
The Scale Effects of Organometal Halide Perovskites

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Crystal Structure of Organometal Halide Perovskite
3. Synthetic Techniques
3.1. Single Crystals
3.2. Polycrystalline Films
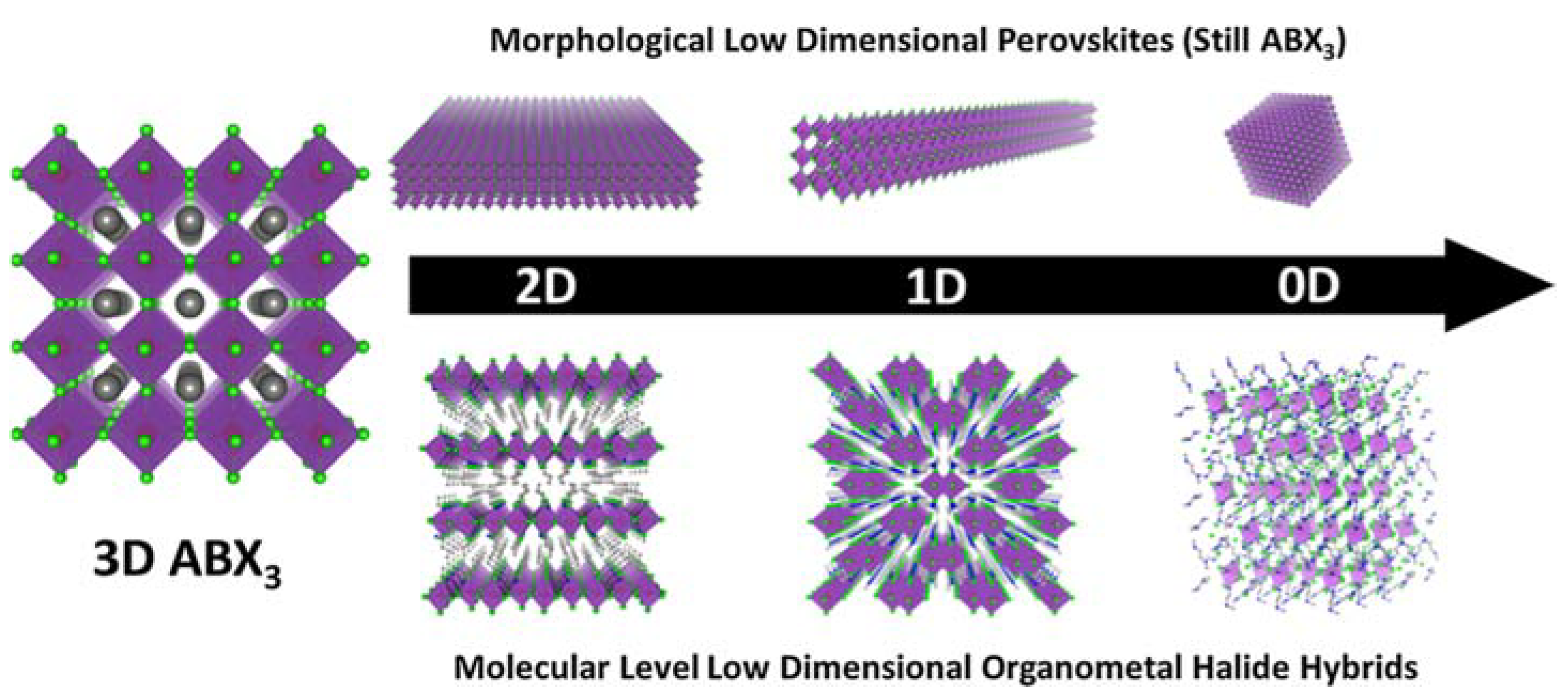
3.3. Morphological Low-Dimensional Perovskites

3.4. Molecular-Level Low-Dimensional Perovskites
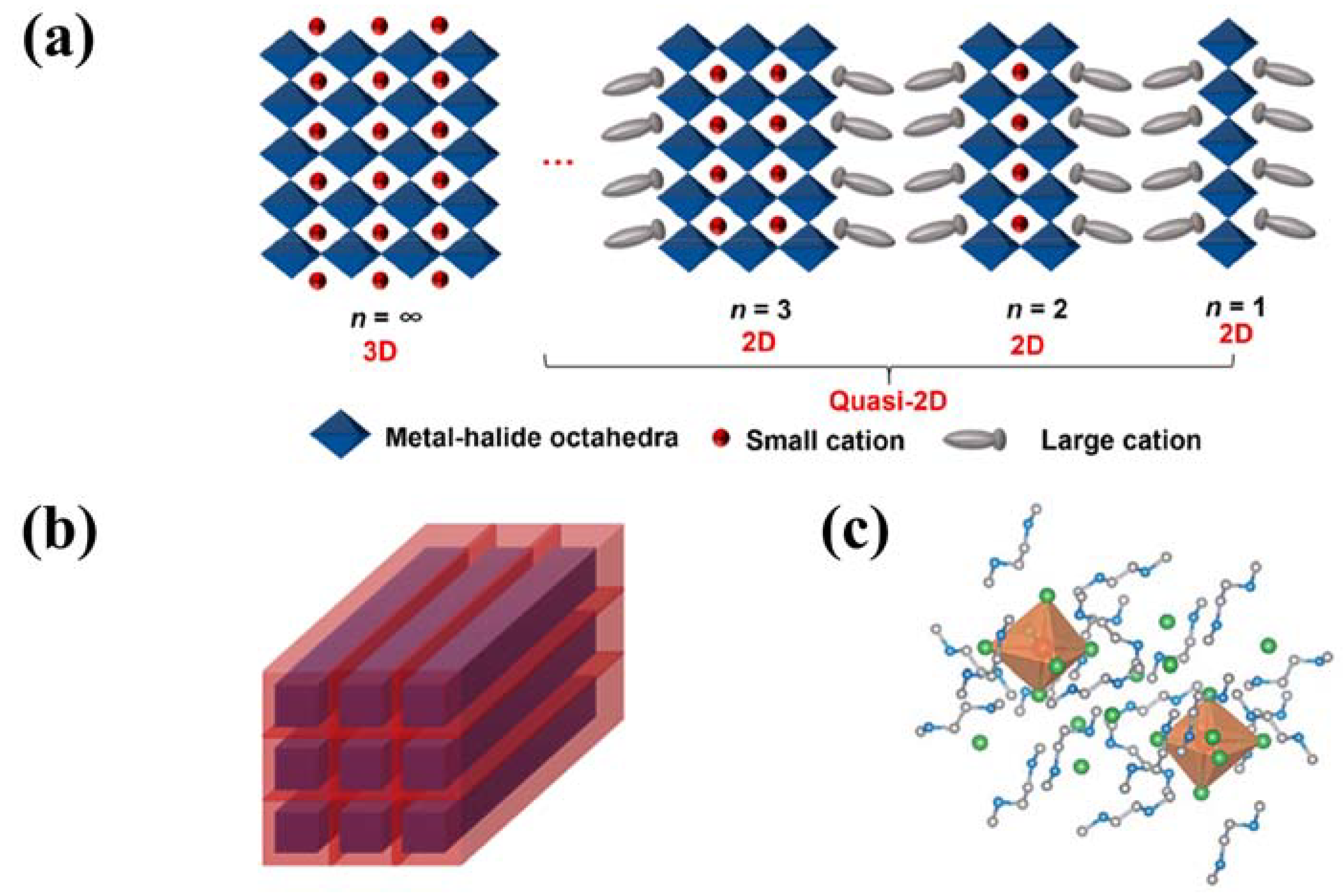
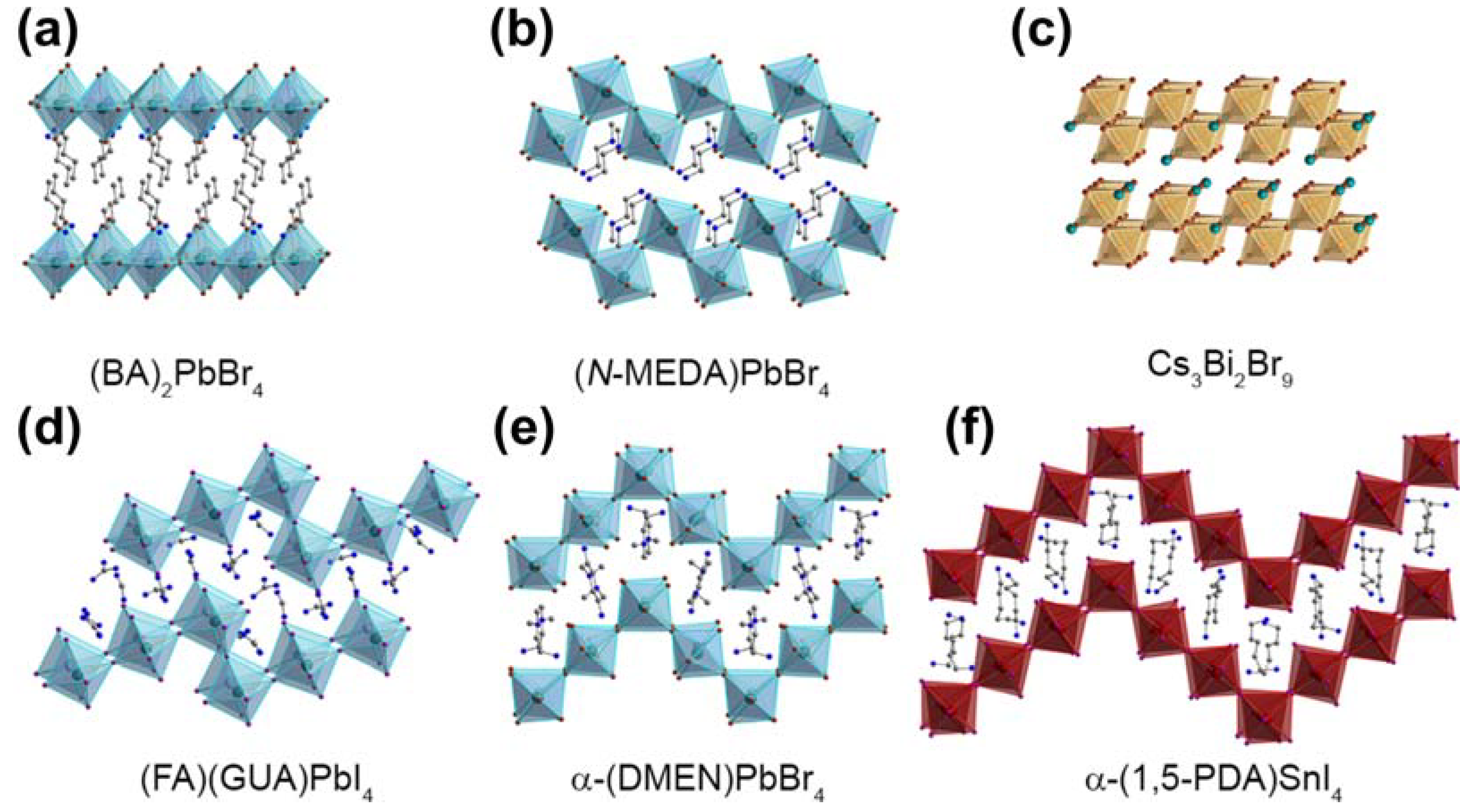
3.4.1. Two-Dimensional Metal Halide Hybrids
3.4.2. Three-Dimensional Metal Halide Hybrids
3.4.3. Zero-Dimensional Metal Halide Hybrids
3.5. Device-Oriented Synthesis
4. Scale-Dependent Effects in Organometal Halide Perovskites
4.1. Scale Effects of Perovskites with Morphological Difference
4.1.1. Carriers Diffusion
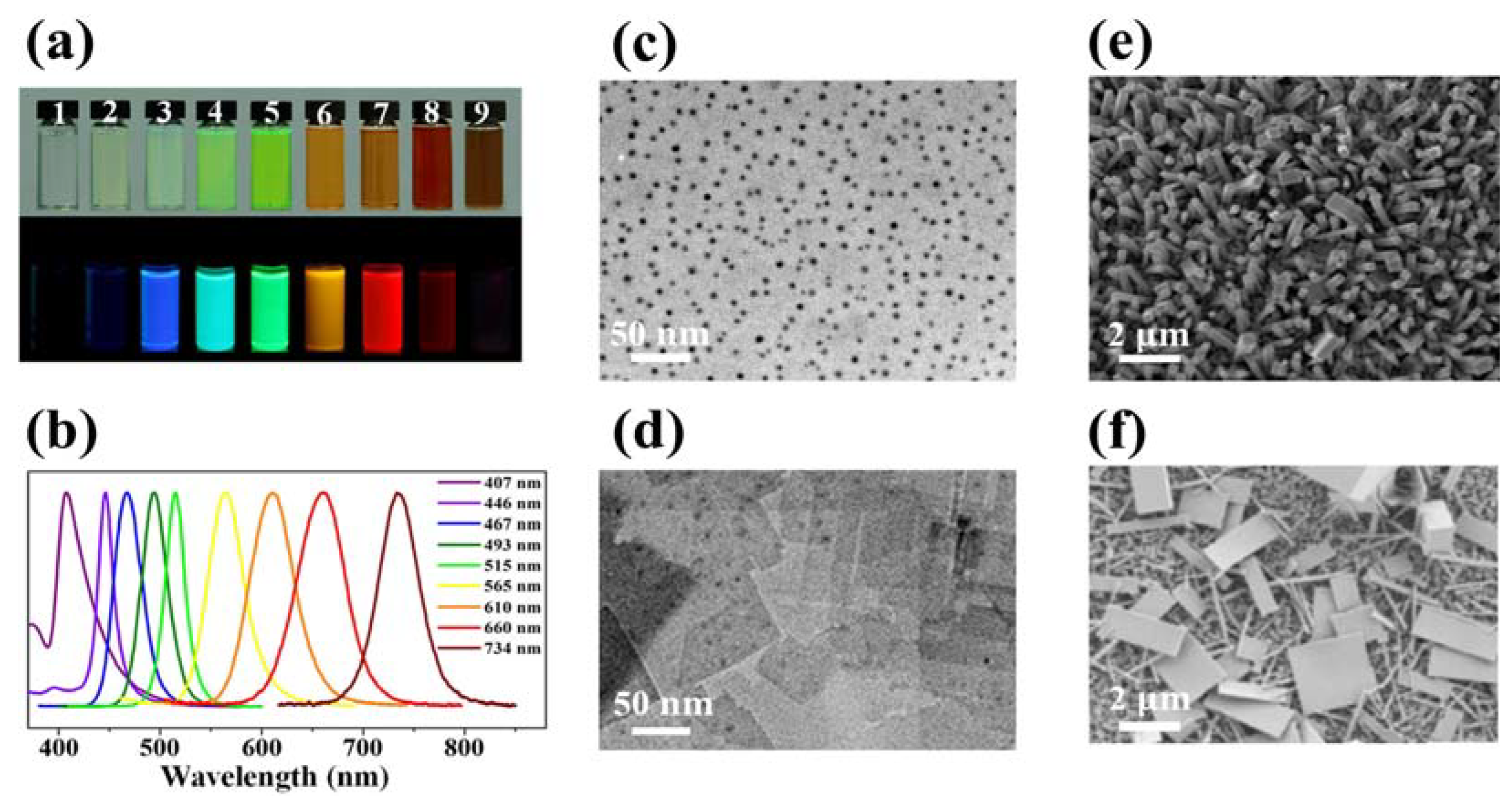
4.1.2. Excitonic Features

4.2. Scale Effects of Perovskites with Molecular-Level Difference
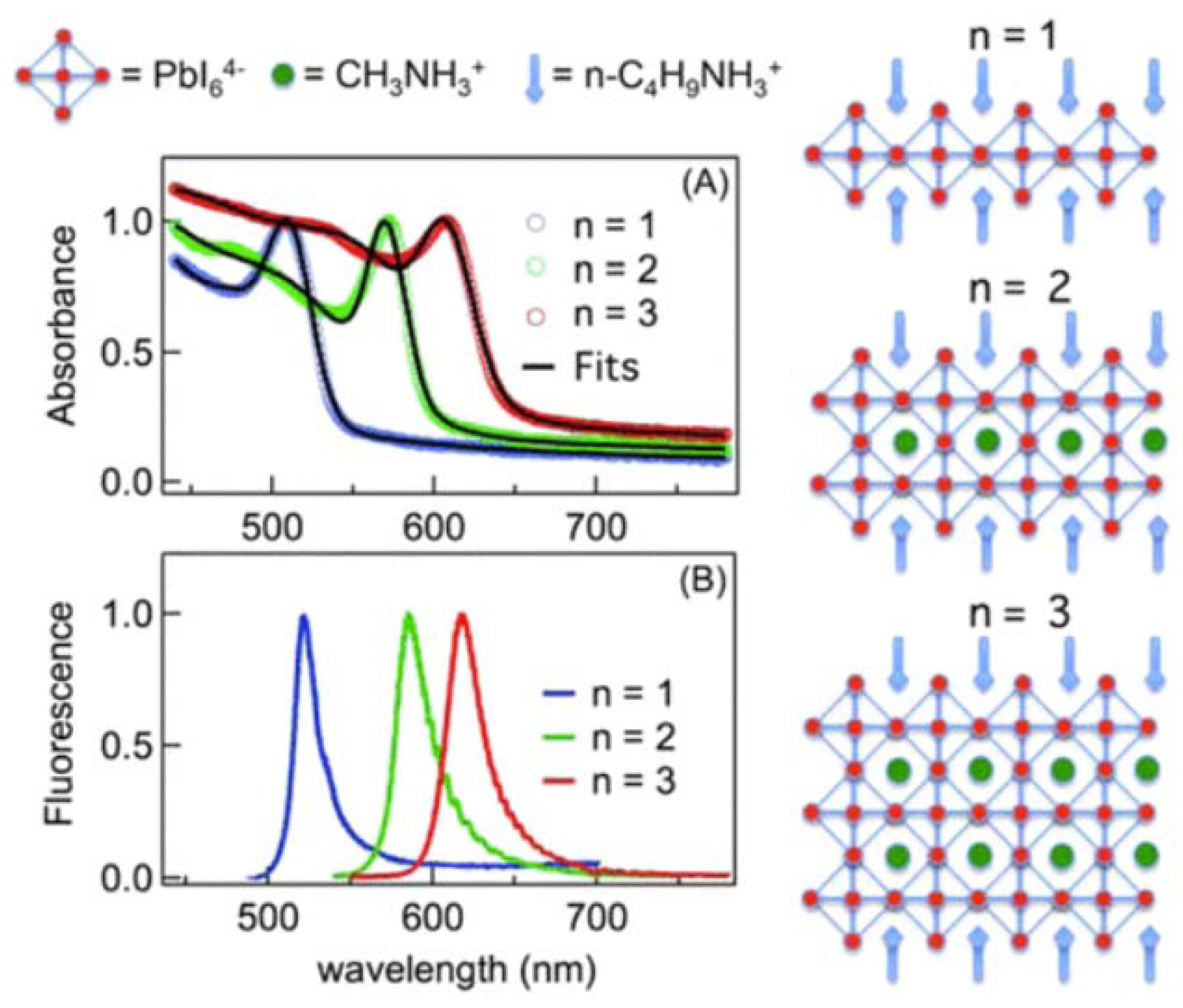
4.2.1. Two-Dimensional and Quasi-Two-Dimensional Metal Halide Hybrids
4.2.2. One-Dimensional Metal Halide Hybrids
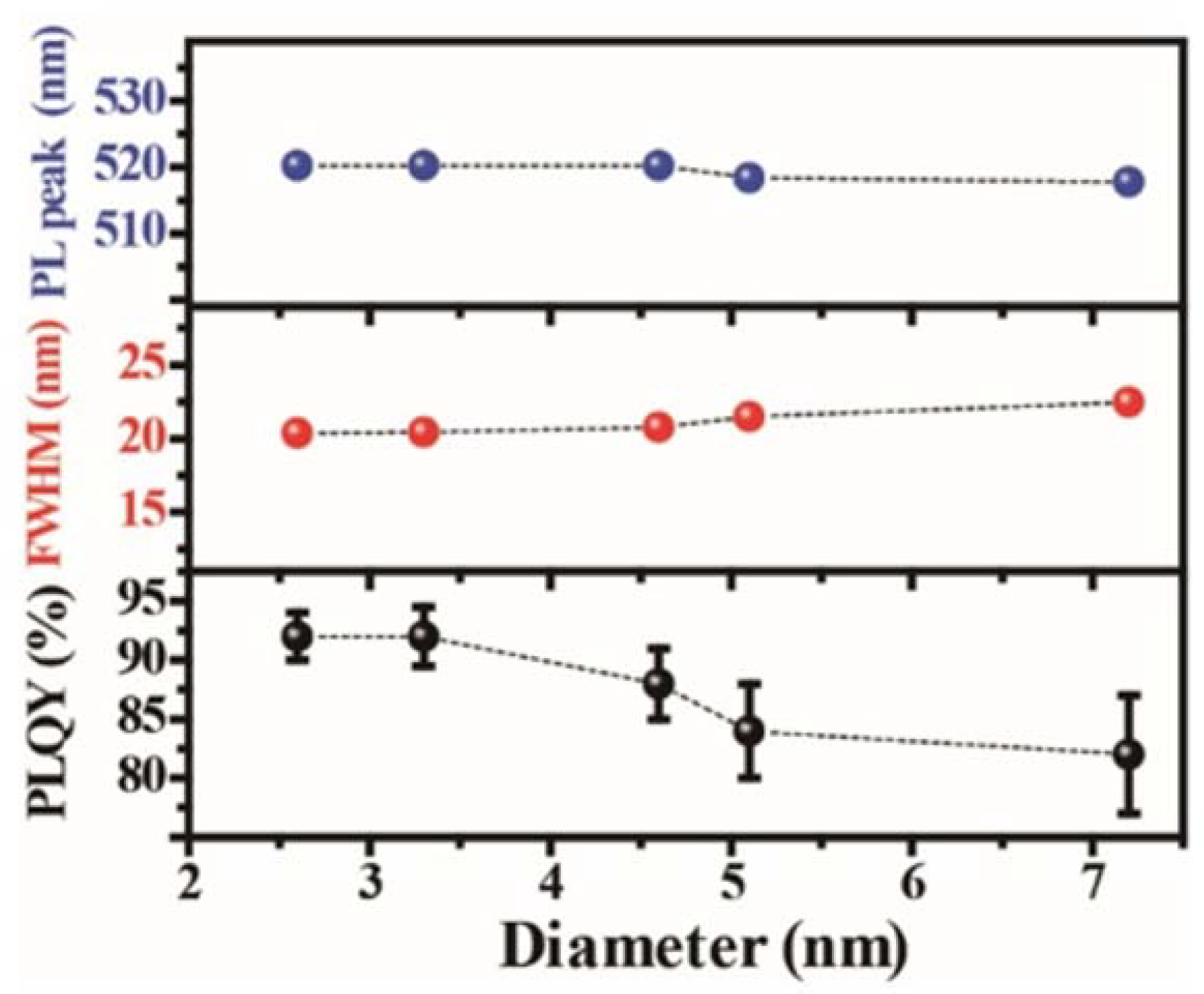
4.2.3. Zero-Dimensional Metal Halide Hybrids
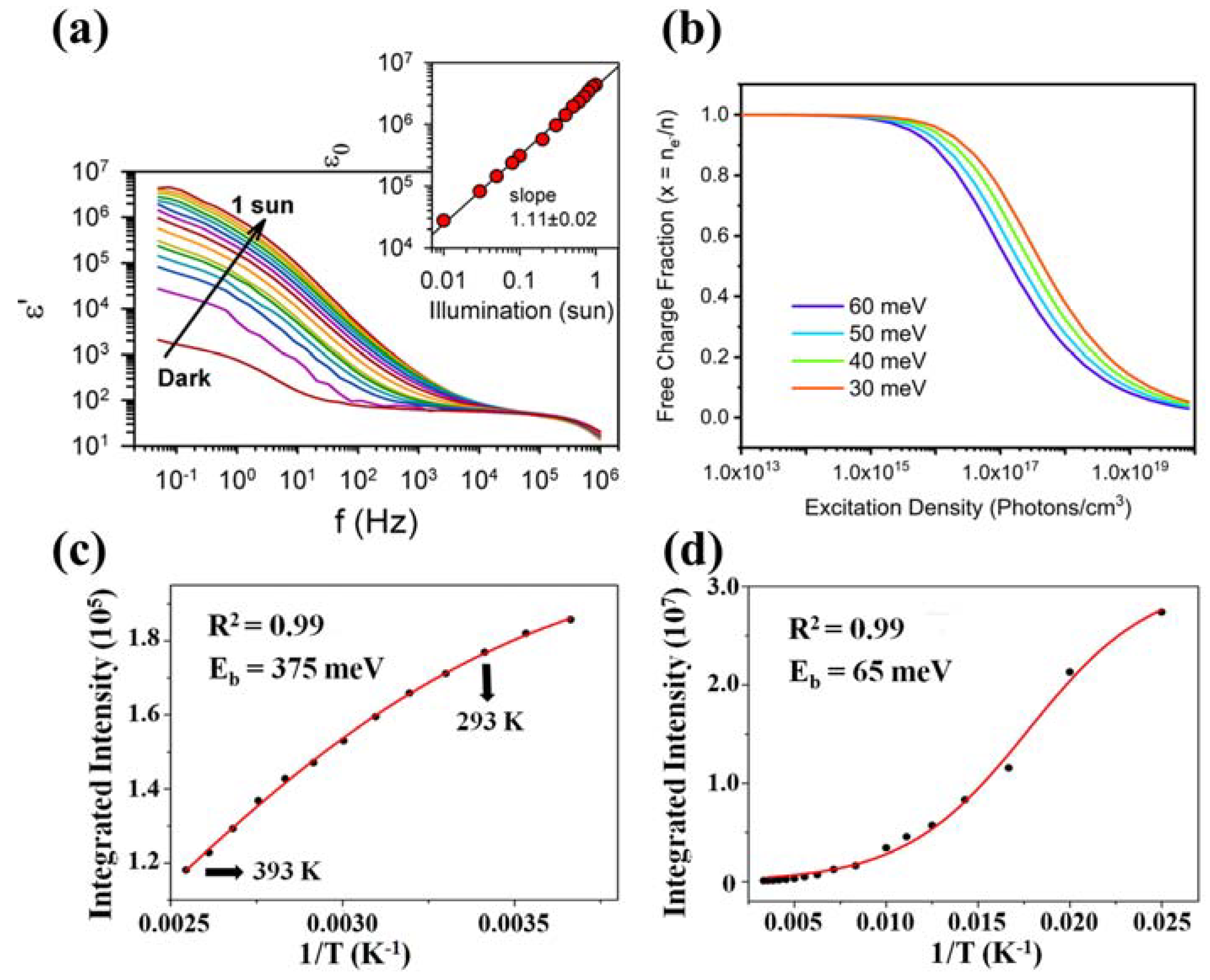
4.3. Defect Effects
4.3.1. Defect Effects in 3D Bulk Perovskites
4.3.2. Defect Effects in Low-Dimensional Perovskites
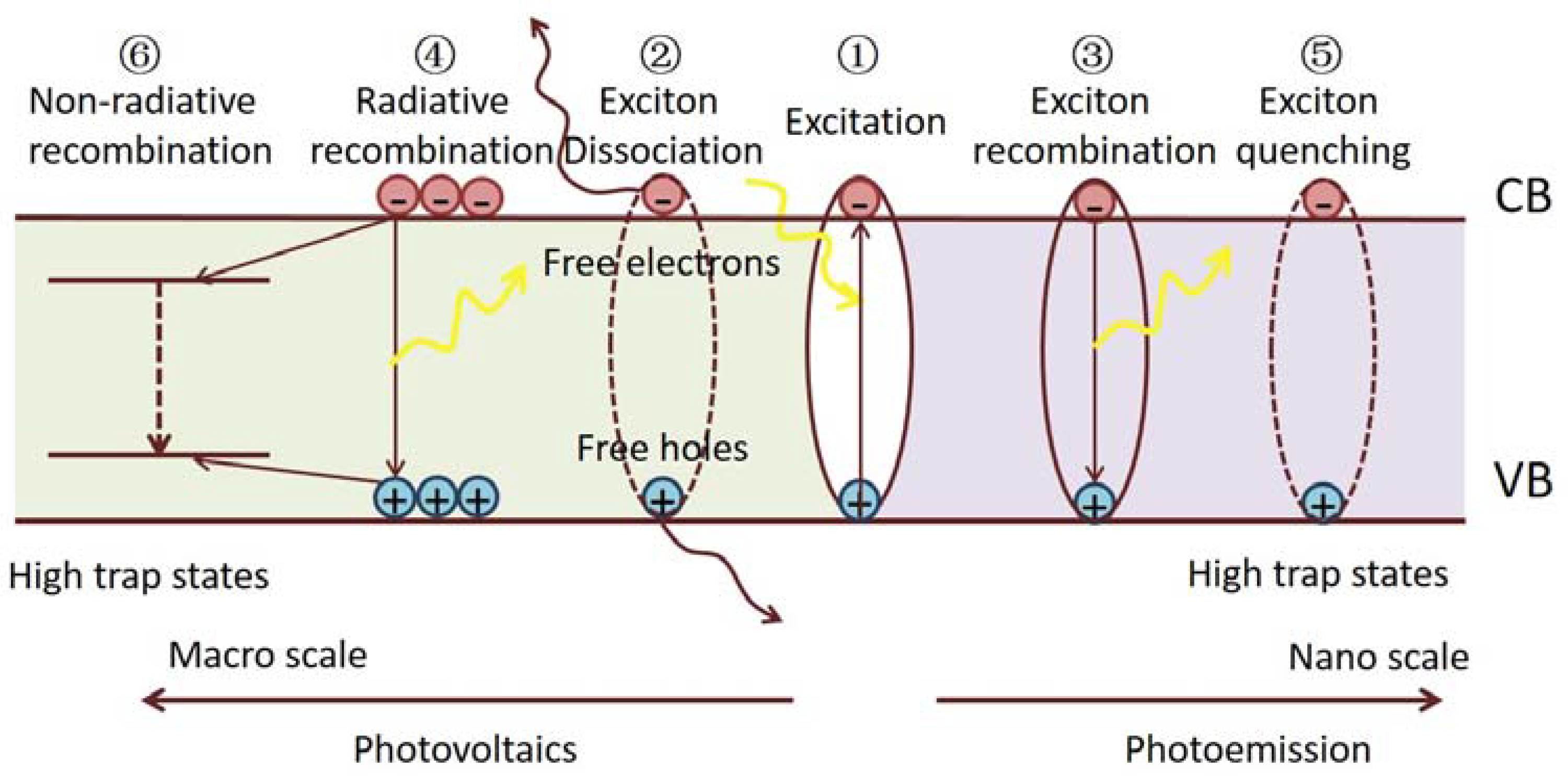
4.3.3. Recombination Kinetics
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.M.; Teuscher, J.; Miyasaka, T.; Murakami, T.N.; Snaith, H.J. Efficient Hybrid Solar Cells Based on Meso-Superstructured Organometal Halide Perovskites. Science 2012, 338, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Burschka, J.; Pellet, N.; Moon, S.J.; Humphry-Baker, R.; Gao, P.; Nazeeruddin, M.K.; Gratzel, M. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 2013, 499, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Noh, J.H.; Jeon, N.J.; Kim, Y.C.; Ryu, S.; Seo, J.; Seok, S.I. High-performance photovoltaic perovskite layers fabricated through intramolecular exchange. Science 2015, 348, 1234–1237. [Google Scholar] [CrossRef]
- Li, G.R.; Tan, Z.K.; Di, D.W.; Lai, M.L.; Jiang, L.; Lim, J.H.W.; Friend, R.H.; Greenham, N.C. Efficient Light-Emitting Diodes Based on Nanocrystalline Perovskite in a Dielectric Polymer Matrix. Nano Lett. 2015, 15, 2640–2644. [Google Scholar] [CrossRef]
- Tan, Z.K.; Moghaddam, R.S.; Lai, M.L.; Docampo, P.; Higler, R.; Deschler, F.; Price, M.; Sadhanala, A.; Pazos, L.M.; Credgington, D.; et al. Bright light-emitting diodes based on organometal halide perovskite. Nat. Nanotechnol. 2014, 9, 687–692. [Google Scholar] [CrossRef]
- Ling, Y.C.; Yuan, Z.; Tian, Y.; Wang, X.; Wang, J.C.; Xin, Y.; Hanson, K.; Ma, B.W.; Gao, H.W. Bright Light-Emitting Diodes Based on Organometal Halide Perovskite Nanoplatelets. Adv. Mater. 2016, 28, 305–311. [Google Scholar] [CrossRef]
