
The Adsorption Mechanism of Hydrogen on FeO Crystal Surfaces: A Density Functional Theory Study



 ,
, 
Abstract
:1. Introduction
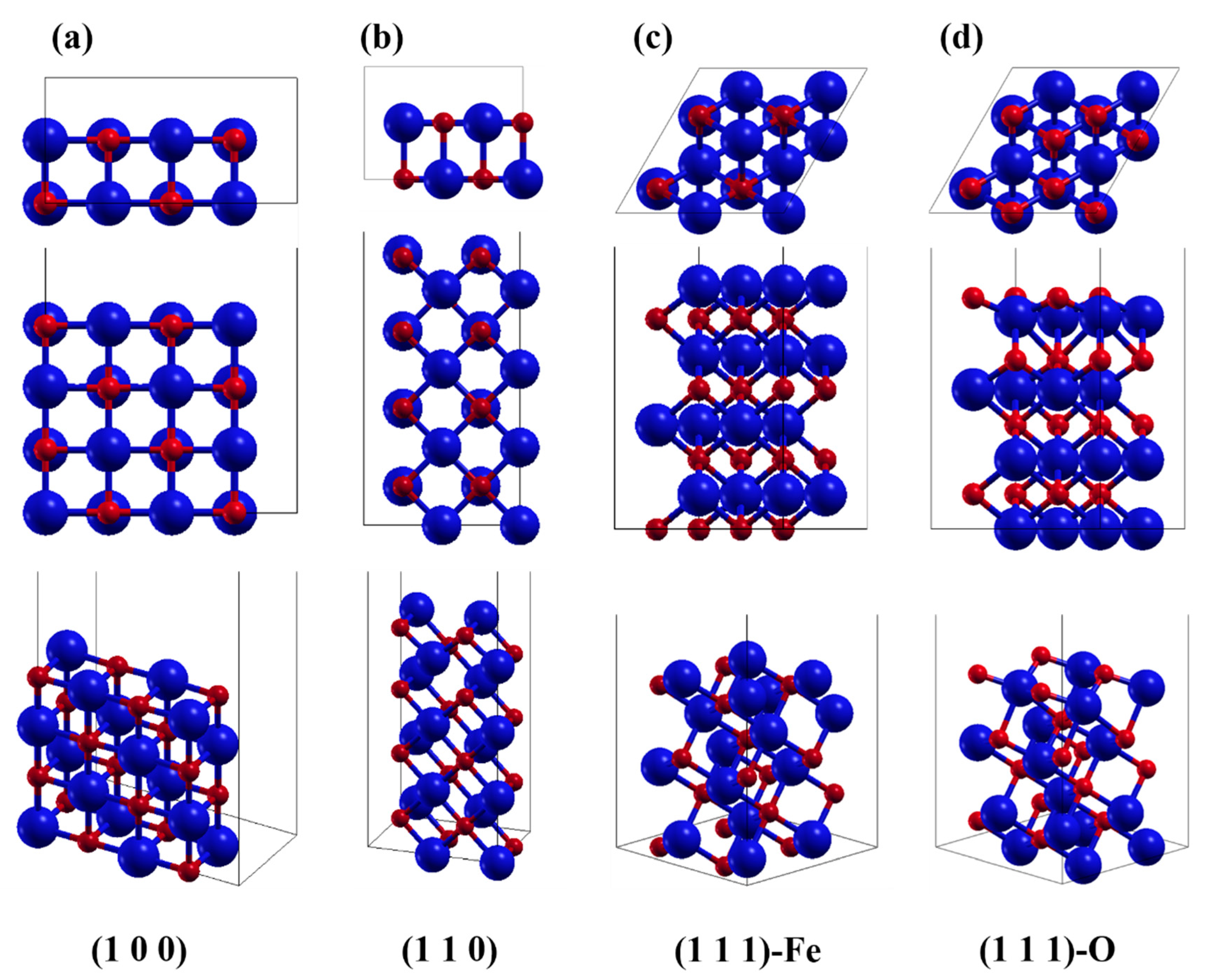
2. Computational Models and Methods
2.1. Surface Energy Calculation
2.2. Adsorption Energy Calculation
2.3. Single-Point Energy Distribution Calculation
2.4. Transition State Calculation
3. Results and Discussion
3.1. Crystal Surface Energy Analysis
3.2. Distribution of Surface Energy with a Single H Atom on Top of the FeO Surfaces
3.3. Bonding Adsorption of H on FeO Surface Site
3.4. Hydrogen Adsorption on FeO Surfaces
3.5. H2 Dissociation and Adsorption on FeO Surfaces
4. Summary
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, Y.; Raipala, K.; Holappa, L. Chapter 1.1—Ironmaking. In Treatise on Process Metallurgy; Seetharaman, S., Ed.; Elsevier: Boston, MA, USA, 2014; pp. 2–88. [Google Scholar]
- Perpiñán, J.; Bailera, M.; Romeo, L.M.; Peña, B.; Eveloy, V. CO2 Recycling in the Iron and Steel Industry via Power-to-Gas and Oxy-Fuel Combustion. Energies 2021, 14, 7090. [Google Scholar] [CrossRef]
- Perpiñán, J.; Peña, B.; Bailera, M.; Eveloy, V.; Kannan, P.; Raj, A.; Lisbona, P.; Romeo, L.M. Integration of carbon capture technologies in blast furnace based steel making: A comprehensive and systematic review. Fuel 2023, 336, 127074. [Google Scholar] [CrossRef]
- Pei, M.; Petäjäniemi, M.; Regnell, A.; Wijk, O. Toward a Fossil Free Future with HYBRIT: Development of Iron and Steelmaking Technology in Sweden and Finland. Metals 2020, 10, 972. [Google Scholar] [CrossRef]
- Zhong, H.; Wen, L.; Li, J.; Xu, J.; Hu, M.; Yang, Z. The adsorption behaviors of CO and H2 on FeO surface: A density functional theory study. Powder Technol. 2016, 303, 100–108. [Google Scholar] [CrossRef]
- Lu, F.; Wen, L.; Zhao, Y.; Zhong, H.; Xu, J.; Zhang, S.; Yang, Z. The competitive adsorption behavior of CO and H2 molecules on FeO surface in the reduction process. Int. J. Hydrogen Energy 2019, 44, 6427–6436. [Google Scholar] [CrossRef]
- Raabe, D. The Materials Science behind Sustainable Metals and Alloys. Chem. Rev. 2023, 123, 2436–2608. [Google Scholar] [CrossRef]
- Lingiardi, O.; Burrai, O.; Fuentealba, C.G.; Etchevarne, P.; Gonzalez, J.M. Natural Gas Injection at Siderar #2 Blast Furnace. In Proceedings of the Ironmaking Conference Proceedings, Chicago, IL, USA, 21–24 March 1999; Volume 58, pp. 135–141. [Google Scholar]
- Knop, K.; Kubiak, H. Process engineering and cost efficiency of steelmaking by means of hydrogen-based direct reduction. Stahl Eisen 1996, 116, 55–64. [Google Scholar]
