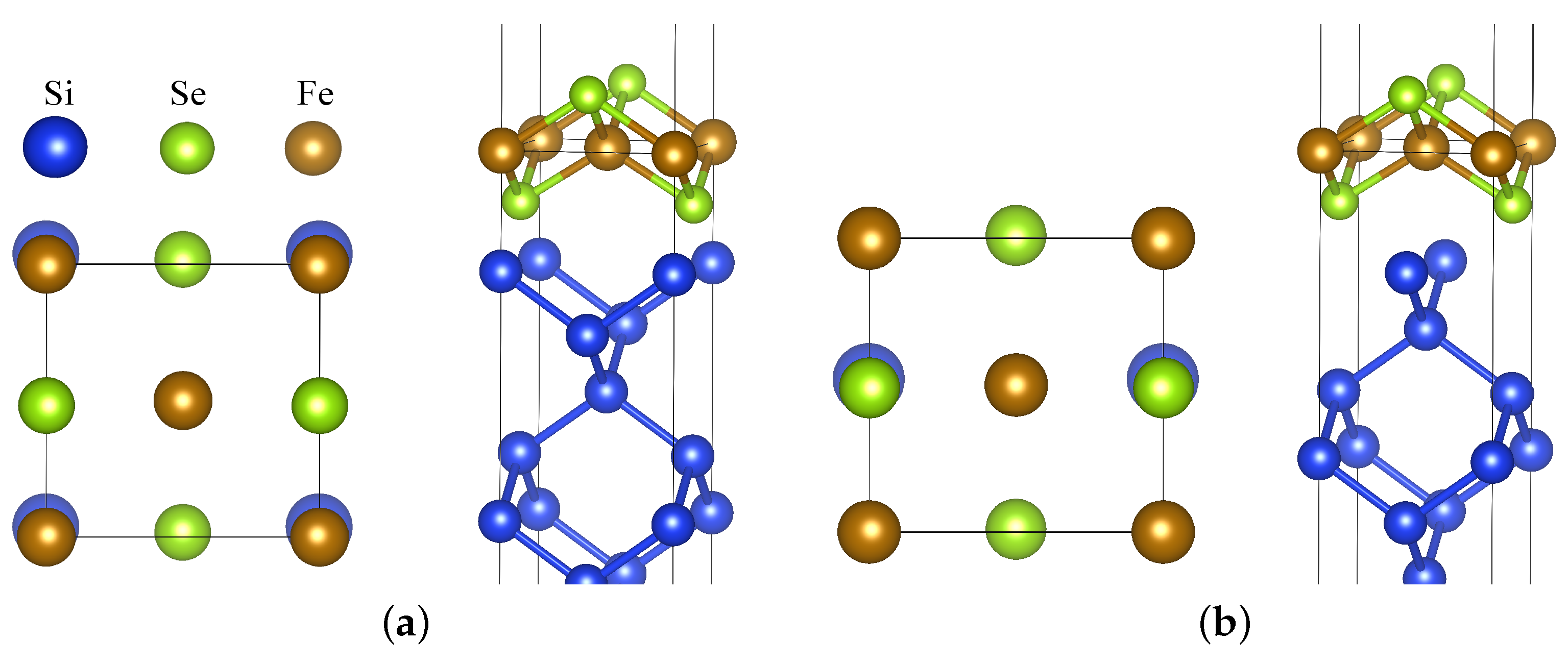
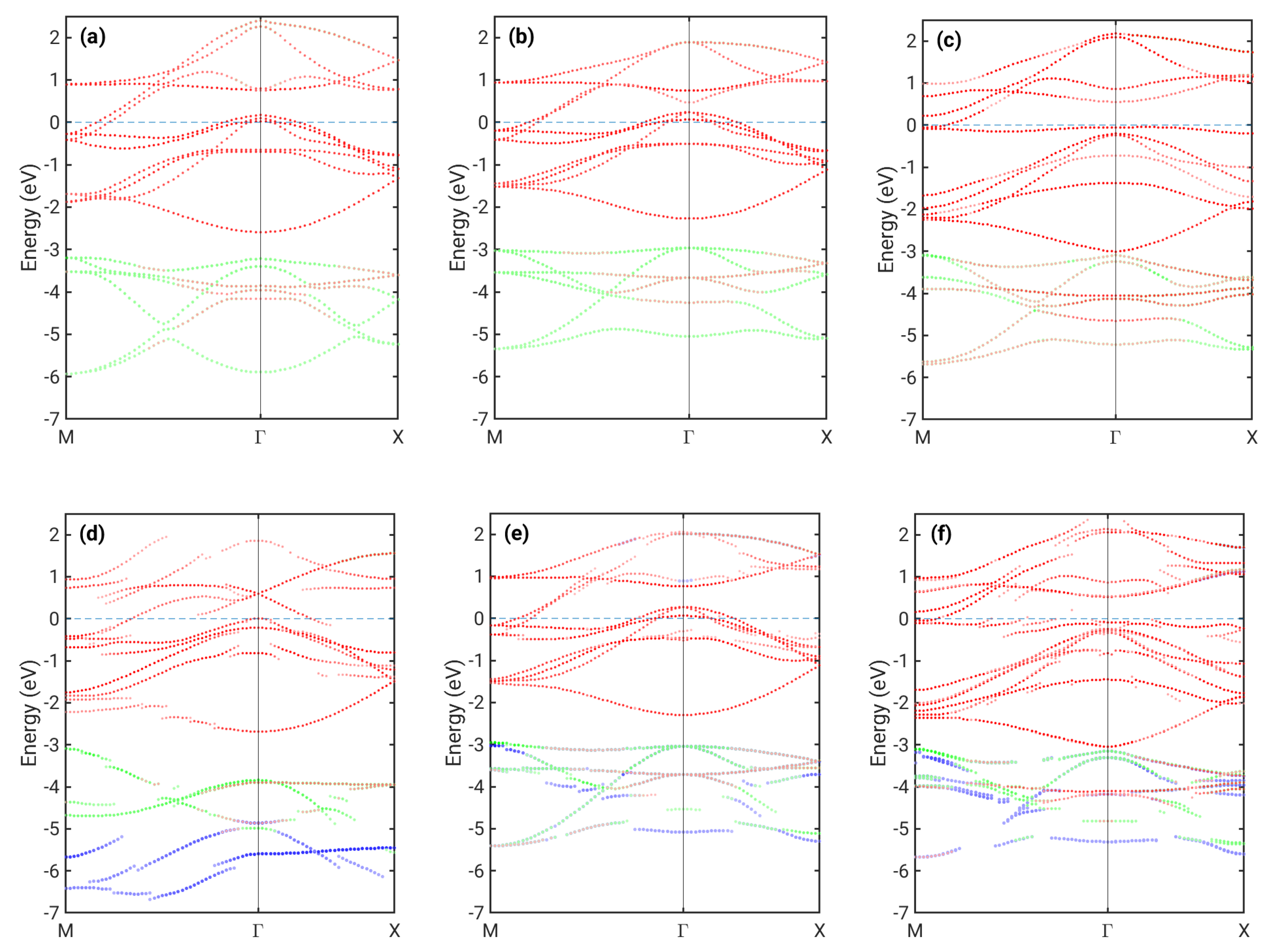
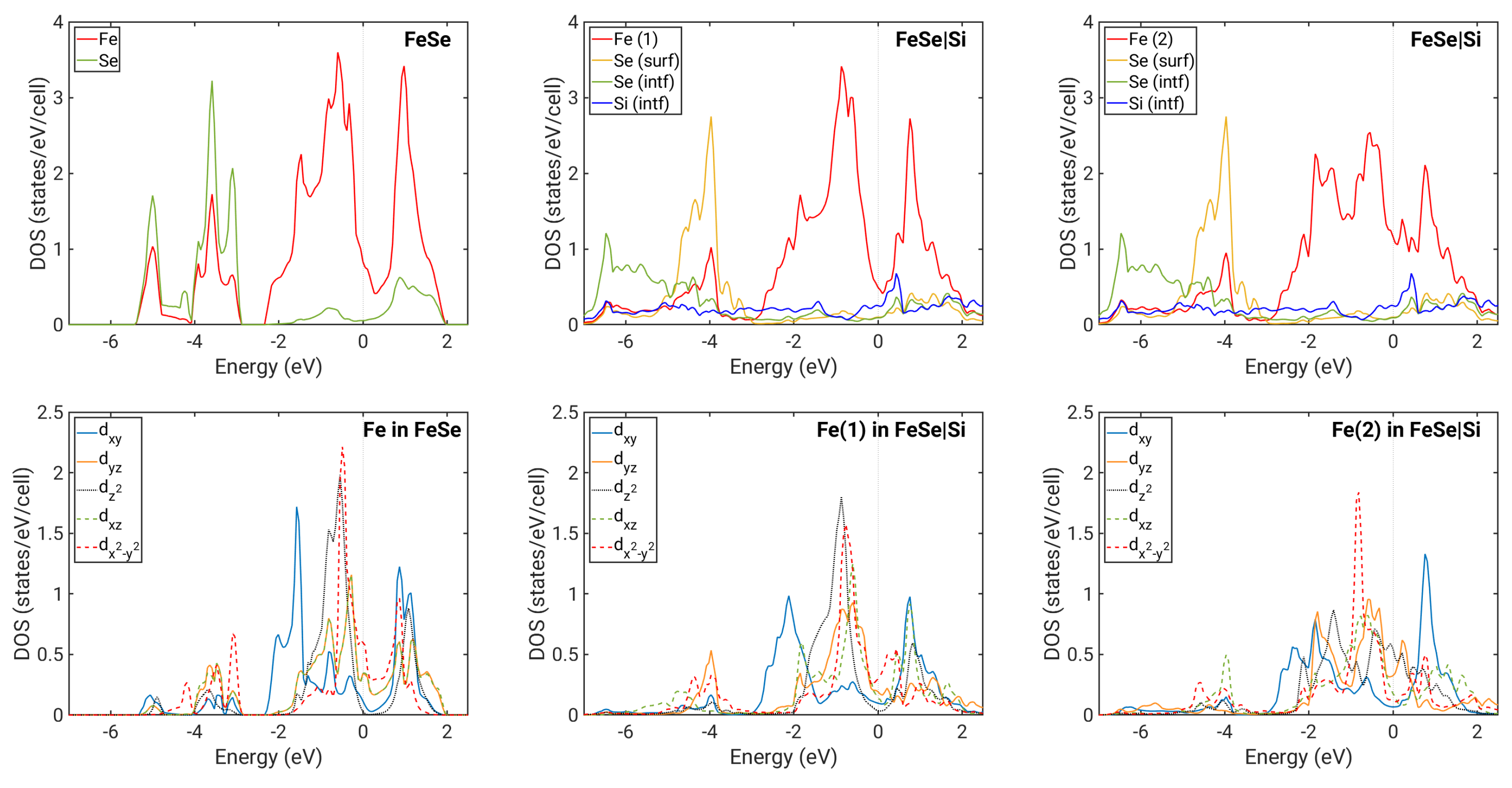
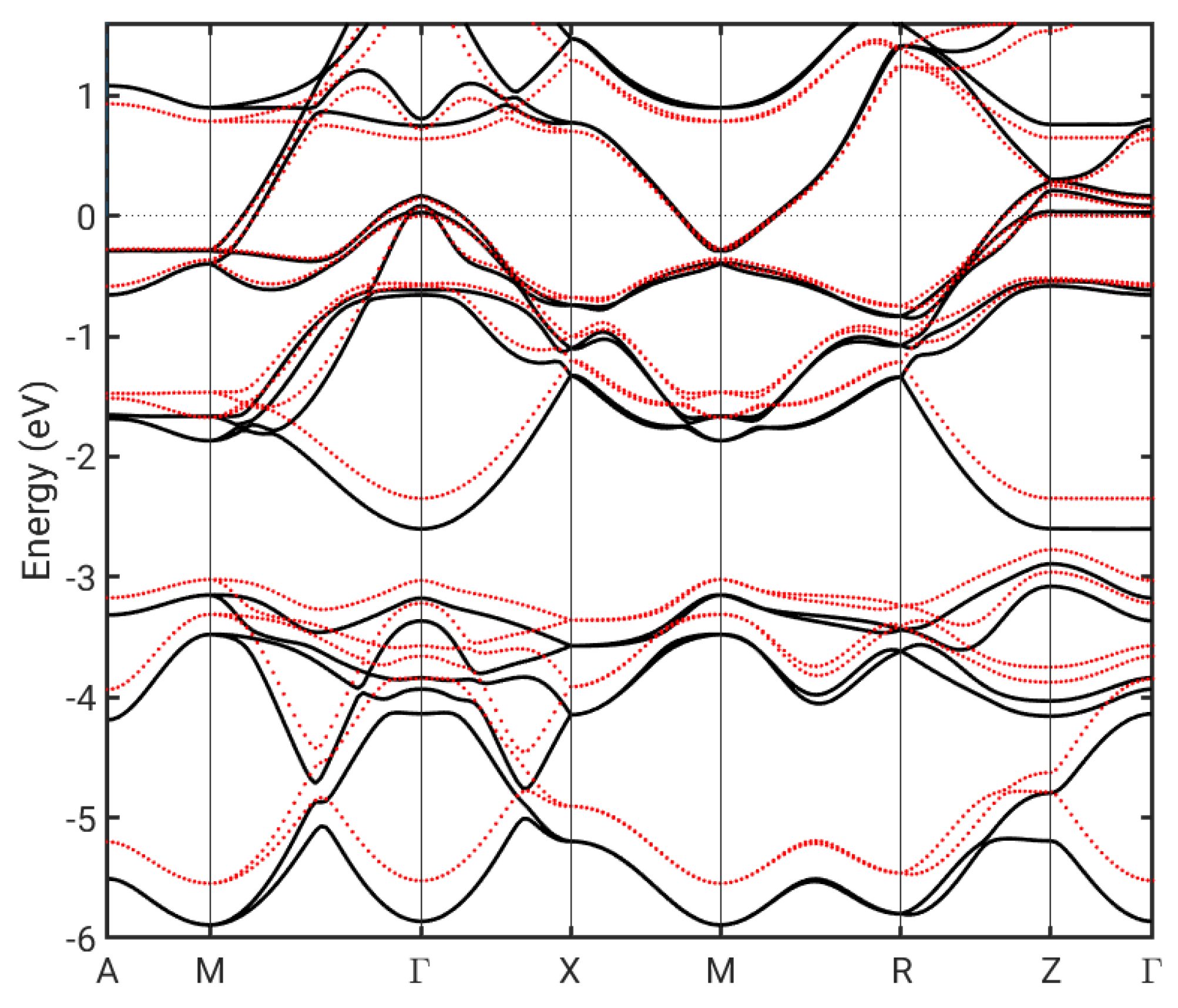
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NM | non-magnetic |
| AFM | anti-ferromagnetic |
| 2D | two-dimensional |
| DFT | density functional theory |
| ML | monolayer |
| SC | superconductor |
| GGA | generalized gradient approximation |
| PBE | Perdew–Burke–Ernzerhof |
| DOS | density of states |
References
- Liu, X.; Zhao, L.; He, S.; He, J.; Liu, D.; Mou, D.; Shen, B.; Hu, Y.; Huang, J.; Zhou, X.J. Electronic structure and superconductivity of FeSe-related superconductors. J. Phys. Condens. Matter 2015, 27, 183201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.C.; Luo, J.Y.; Yeh, K.W.; Chen, T.K.; Huang, T.W.; Wu, P.M.; Lee, Y.C.; Huang, Y.L.; Chu, Y.Y.; Yan, D.C.; et al. Superconductivity in the PbO-type structure -FeSe. Proc. Natl. Acad. Sci. USA 2008, 105, 14262–14264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.F.; Liu, Z.L.; Liu, C.; Gao, C.L.; Qian, D.; Xue, Q.K.; Liu, Y.; Jia, J.F. Superconductivity above 100 K in single-layer FeSe films on doped SrTiO3. Nat. Mater. 