- Bai, Z.L.; Zhong, H.Z. Halide perovskite quantum dots: Potential candidates for display technology. Sci. Bull. 2015, 60, 1622–1624. [Google Scholar] [CrossRef]
- Stoumpos, C.C.; Cao, D.H.; Clark, D.J.; Young, J.; Rondinelli, J.M.; Jang, J.I.; Hupp, J.T.; Kanatzidis, M.G. Ruddlesden–Popper Hybrid Lead Iodide Perovskite 2D Homologous Semiconductors. Chem. Mater. 2016, 28, 2852–2867. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, D.; Wang, J.; Wang, N. Large Organic Cations in Quasi-2D Perovskites for High-Performance Light-Emitting Diodes. J. Phys. Chem. Lett. 2020, 11, 8502–8510. [Google Scholar] [CrossRef]
- Deschler, F.; Price, M.; Pathak, S.; Klintberg, L.E.; Jarausch, D.D.; Higler, R.; Huttner, S.; Leijtens, T.; Stranks, S.D.; Snaith, H.J.; et al. High Photoluminescence Efficiency and Optically Pumped Lasing in Solution-Processed Mixed Halide Perovskite Semiconductors. J. Phys. Chem. Lett. 2014, 5, 1421–1426. [Google Scholar] [CrossRef]
- Xing, G.C.; Mathews, N.; Lim, S.S.; Yantara, N.; Liu, X.F.; Sabba, D.; Gratzel, M.; Mhaisalkar, S.; Sum, T.C. Low-temperature solution-processed wavelength-tunable perovskites for lasing. Nat. Mater. 2014, 13, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ha, S.T.; Liu, X.F.; Sum, T.C.; Xiong, Q.H. Room-Temperature Near-Infrared High-Q Perovskite Whispering-Gallery Planar Nano lasers. Nano Lett. 2014, 14, 5995–6001. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.M.; Fu, Y.P.; Meng, F.; Wu, X.X.; Gong, Z.Z.; Ding, Q.; Gustafsson, M.V.; Trinh, M.T.; Jin, S.; Zhu, X.Y. Lead halide perovskite nanowire lasers with low lasing thresholds and high quality factors. Nat. Mater. 2015, 14, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Hu, K.; Zhang, H.H.; Wang, X.D.; Yao, J.N.; Fu, H.B. Perovskite Microdisk Microlasers Self-Assembled from Solution. Adv. Mater. 2015, 27, 3405–3410. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Liu, X.F.; Zhang, Q.; Ha, S.T.; Yuan, Y.W.; Shen, C.; Sum, T.C.; Xiong, Q.H. Vapor Phase Synthesis of Organometal Halide Perovskite Nanowires for Tunable Room-Temperature Nanolasers. Nano Lett. 2015, 15, 4571–4577. [Google Scholar] [CrossRef]
- Dou, L.T.; Yang, Y.; You, J.B.; Hong, Z.R.; Chang, W.H.; Li, G.; Yang, Y. Solution-processed hybrid perovskite photodetectors with high detectivity. Nat. Commun. 2014, 5, 5404. [Google Scholar] [CrossRef]
- Lin, Q.Q.; Armin, A.; Burn, P.L.; Meredith, P. Filterless narrowband visible photodetectors. Nat. Photonics 2015, 9, 687–694. [Google Scholar] [CrossRef]