- Shen, Z.; Xu, J.; Liu, H.; Liang, Q. Modeling study for the effect of particle size on char gasification with CO2. AIChE J. 2017, 63, 716–724. [Google Scholar] [CrossRef]
- Liu, W.; Zuo, H.; Wang, J.; Xue, Q.; Ren, B.; Yang, F. The production and application of hydrogen in steel industry. Int. J. Hydrogen Energy 2021, 46, 10548–10569. [Google Scholar] [CrossRef]
- Germeshuizen, L.M.; Blom, P.W.E. A techno-economic evaluation of the use of hydrogen in a steel production process, utilizing nuclear process heat. Int. J. Hydrogen Energy 2013, 38, 10671–10682. [Google Scholar] [CrossRef]
- Zhang, J.; Schenk, J.; Liu, Z.; Li, K. Editorial for special issue on hydrogen metallurgy. Int. J. Miner. Metall. Mater. 2022, 29, 1817–1819. [Google Scholar] [CrossRef]
- Ohno, K.-I.; Maeda, T.; Kunitomo, K.; Hara, M. Effect of FeO concentration in sinter iron ore on reduction behavior in a hydrogen-enriched blast furnace. Int. J. Miner. Metall. Mater. 2022, 29, 1820–1829. [Google Scholar] [CrossRef]
- Lan, C.; Zhang, S.; Liu, X.; Lyu, Q.; Jiang, M. Change and mechanism analysis of the softening-melting behavior of the iron-bearing burden in a hydrogen-rich blast furnace. Int. J. Hydrogen Energy 2020, 45, 14255–14265. [Google Scholar] [CrossRef]
- Fridman, A. Plasma Chemistry; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Zvetkov, Y.V.; Panfilov, S. Low-Temperature Plasma in Reduction Processes; Nauka: Moscow, Russia, 1980. [Google Scholar]
- Sabat, K.C.; Rajput, P.; Paramguru, R.K.; Bhoi, B.; Mishra, B.K. Reduction of Oxide Minerals by Hydrogen Plasma: An Overview. Plasma Chem. Plasma Process. 2014, 34, 1–23. [Google Scholar] [CrossRef]
- Kamiya, K.; Kitahara, N.; Morinaka, I.; Sakuraya, K.; Ozawa, M.; Tanaka, M. Reduction of Molten Iron Oxide and FeO Bearing Slags by H2-Ar Plasma. Trans. Iron Steel Inst. Jpn. 1984, 24, 7–16. [Google Scholar] [CrossRef]
- Degout, D.; Kassabji, F.; Fauchais, P. Titanium dioxide plasma treatment. Plasma Chem. Plasma Process. 1984, 4, 179–198. [Google Scholar] [CrossRef]
- Kitamura, T.; Shibata, K.; Takeda, K. In-flight reduction of Fe2O3, Cr2O3, TiO2 and Al2O3 by Ar-H2 and Ar-CH4 plasma. ISIJ Int. 1993, 33, 1150–1158. [Google Scholar] [CrossRef] [Green Version]
- Mohai, I.; Szépvölgyi, J.; Karoly, Z.; Mohai, M.; Toth, M.; Babievskaya, I.; Krenev, V. Reduction of metallurgical wastes in an RF thermal plasma reactor. Plasma Chem. Plasma Process. 2001, 21, 547–563. [Google Scholar] [CrossRef]