2014, 14, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Barends, R.; Fowler, A.G.; Megrant, A.; Jeffrey, E.; White, T.C.; Sank, D.; Mutus, J.Y.; Campbell, B.; Chen, Y.; et al. State preservation by repetitive error detection in a superconducting quantum circuit. Nature 2015, 519, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Arute, F.; Arya, K.; Babbush, R.; Bacon, D.; Bardin, J.C.; Barends, R.; Biswas, R.; Boixo, S.; Brandao, F.G.S.L.; Buell, D.A.; et al. Quantum supremacy using a programmable superconducting processor. Nature 2019, 574, 505–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyatti, M.; Wolff, M.A.; Gundareva, I.; Kruth, M.; Ferrari, S.; Dunin-Borkowski, R.E.; Schuck, C. Energy-level quantization and single-photon control of phase slips in YBa2Cu3O7–x nanowires. Nat. Commun. 2020, 11, 763. [Google Scholar] [CrossRef] [Green Version]
- Hirschfeld, P.J.; Korshunov, M.M.; Mazin, I.I. Gap symmetry and structure of Fe-based superconductors. Rep. Prog. Phys. 2011, 74, 124508. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Nakayama, K.; Sugawara, K.; Sato, T.; Takahashi, T. High-temperature superconductivity in potassium-coated multilayer FeSe thin films. Nat. Mater. 2015, 14, 775–779. [Google Scholar] [CrossRef]