- Fang, Y.J.; Dong, Q.F.; Shao, Y.C.; Yuan, Y.B.; Huang, J.S. Highly narrowband perovskite single-crystal photodetectors enabled by surface-charge recombination. Nat. Photonics 2015, 9, 679–686. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Zhu, X.; Yang, Z.; Ke, W.; Feng, J.; Ren, X.; Zhao, K.; Liu, M.; Kanatzidis, M.G.; et al. Inch-sized high-quality perovskite single crystals by suppressing phase segregation for light-powered integrated circuits. Sci. Adv. 2021, 7, eabc8844. [Google Scholar] [CrossRef]
- Chin, X.Y.; Cortecchia, D.; Yin, J.; Bruno, A.; Soci, C. Lead iodide perovskite light-emitting field-effect transistor. Nat. Commun. 2015, 6, 7383. [Google Scholar] [CrossRef] [PubMed]
- Srimath Kandada, A.R.; Petrozza, A. Photophysics of Hybrid Lead Halide Perovskites: The Role of Microstructure. Acc. Chem. Res. 2016, 49, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Lin, H.; Tian, Y.; Yuan, Z.; Clark, R.; Chen, B.; van de Burgt, L.J.; Wang, J.C.; Zhou, Y.; Hanson, K.; et al. Luminescent zero-dimensional organic metal halide hybrids with near-unity quantum efficiency. Chem. Sci. 2018, 9, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhou, C.; Tian, Y.; Siegrist, T.; Ma, B. Low-Dimensional Organometal Halide Perovskites. ACS Energy Lett. 2018, 3, 54–62. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, H.Z.; Chen, C.; Wu, X.G.; Hu, X.M.; Huang, H.L.; Han, J.B.; Zou, B.S.; Dong, Y.P. Brightly Luminescent and Color-Tunable Colloidal CH3NH3PbX3 (X = Br, I, Cl) Quantum Dots: Potential Alternatives for Display Technology. ACS Nano 2015, 9, 4533–4542. [Google Scholar] [CrossRef]
- Yuan, Z.; Shu, Y.; Xin, Y.; Ma, B. Highly luminescent nanoscale quasi-2D layered lead bromide perovskites with tunable emissions. Chem. Commun. 2016, 52, 3887–3890. [Google Scholar] [CrossRef]
- Docampo, P.; Bein, T. A Long-Term View on Perovskite Optoelectronics. Acc. Chem. Res. 2016, 49, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.F.; Fang, Y.J.; Shao, Y.C.; Mulligan, P.; Qiu, J.; Cao, L.; Huang, J.S. Electron-hole diffusion lengths > 175 mu m in solution-grown CH3NH3PbI3 single crystals. Science 2015, 347, 967–970. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhou, C.; Tian, Y.; Shu, Y.; Messier, J.; Wang, J.C.; van de Burgt, L.J.; Kountouriotis, K.; Xin, Y.; Holt, E.; et al. One-dimensional organic lead halide perovskites with efficient bluish white-light emission. Nat. Commun. 2017, 8, 14051. [Google Scholar] [CrossRef]
- Zhou, C.; Lin, H.; Shi, H.; Tian, Y.; Pak, C.; Shatruk, M.; Zhou, Y.; Djurovich, P.; Du, M.-H.; Ma, B. A Zero-Dimensional Organic Seesaw-Shaped Tin Bromide with Highly Efficient Strongly Stokes-Shifted Deep-Red Emission. Angew. Chem. Int. Ed. 2018, 57, 1021–1024. [Google Scholar] [CrossRef]
- Chen, P.; Bai, Y.; Lyu, M.; Yun, J.-H.; Hao, M.; Wang, L. Progress and Perspective in Low-Dimensional Metal Halide Perovskites for Optoelectronic Applications. Sol. RRL 2018, 2, 1700186. [Google Scholar] [CrossRef]