- Gilles, H.L.; Clump, C.W. Reduction of iron ore with hydrogen in a direct current plasma jet. Ind. Eng. Chem. Process Des. Dev. 1970, 9, 194–207. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ito, M.; Ishikawa, H. Reduction and dephosphorization of molten iron oxide with hydrogen-argon plasma. Plasma Chem. Plasma Process. 1981, 1, 149–160. [Google Scholar] [CrossRef]
- Meng, Y.; Liu, X.-Y.; Bai, M.-M.; Chen, J.; Ma, Y.-J.; Wen, X.-D. Adsorption or deoxidation of H2 interacted with Fe3O4 surface under different H coverage: A DFT study. Appl. Surf. Sci. 2020, 502, 144097. [Google Scholar] [CrossRef]
- Yilmaz, C.; Wendelstorf, J.; Turek, T. Modeling and simulation of hydrogen injection into a blast furnace to reduce carbon dioxide emissions. J. Clean. Prod. 2017, 154, 488–501. [Google Scholar] [CrossRef]
- Ma, Y.; Souza Filho, I.R.; Bai, Y.; Schenk, J.; Patisson, F.; Beck, A.; van Bokhoven, J.A.; Willinger, M.G.; Li, K.; Xie, D.; et al. Hierarchical nature of hydrogen-based direct reduction of iron oxides. Scr. Mater. 2022, 213, 114571. [Google Scholar] [CrossRef]
- Souza Filho, I.R.; Springer, H.; Ma, Y.; Mahajan, A.; da Silva, C.C.; Kulse, M.; Raabe, D. Green steel at its crossroads: Hybrid hydrogen-based reduction of iron ores. J. Clean. Prod. 2022, 340, 130805. [Google Scholar] [CrossRef]
- Ma, Y.; Souza Filho, I.R.; Zhang, X.; Nandy, S.; Barriobero-Vila, P.; Requena, G.; Vogel, D.; Rohwerder, M.; Ponge, D.; Springer, H.; et al. Hydrogen-based direct reduction of iron oxide at 700 °C: Heterogeneity at pellet and microstructure scales. Int. J. Miner. Metall. Mater. 2022, 29, 1901–1907. [Google Scholar] [CrossRef]
- Khanchandani, H.; Stephenson, L.T.; Raabe, D.; Zaefferer, S.; Gault, B. Hydrogen/Deuterium Charging Methods for the Investigation of Site-Specific Microstructural Features by Atom Probe Tomography. Microsc. Microanal. 2022, 28, 1664. [Google Scholar] [CrossRef]
- Bai, Y.; Mianroodi, J.R.; Ma, Y.; da Silva, A.K.; Svendsen, B.; Raabe, D. Chemo-mechanical phase-field modeling of iron oxide reduction with hydrogen. Acta Mater. 2022, 231, 117899. [Google Scholar] [CrossRef]
- Li, Z.; Huang, W. Reactivity of hydrogen species on oxide surfaces. Sci. China Chem. 2021, 64, 1076–1087. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, W.; Zhang, Y.; Wu, Z.; Chen, B.; Jiang, Z.; Ma, Y.; Yang, J.; Huang, W. Oxygen Vacancy-Controlled Reactivity of Hydroxyls on an FeO(111) Monolayer Film. J. Phys. Chem. C 2011, 115, 6815–6824. [Google Scholar] [CrossRef]