- Shi, X.; Han, Z.Q.; Peng, X.L.; Richard, P.; Qian, T.; Wu, X.X.; Qiu, M.W.; Wang, S.C.; Hu, J.P.; Sun, Y.J.; et al. Enhanced superconductivity accompanying a Lifshitz transition in electron-doped FeSe monolayer. Nat. Commun. 2017, 8, 14988. [Google Scholar] [CrossRef] [Green Version]
- Kordyuk, A.A. Iron-based superconductors: Magnetism, superconductivity, and electronic structure (Review Article). Low Temp. Phys. 2012, 38, 888–899. [Google Scholar] [CrossRef] [Green Version]
- Coldea, A.I.; Watson, M.D. The Key Ingredients of the Electronic Structure of FeSe. Annu. Rev. Condens. Matter Phys. 2018, 9, 125–146. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lu, Z.Y.; Xiang, T. Atomic and electronic structures of FeSe monolayer and bilayer thin films on SrTiO3(001): First-principles study. Phys. Rev. B 2012, 85, 235123. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Ji, W.; Hu, J.; Lu, Z.Y.; Xiang, T. First-Principles Calculations of the Electronic Structure of Tetragonal α-FeTe and α-FeSe Crystals: Evidence for a Bicollinear Antiferromagnetic Order. Phys. Rev. Lett. 2009, 102, 