- Yu, G.; Lin, F.; Zhou, K.; Fang, S.; Shi, Y.; Liu, W.; Hu, H.; Ma, B.; Lin, H. One-Dimensional Organic–Metal Halide with Highly Efficient Warm White-Light Emission and Its Moisture-Induced Structural Transformation. Chem. Mater. 2021, 33, 5668–5674. [Google Scholar] [CrossRef]
- Lin, F.; Wang, H.; Liu, W.; Li, J. Zero-dimensional ionic antimony halide inorganic–organic hybrid with strong greenish yellow emission. J. Mater. Chem. C 2020, 8, 7300–7303. [Google Scholar] [CrossRef]
- Wu, X.X.; Trinh, M.T.; Zhu, X.Y. Excitonic Many-Body Interactions in Two-Dimensional Lead Iodide Perovskite Quantum Wells. J. Phys. Chem. C 2015, 119, 14714–14721. [Google Scholar] [CrossRef]
- Ponseca, C.S.; Savenije, T.J.; Abdellah, M.; Zheng, K.B.; Yartsev, A.; Pascher, T.; Harlang, T.; Chabera, P.; Pullerits, T.; Stepanov, A.; et al. Organometal Halide Perovskite Solar Cell Materials Rationalized: Ultrafast Charge Generation, High and Microsecond-Long Balanced Mobilities, and Slow Recombination. J. Am. Chem. Soc. 2014, 136, 5189–5192. [Google Scholar] [CrossRef]
- Stranks, S.D.; Burlakov, V.M.; Leijtens, T.; Ball, J.M.; Goriely, A.; Snaith, H.J. Recombination Kinetics in Organic-Inorganic Perovskites: Excitons, Free Charge, and Subgap States. Phys. Rev. Appl. 2014, 2, 034007. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.H.; Lee, J.H.; Hong, K.H. The Role of Intrinsic Defects in Methylammonium Lead Iodide Perovskite. J. Phys. Chem. Lett. 2014, 5, 1312–1317. [Google Scholar] [CrossRef]
- Wu, X.X.; Trinh, M.T.; Niesner, D.; Zhu, H.M.; Norman, Z.; Owen, J.S.; Yaffe, O.; Kudisch, B.J.; Zhu, X.Y. Trap States in Lead Iodide Perovskites. J. Am. Chem. Soc. 2015, 137, 2089–2096. [Google Scholar] [CrossRef]
- Stranks, S.D.; Eperon, G.E.; Grancini, G.; Menelaou, C.; Alcocer, M.J.P.; Leijtens, T.; Herz, L.M.; Petrozza, A.; Snaith, H.J. Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber. Science 2013, 342, 341–344. [Google Scholar] [CrossRef]
- Luo, B.B.; Pu, Y.C.; Yang, Y.; Lindley, S.A.; Abdelmaged, G.; Ashry, H.; Li, Y.; Li, X.M.; Zhang, J.Z. Synthesis, Optical Properties, and Exciton Dynamics of Organolead Bromide Perovskite Nanocrystals. J. Phys. Chem. C 2015, 119, 26672–26682. [Google Scholar] [CrossRef]
- Tian, Y.X.; Merdasa, A.; Unger, E.; Abdellah, M.; Zheng, K.B.; McKibbin, S.; Mikkelsen, A.; Pullerits, T.; Yartsev, A.; Sundstrom, V.; et al. Enhanced Organo-Metal Halide Perovskite Photoluminescence from Nanosized Defect-Free Crystallites and Emitting Sites. J. Phys. Chem. Lett. 2015, 6, 4171–4177. [Google Scholar] [CrossRef] [PubMed]
- Mitzi, D.B. Synthesis, structure, and properties of organic-inorganic perovskites and related materials. In Progress in Inorganic Chemistry; Wiley: Hoboken, NJ, USA, 1999; Volume 48, pp. 1–121. [Google Scholar] [CrossRef]