- Xu, L.; Wu, Z.; Zhang, W.; Jin, Y.; Yuan, Q.; Ma, Y.; Huang, W. Oxygen Vacancy-Induced Novel Low-Temperature Water Splitting Reactions on FeO(111) Monolayer-Thick Film. J. Phys. Chem. C 2012, 116, 22921–22929. [Google Scholar] [CrossRef]
- Neufeld, O.; Caspary Toroker, M. Play the heavy: An effective mass study for α-Fe2O3 and corundum oxides. J. Chem. Phys. 2016, 144, 432–449. [Google Scholar] [CrossRef]
- Pekdur, Z.S.; Ydrm, S.Z.; Büyükmumcu, Z. Synthesis and Thermal Properties of Magnetite Nano Structures and DFT Analysis of Fe3O4 Cluster as Its Smallest Representative Unit. J. Mol. Struct. 2020, 1222, 128895. [Google Scholar] [CrossRef]
- Liu, S.; Xiang, D.; Xu, Y.; Sun, Z.; Cao, Y. Relationship between electronic properties of Fe3O4 substituted by Ca and Ba and their reactivity in chemical looping process: A first-principles study. Appl. Energy 2017, 202, 550–557. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, W.; Chang, X.; Ding, D.; Sun, L. DFT insights to mercury species mechanism on pure and Mn doped Fe3O4(111) surfaces. Appl. Surf. Sci. 2020, 514, 145876. [Google Scholar] [CrossRef]
- Santos-Carballal, D.; Roldan, A.; Grau-Crespo, R.; de Leeuw, N.H. A DFT study of the structures, stabilities and redox behaviour of the major surfaces of magnetite Fe3O4. Phys. Chem. Chem. Phys. 2014, 16, 21082–21097. [Google Scholar] [CrossRef] [Green Version]
- Tran, F.; Blaha, P.; Schwarz, K.; Novak, P. Hybrid exchange-correlation energy functionals for strongly correlated electrons: Applications to transition-metal monoxides. Phys. Review. B Condens. Matter Mater. Phys. 2006, 74, 155108. [Google Scholar] [CrossRef] [Green Version]
- Ossowski, T.; Wang, Y.; Carraro, G.; Kiejna, A.; Lewandowski, M. Structure of mono- and bilayer FeO on Ru(0001): STM and DFT study. J. Magn. Magn. Mater. 2022, 546, 168832. [Google Scholar] [CrossRef]
- Giordano, L.; Pacchioni, G.; Goniakowski, J.; Nilius, N.; Freund, H.J. Interplay between structural, magnetic, and electronic properties in a FeO/Pt(111) ultrathin film. Phys. Rev. B 2007, 37, 075416. [Google Scholar] [CrossRef] [Green Version]
- Rowan, A.D.; Patterson, C.H.; Gasparov, L.V. Hybrid density functional theory applied to magnetite: Crystal structure, charge order, and phonons. Phys. Rev. B Condens. Matter 2009, 79, 205103. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.J.; Chang, C.F.; Jeng, H.T.; Guo, G.Y.; Lin, H.J.; Wu, W.B.; Ku, H.C.; Fujimori, A.; Takahashi, Y.; Chen, C.T. Spin and orbital magnetic moments of Fe3O4. Phys. Rev. Lett. 2004, 93, 077204. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Z. A DFT-based microkinetic theory for Fe2O3 reduction by CO in chemical looping. Proc. Combust. Inst. 2022, 39, 4447–4455. [Google Scholar] [CrossRef]