177003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazhirov, T.; Cohen, M.L. Effects of charge doping and constrained magnetization on the electronic structure of an FeSe monolayer. J. Phys. Condens. Matter 2013, 25, 105506. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Wang, Z.; Kang, W.; Zhang, P. Antiferromagnetic FeSe monolayer on SrTiO3: The charge doping and electric field effects. Sci. Rep. 2013, 3, 2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Zhang, B.J.; Lu, Z.Y. First-principles study of magnetic frustration in FeSe epitaxial films onSrTiO3. Phys. Rev. B 2015, 91, 045107. [Google Scholar] [CrossRef] [Green Version]
- Glasbrenner, J.K.; Mazin, I.I.; Jeschke, H.O.; Hirschfeld, P.J.; Fernandes, R.M.; Valentí, R. Effect of magnetic frustration on nematicity and superconductivity in iron chalcogenides. Nat. Phys. 2015, 11, 953–958. [Google Scholar] [CrossRef]
- Zhou, Y.; Miao, L.; Wang, P.; Zhu, F.; Jiang, W.; Jiang, S.; Zhang, Y.; Lei, B.; Chen, X.; Ding, H.; et al. Antiferromagnetic Order in Epitaxial FeSe Films on SrTiO3. Phys. Rev. Lett. 2018, 120, 097001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fikáček, J.; Procházka, P.; Stetsovych, V.; Průša, S.; Vondráček, M.; Kormoš, L.; Skála, T.; Vlaic, P.; Caha, O.; Carva, K.; et