- Cheng, Z.Y.; Lin, J. Layered organic-inorganic hybrid perovskites: Structure, optical properties, film preparation, patterning and templating engineering. CrystEngComm 2010, 12, 2646–2662. [Google Scholar] [CrossRef]
- Zhou, C.; Lin, H.; He, Q.; Xu, L.; Worku, M.; Chaaban, M.; Lee, S.; Shi, X.; Du, M.-H.; Ma, B. Low dimensional metal halide perovskites and hybrids. Mater. Sci. Eng. R Rep. 2019, 137, 38–65. [Google Scholar] [CrossRef]
- Chen, J.N.; Zhou, S.S.; Jin, S.Y.; Li, H.Q.; Zhai, T.Y. Crystal organometal halide perovskites with promising optoelectronic applications. J. Mater. Chem. C 2016, 4, 11–27. [Google Scholar] [CrossRef]
- Bai, S.; Yuan, Z.; Gao, F. Colloidal metal halide perovskite nanocrystals: Synthesis, characterization, and applications. J. Mater. Chem. C 2016, 4, 3898–3904. [Google Scholar] [CrossRef]
- Dang, Y.Y.; Liu, Y.; Sun, Y.X.; Yuan, D.S.; Liu, X.L.; Lu, W.Q.; Liu, G.F.; Xia, H.B.; Tao, X.T. Bulk crystal growth of hybrid perovskite material CH3NH3PbI3. CrystEngComm 2015, 17, 665–670. [Google Scholar] [CrossRef]
- Shi, D.; Adinolfi, V.; Comin, R.; Yuan, M.J.; Alarousu, E.; Buin, A.; Chen, Y.; Hoogland, S.; Rothenberger, A.; Katsiev, K.; et al. Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals. Science 2015, 347, 519–522. [Google Scholar] [CrossRef]
- Park, J.S.; Choi, S.; Yan, Y.; Yang, Y.; Luther, J.M.; Wei, S.H.; Parilla, P.; Zhu, K. Electronic Structure and Optical Properties of alpha-CH3NH3PbBr3 Perovskite Single Crystal. J. Phys. Chem. Lett. 2015, 6, 4304–4308. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, Y.; Yang, M.J.; Choi, S.; Zhu, K.; Luther, J.M.; Beard, M.C. Low surface recombination velocity in solution-grown CH3NH3PbBr3 perovskite single crystal. Nat. Commun. 2015, 6, 7961. [Google Scholar] [CrossRef]
- Saidaminov, M.I.; Abdelhady, A.L.; Murali, B.; Alarousu, E.; Burlakov, V.M.; Peng, W.; Dursun, I.; Wang, L.F.; He, Y.; Maculan, G.; et al. High-quality bulk hybrid perovskite single crystals within minutes by inverse temperature crystallization. Nat. Commun. 2015, 6, 7586. [Google Scholar] [CrossRef]
- Maculan, G.; Sheikh, A.D.; Abdelhady, A.L.; Saidaminov, M.I.; Hague, M.A.; Murali, B.; Alarousu, E.; Mohammed, O.F.; Wu, T.; Bakr, O.M. CH3NH3PbCl3 Single Crystals: Inverse Temperature Crystallization and Visible-Blind UV-Photodetector. J. Phys. Chem. Lett. 2015, 6, 3781–3786. [Google Scholar] [CrossRef] [PubMed]
- Etgar, L.; Gao, P.; Xue, Z.S.; Peng, Q.; Chandiran, A.K.; Liu, B.; Nazeeruddin, M.K.; Gratzel, M. Mesoscopic CH3NH3PbI3/TiO2 Heterojunction Solar Cells. J. Am. Chem. Soc. 2012, 134, 17396–17399. [Google Scholar] [CrossRef] [PubMed]