- Gavarri, J.-R.; Carel, C. The complex nonstoichiometry of wüstite Fe1−xO: Review and comments. Prog. Solid State Chem. 2019, 53, 27–49. [Google Scholar] [CrossRef]
- He, S.; Sun, H.; Hu, C.; Li, J.; Zhu, Q.; Li, H.-Z. Direct reduction of fine iron ore concentrate in a conical fluidized bed. Powder Technol. 2017, 313, 161–168. [Google Scholar] [CrossRef]
- Aireddy, D.R.; Ding, K. Heterolytic Dissociation of H2 in Heterogeneous Catalysis. ACS Catal. 2022, 12, 4707–4723. [Google Scholar] [CrossRef]
- Zhang, S.; Li, K.; Ma, Y.; Guo, F.; Jiang, C.; Liang, Z.; Bu, Y.; Zhang, J. Density Functional Studies on the Atomistic Structure and Properties of Iron Oxides: A Parametric Study. Materials 2022, 15, 8316. [Google Scholar] [CrossRef]
- Zheng, X.; Paul, S.; Moghimi, L.; Wang, Y.; Vil’a, R.A.; Zhang, F.; Gao, X.; Deng, J.; Jiang, Y.; Xiao, X.; et al. Correlating Chemical Reaction and Mass Transport in Hydrogen-based Direct Reduction of Iron Oxide. arXiv 2023, arXiv:2302.14215. [Google Scholar]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I. Quantum ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Scandolo, S.; Giannozzi, P.; Cavazzoni, C.; Gironcoli, S.D.; Pasquarello, A.; Baroni, S. First-principles codes for computational crystallography in the Quantum-ESPRESSO package. Z. Für Krist. 2005, 220, 574–579. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Piccinin, S. The band structure and optical absorption of hematite (α-Fe2O3): A first-principles GW-BSE study. Phys. Chem. Chem. Phys. 2019, 21, 2957–2967. [Google Scholar] [CrossRef]
- Eglitis, R.I. Ab initio hybrid DFT calculations of BaTiO3, PbTiO3, SrZrO3 and PbZrO3 (111) surfaces. Appl. Surf. Sci. 2015, 358, 556–562. [Google Scholar] [CrossRef]
- Tian, X.; Wang, T.; Fan, L.; Wang, Y.; Lu, H.; Mu, Y. A DFT based method for calculating the surface energies of asymmetric MoP facets. Appl. Surf. Sci. 2018, 427, 357–362. [Google Scholar] [CrossRef]
- Robertson, A.W.; Lee, G.-D.; He, K.; Yoon, E.; Kirkland, A.I.; Warner, J.H. Stability and Dynamics of the Tetravacancy in Graphene. Nano Lett. 2014, 14, 1634–1642. [Google Scholar] [CrossRef]