al. Step-edge assisted large scale FeSe monolayer growth on epitaxial Bi2Se3 thin films. New J. Phys. 2020, 22, 073050. [Google Scholar] [CrossRef]
- Manna, S.; Kamlapure, A.; Cornils, L.; Hänke, T.; Hedegaard, E.M.J.; Bremholm, M.; Iversen, B.B.; Hofmann, P.; Wiebe, J.; Wiesendanger, R. Interfacial superconductivity in a bi-collinear antiferromagnetically ordered FeTe monolayer on a topological insulator. Nat. Commun. 2017, 8, 14074. [Google Scholar] [CrossRef] [Green Version]
- The ELK Code. Available online: http://elk.sourceforge.net/ (accessed on 1 December 2021).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, R.M.; Vafek, O. Distinguishing spin-orbit coupling and nematic order in the electronic spectrum of iron-based superconductors. Phys. Rev. B 2014, 90, 214514. [Google Scholar] [CrossRef] [Green Version]
- Borisenko, S.V.; Evtushinsky, D.V.; Liu, Z.H.; Morozov, I.; Kappenberger, R.; Wurmehl, S.; Büchner, B.; Yaresko, A.N.; Kim, T.K.; Hoesch, M.; et al. Direct observation of spin–orbit coupling in iron-based superconductors. Nat. Phys. 2015, 12, 311–317. [Google Scholar] [CrossRef]