- Jeon, N.J.; Noh, J.H.; Kim, Y.C.; Yang, W.S.; Ryu, S.; Il Seol, S. Solvent engineering for high-performance inorganic-organic hybrid perovskite solar cells. Nat. Mater. 2014, 13, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.Z.; Johnston, M.B.; Snaith, H.J. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 2013, 501, 395–398. [Google Scholar] [CrossRef]
- Malinkiewicz, O.; Yella, A.; Lee, Y.H.; Espallargas, G.M.; Graetzel, M.; Nazeeruddin, M.K.; Bolink, H.J. Perovskite solar cells employing organic charge-transport layers. Nat. Photonics 2014, 8, 128–132. [Google Scholar] [CrossRef]
- Chen, Q.; Zhou, H.P.; Hong, Z.R.; Luo, S.; Duan, H.S.; Wang, H.H.; Liu, Y.S.; Li, G.; Yang, Y. Planar Heterojunction Perovskite Solar Cells via Vapor-Assisted Solution Process. J. Am. Chem. Soc. 2014, 136, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Carrero, S.; Galian, R.E.; Perez-Prieto, J. Organometal Halide Perovskites: Bulk Low-Dimension Materials and Nanoparticles. Part. Part. Syst. Charact. 2015, 32, 709–720. [Google Scholar] [CrossRef]
- Yuan, Z.; Shu, Y.; Tian, Y.; Xin, Y.; Ma, B.W. A facile one-pot synthesis of deep blue luminescent lead bromide perovskite microdisks. Chem. Commun. 2015, 51, 16385–16388. [Google Scholar] [CrossRef]
- Jang, D.M.; Park, K.; Kim, D.H.; Park, J.; Shojaei, F.; Kang, H.S.; Ahn, J.P.; Lee, J.W.; Song, J.K. Reversible Halide Exchange Reaction of Organometal Trihalide Perovskite Colloidal Nanocrystals for Full-Range Band Gap Tuning. Nano Lett. 2015, 15, 5191–5199. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, F.; Liu, L.; Zhang, F.; Wu, X.-g.; Shi, L.; Zou, B.; Pei, Q.; Zhong, H. Emulsion Synthesis of Size-Tunable CH3NH3PbBr3 Quantum Dots: An Alternative Route toward Efficient Light-Emitting Diodes. ACS Appl. Mater. Interfaces 2015, 7, 28128–28133. [Google Scholar] [CrossRef]
- Hassan, Y.; Song, Y.; Pensack, R.D.; Abdelrahman, A.I.; Kobayashi, Y.; Winnik, M.A.; Scholes, G.D. Structure-Tuned Lead Halide Perovskite Nanocrystals. Adv. Mater. 2016, 28, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Eiden, A.; Prakash, G.V.; Baumberg, J.J. Exfoliation of self-assembled 2D organic-inorganic perovskite semiconductors. Appl. Phys. Lett. 2014, 104, 171111. [Google Scholar] [CrossRef]
- Yaffe, O.; Chernikov, A.; Norman, Z.M.; Zhong, Y.; Velauthapillai, A.; van der Zande, A.; Owen, J.S.; Heinz, T.F. Excitons in ultrathin organic-inorganic perovskite crystals. Phys. Rev. B 2015, 92, 045414. [Google Scholar] [CrossRef]
- Dou, L.T.; Wong, A.B.; Yu, Y.; Lai, M.L.; Kornienko, N.; Eaton, S.W.; Fu, A.; Bischak, C.G.; Ma, J.; Ding, T.N.; et al. Atomically thin two-dimensional organic-inorganic hybrid perovskites. Science 2015, 349, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.B.; Lai, M.L.; Eaton, S.W.; Yu, Y.; Lin, E.; Dou, L.; Fu, A.; Yang, P.D. Growth and Anion Exchange Conversion of CH3NH3PbX3 Nanorod Arrays for Light-Emitting Diodes. Nano Lett. 2015, 15, 5519–5524. [Google Scholar] [CrossRef]