- Zheng, P.; Zhang, X.; Duan, Y.; Yan, M.; Chapman, R.; Jiang, Y.; Li, H. Oxidation of graphene with variable defects: Alternately symmetrical escape and self-restructuring of carbon rings. Nanoscale 2020, 12, 10140–10148. [Google Scholar] [CrossRef]
- Hashimoto, A.; Suenaga, K.; Gloter, A.; Urita, K.; Iijima, S. Direct evidence for atomic defects in graphene layers. Nature 2004, 430, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.-Q.; Shi, J.-J.; Yang, M.; Zhang, S.; Zhang, M. Magnetism induced by D3-symmetry tetra-vacancy defects in graphene. Chem. Phys. Lett. 2011, 510, 246–251. [Google Scholar] [CrossRef]
- Yamashita, K.; Saito, M.; Oda, T. Atomic geometry and stability of mono-, di-, and trivacancies in graphene. Jpn. J. Appl. Phys. 2006, 45, 6534. [Google Scholar] [CrossRef]
- Hwang, D.G.; Jeong, E.; Lee, S.G. Density functional theory study of CH4 and CO2 adsorption by fluorinated graphene. Carbon Lett. 2016, 20, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Tit, N.; Said, K.; Mahmoud, N.M.; Kouser, S.; Yamani, Z.H. Ab-initio investigation of adsorption of CO and CO2 molecules on graphene: Role of intrinsic defects on gas sensing. Appl. Surf. Sci. 2017, 394, 219–230. [Google Scholar] [CrossRef]
- Meng, Y.; Liu, X.-W.; Bai, M.; Guo, W.-P.; Cao, D.-B.; Yang, Y.; Li, Y.-W.; Wen, X.-D. Prediction on morphologies and phase equilibrium diagram of iron oxides nanoparticles. Appl. Surf. Sci. 2019, 480, 478–486. [Google Scholar] [CrossRef]
- Demaison, J.; Herman, M.; Liévin, J. The equilibrium OH bond length. Int. Rev. Phys. Chem. 2007, 26, 391–420. [Google Scholar] [CrossRef]
- Li, S.-Y.; Zhao, W.-M.; Qiao, J.-H.; Wang, Y. Competitive adsorption of CO and H2 on strained Fe(110) surface. Acta Phys. Sin. 2019, 68, 217103. [Google Scholar] [CrossRef]
- Jr, N.B.A.; Kasai, H.; Diño, W.A.; Nakanishi, H. Potential Energy of H2 Dissociation and Adsorption on Pt(111) Surface: First-Principles Calculation. Jpn. J. Appl. Phys. 2007, 46, 4233. [Google Scholar] [CrossRef]
- Cheng, Q.; Conejo, A.N.; Wang, Y.; Zhang, J.; Zheng, A.; Liu, Z. Adsorption properties of hydrogen with iron oxides (FeO, Fe2O3): A ReaxFF molecular dynamics study. Comput. Mater. Sci. 2023, 218, 111926. [Google Scholar] [CrossRef]
- Staykov, A.T.; Yamabe, J.; Somerday, B.p. Effect of hydrogen gas impurities on the hydrogen dissociation on iron surface. Int. J. Quantum Chem. 2014, 114, 626–635. [Google Scholar] [CrossRef]