- dos Santos, R.B.; Rivelino, R.; Gueorguiev, G.K.; Kakanakova-Georgieva, A. Exploring 2D structures of indium oxide of different stoichiometry. CrystEngComm 2021, 23, 6661–6667. [Google Scholar] [CrossRef]
- Freitas, R.R.Q.; de Brito Mota, F.; Rivelino, R.; de Castilho, C.M.C.; Kakanakova-Georgieva, A.; Gueorguiev, G.K. Tuning band inversion symmetry of buckled III-Bi sheets by halogenation. Nanotechnology 2016, 27, 055704. [Google Scholar] [CrossRef] [PubMed]
- Haastrup, S.; Strange, M.; Pandey, M.; Deilmann, T.; Schmidt, P.S.; Hinsche, N.F.; Gjerding, M.N.; Torelli, D.; Larsen, P.M.; Riis-Jensen, A.C.; et al. The Computational 2D Materials Database: High-throughput modeling and discovery of atomically thin crystals. 2D Mater. 2018, 5, 042002. [Google Scholar] [CrossRef]
- Sims, H.; Leonard, D.N.; Birenbaum, A.Y.; Ge, Z.; Berlijn, T.; Li, L.; Cooper, V.R.; Chisholm, M.F.; Pantelides, S.T. Intrinsic interfacial van der Waals monolayers and their effect on the high-temperature superconductor FeSe/SrTiO3. Phys. Rev. B 2019, 100, 144103. [Google Scholar] [CrossRef] [Green Version]
- VESTA - Visualization for Electronic and STructural Analysis. Available online: https://jp-minerals.org/vesta (accessed on 3 December 2021).