- Fu, Y.P.; Meng, F.; Rowley, M.B.; Thompson, B.J.; Shearer, M.J.; Ma, D.W.; Hamers, R.J.; Wright, J.C.; Jin, S. Solution Growth of Single Crystal Methylammonium Lead Halide Perovskite Nanostructures for Optoelectronic and Photovoltaic Applications. J. Am. Chem. Soc. 2015, 137, 5810–5818. [Google Scholar] [CrossRef]
- Dohner, E.R.; Jaffe, A.; Bradshaw, L.R.; Karunadasa, H.I. Intrinsic White-Light Emission from Layered Hybrid Perovskites. J. Am. Chem. Soc. 2014, 136, 13154–13157. [Google Scholar] [CrossRef]
- Dohner, E.R.; Hoke, E.T.; Karunadasa, H.I. Self-Assembly of Broadband White-Light Emitters. J. Am. Chem. Soc. 2014, 136, 1718–1721. [Google Scholar] [CrossRef]
- Lee, S.; Karkee, R.; Ben-Akacha, A.; Luong, D.; Vellore Winfred, J.S.R.; Lin, X.; Strubbe, D.A.; Ma, B. One-dimensional organic metal halide nanoribbons with dual emission. Chem. Commun. 2023, 59, 3711–3714. [Google Scholar] [CrossRef]
- Zhou, C.; Xu, L.-J.; Lee, S.; Lin, H.; Ma, B. Recent Advances in Luminescent Zero-Dimensional Organic Metal Halide Hybrids. Adv. Opt. Mater. 2021, 9, 2001766. [Google Scholar] [CrossRef]
- Ben-Akacha, A.; Zhou, C.; Chaaban, M.; Beery, D.; Lee, S.; Worku, M.; Lin, X.; Westphal, R.; Ma, B. Mechanochemical Synthesis of Zero Dimensional Organic-Inorganic Metal Halide Hybrids. ChemPhotoChem 2021, 5, 326–329. [Google Scholar] [CrossRef]
- deQuilettes, D.W.; Vorpahl, S.M.; Stranks, S.D.; Nagaoka, H.; Eperon, G.E.; Ziffer, M.E.; Snaith, H.J.; Ginger, D.S. Impact of microstructure on local carrier lifetime in perovskite solar cells. Science 2015, 348, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni, S.; Heinz, F.D.; Im, J.H.; Veurman, W.; Padilla, M.; Schubert, M.C.; Wurfel, U.; Gratzel, M.; Park, N.G.; Hinsch, A. Analysing the effect of crystal size and structure in highly efficient CH3NH3PbI3 perovskite solar cells by spatially resolved photo- and electroluminescence imaging. Nanoscale 2015, 7, 19653–19662. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Ohkita, H.; Benten, H.; Ito, S. Photovoltaic Performance of Perovskite Solar Cells with Different Grain Sizes. Adv. Mater. 2016, 28, 917–922. [Google Scholar] [CrossRef]
- Nie, W.Y.; Tsai, H.H.; Asadpour, R.; Blancon, J.C.; Neukirch, A.J.; Gupta, G.; Crochet, J.J.; Chhowalla, M.; Tretiak, S.; Alam, M.A.; et al. High-efficiency solution-processed perovskite solar cells with millimeter-scale grains. Science 2015, 347, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Wang, Q.; Shao, Y.C.; Yuan, Y.B.; Xiao, Z.G.; Huang, J.S. Non-wetting surface-driven high-aspect-ratio crystalline grain growth for efficient hybrid perovskite solar cells. Nat. Commun. 2015, 6, 7747. [Google Scholar] [CrossRef]