- Wang, T.; Tian, X.; Yang, Y.; Li, Y.-W.; Wang, J.; Beller, M.; Jiao, H. Co-adsorption and mutual interaction of nCO+mH2 on the Fe(110) and Fe(111) surfaces. Catal. Today 2016, 261, 82–92. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. First-principles study of the adsorption of atomic H on Ni (111), (100) and (110). Surf. Sci. 2000, 459, 287–302. [Google Scholar] [CrossRef]
- Li, Z.; Huang, W. Hydride species on oxide catalysts. J. Phys. Condens. Matter 2021, 33, 433001. [Google Scholar] [CrossRef]
- Matz, O.; Calatayud, M. H2 Dissociation and Oxygen Vacancy Formation on Ce2O3 Surfaces. Top. Catal. 2019, 62, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Nobuhara, K.; Kasai, H.; Diño, W.A.; Nakanishi, H. H2 dissociative adsorption on Mg, Ti, Ni, Pd and La Surfaces. Surf. Sci. 2004, 566–568, 703–707. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | (1 0 0) | (1 1 0) | (1 1 1)-Fe | (1 1 1)-O |
|---|---|---|---|---|
| Surface energy (J/m2) | 0.6871 | 1.4070 | 2.3993 | 1.3817 |
| Surface | Energy/eV | H Bonding Length/Å | |
|---|---|---|---|
| Fetop | (1 0 0) | −1.8288 | 1.582 |
| (1 1 0) | −2.4052 | 1.562 | |
| (1 1 1)-Fe | −2.2609 | 1.557 | |
| Otop | (1 0 0) | −2.7816 | 0.982 |
| (1 1 0) | −3.0680 | 0.979 | |
| (1 1 1)-O | −3.7424 | 0.972 |
| Horizontal | Perpendicular | |||||
|---|---|---|---|---|---|---|
| H2-Fetop | H2-Otop | Bridge | Hole | H2-Fetop | H2-Otop | |
| (1 0 0) | −0.5249 | −0.4838 | −0.5017 | −0.4957 | −0.5036 | |
| (1 1 0) | −0.5194 | −0.4268 | −0.4854 | −0.4607 | ||
| (1 1 1)-O | −0.1937 | −0.2400 | −0.2167 | |||
| (1 1 1)-Fe | −0.4781 | −0.4381 | −0.4660 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Li, K.; Ma, Y.; Bu, Y.; Liang, Z.; Yang, Z.; Zhang, J. The Adsorption Mechanism of Hydrogen on FeO Crystal Surfaces: A Density Functional Theory Study. Nanomaterials 2023, 13, 2051. https://doi.org/10.3390/nano13142051
Zhang S, Li K, Ma Y, Bu Y, Liang Z, Yang Z, Zhang J. The Adsorption Mechanism of Hydrogen on FeO Crystal Surfaces: A Density Functional Theory Study. Nanomaterials. 2023; 13(14):2051. https://doi.org/10.3390/nano13142051
Chicago/Turabian StyleZhang, Shujie, Kejiang Li, Yan Ma, Yushan Bu, Zeng Liang, Zonghao Yang, and Jianliang Zhang. 2023. "The Adsorption Mechanism of Hydrogen on FeO Crystal Surfaces: A Density Functional Theory Study" Nanomaterials 13, no. 14: 2051. https://doi.org/10.3390/nano13142051
APA StyleZhang, S., Li, K., Ma, Y., Bu, Y., Liang, Z., Yang, Z., & Zhang, J. (2023). The Adsorption Mechanism of Hydrogen on FeO Crystal Surfaces: A Density Functional Theory Study. Nanomaterials, 13(14), 2051. https://doi.org/10.3390/nano13142051