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Jiang, H.; Zhao, T.; Ren, Y.; Zhang, R.; Wu, M. Ab initio prediction and characterization of phosphorene-like SiS and SiSe as anode materials for sodium-ion batteries. Sci. Bull. 2017, 62, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Subedi, A.; Zhang, L.; Singh, D.J.; Du, M.H. Density functional study of FeS, FeSe, and FeTe: Electronic structure, magnetism, phonons, and superconductivity. Phys. Rev. B 2008, 78, 134514. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Miyata, Y.; Phan, G.; Sato, T.; Tanabe, Y.; Urata, T.; Tanigaki, K.; Takahashi, T. Reconstruction of Band Structure Induced by Electronic Nematicity in an FeSe Superconductor. Phys. Rev. Lett. 2014, 113, 237001. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.D.; Kim, T.K.; Haghighirad, A.A.; Davies, N.R.; McCollam, A.; Narayanan, A.; Blake, S.F.; Chen, Y.L.; Ghannadzadeh, S.; Schofield, A.J.; et al. Emergence of the nematic electronic state in FeSe. Phys. Rev. B 2015, 91, 155106. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Zhang, Y.; Xia, M.; Ye, Z.; Chen, F.; Xie, X.; Peng, R.; Xu, D.; Fan, Q.; Xu, H.; et al. Interface-induced superconductivity and strain-dependent spin density waves in FeSe/SrTiO3 thin films. Nat. Mater. 2013, 12, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.Y.; Tan, S.; Xiang, H.; Feng, D.L.; Gong, X.G. Interfacial effects on the spin density wave in FeSe/SrTiO3 thin films. Phys. Rev. B 2014, 89, 014501. [Google Scholar] [CrossRef] [Green Version]
- Linscheid, A.; Maiti, S.; Wang, Y.; Johnston, S.; Hirschfeld, P. HighTcvia Spin Fluctuations from Incipient Bands: Application to Monolayers and Intercalates of FeSe. Phys. Rev. Lett. 2016, 117, 077003. [Google Scholar] [CrossRef] [Green Version]
- Berlijn, T.; Cheng, H.P.; Hirschfeld, P.J.; Ku, W. Doping effects of Se vacancies in monolayer FeSe. Phys. Rev. B 2014, 89, 020501. [Google Scholar] [CrossRef] [Green Version]
- Coh, S.; Lee, D.H.; Louie, S.G.; Cohen, M.L. Proposal for a bulk material based on a monolayer FeSe onSrTiO3 high-temperature superconductor. Phys. Rev. B 2016, 93, 245138. [Google Scholar] [CrossRef] [Green Version]
- Castelvecchi, D. Quantum computers ready to leap out of the lab in 2017. Nature 2017, 541, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Wang, X.; Zhang, H.; Yuan, S. Reactive Molecular Dynamics on the Oxidation of H–Si(100) Surface: Effect of Humidity and Temperature. J. Phys. Chem. C 2019, 124, 1932–1940. [Google Scholar] [CrossRef]
- Bozovic, I.; Ahn, C. A new frontier for superconductivity. Nat. Phys. 2014, 10, 892–895. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe in free-standing FeSe | |||||
| Fe(1) in FeSe on Si, IC1 | |||||
| Fe(2) in FeSe on Si, IC1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carva, K.; Vlaic, P.; Honolka, J. Electronic Structure of Monolayer FeSe on Si(001) from First Principles. Nanomaterials 2022, 12, 270. https://doi.org/10.3390/nano12020270
Carva K, Vlaic P, Honolka J. Electronic Structure of Monolayer FeSe on Si(001) from First Principles. Nanomaterials. 2022; 12(2):270. https://doi.org/10.3390/nano12020270
Chicago/Turabian StyleCarva, Karel, Petru Vlaic, and Jan Honolka. 2022. "Electronic Structure of Monolayer FeSe on Si(001) from First Principles" Nanomaterials 12, no. 2: 270. https://doi.org/10.3390/nano12020270
APA StyleCarva, K., Vlaic, P., & Honolka, J. (2022). Electronic Structure of Monolayer FeSe on Si(001) from First Principles. Nanomaterials, 12(2), 270. https://doi.org/10.3390/nano12020270