- Hu, M.; Bi, C.; Yuan, Y.; Bai, Y.; Huang, J. Stabilized Wide Bandgap MAPbBrxI3–x Perovskite by Enhanced Grain Size and Improved Crystallinity. Adv. Sci. 2015, 3, 1500301. [Google Scholar] [CrossRef]
- Wang, B.; Wong, K.Y.; Yang, S.; Chen, T. Crystallinity and defect state engineering in organo-lead halide perovskite for high-efficiency solar cells. J. Mater. Chem. A 2016, 4, 3806–3812. [Google Scholar] [CrossRef]
- Im, J.H.; Jang, I.H.; Pellet, N.; Gratzel, M.; Park, N.G. Growth of CH3NH3PbI3 cuboids with controlled size for high-efficiency perovskite solar cells. Nat. Nanotechnol. 2014, 9, 927–932. [Google Scholar] [CrossRef]
- Cho, H.; Jeong, S.-H.; Park, M.-H.; Kim, Y.-H.; Wolf, C.; Lee, C.-L.; Heo, J.H.; Sadhanala, A.; Myoung, N.; Yoo, S.; et al. Overcoming the electroluminescence efficiency limitations of perovskite light-emitting diodes. Science 2015, 350, 1222–1225. [Google Scholar] [CrossRef]
- Kerner, R.A.; Zhao, L.; Xiao, Z.; Rand, B.P. Ultrasmooth metal halide perovskite thin films via sol-gel processing. J. Mater. Chem. A 2016, 4, 8308–8315. [Google Scholar] [CrossRef]
- Li, J.Q.; Bade, S.G.R.; Shan, X.; Yu, Z.B. Single-Layer Light-Emitting Diodes Using Organometal Halide Perovskite/Poly(ethylene oxide) Composite Thin Films. Adv. Mater. 2015, 27, 5196–5202. [Google Scholar] [CrossRef]
- Bade, S.G.R.; Li, J.; Shan, X.; Ling, Y.; Tian, Y.; Dilbeck, T.; Besara, T.; Geske, T.; Gao, H.; Ma, B.; et al. Fully Printed Halide Perovskite Light-Emitting Diodes with Silver Nanowire Electrodes. ACS Nano 2015, 10, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.C.; Mathews, N.; Sun, S.Y.; Lim, S.S.; Lam, Y.M.; Gratzel, M.; Mhaisalkar, S.; Sum, T.C. Long-Range Balanced Electron- and Hole-Transport Lengths in Organic-Inorganic CH3NH3PbI3. Science 2013, 342, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Wehrenfennig, C.; Eperon, G.E.; Johnston, M.B.; Snaith, H.J.; Herz, L.M. High Charge Carrier Mobilities and Lifetimes in Organolead Trihalide Perovskites. Adv. Mater. 2014, 26, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.G.; Dong, Q.F.; Bi, C.; Shao, Y.C.; Yuan, Y.B.; Huang, J.S. Solvent Annealing of Perovskite-Induced Crystal Growth for Photovoltaic-Device Efficiency Enhancement. Adv. Mater. 2014, 26, 6503–6509. [Google Scholar] [CrossRef]
- Ayres, J.R. Characterization Of Trapping States In Polycrystalline-Silicon Thin-Film Transistors by Deep-Level Transient Spectroscopy. J. Appl. Phys. 1993, 74, 1787–1792. [Google Scholar] [CrossRef]
- Balcioglu, A.; Ahrenkiel, R.K.; Hasoon, F. Deep-level impurities in CdTe/CdS thin-film solar cells. J. Appl. Phys. 2000, 88, 7175–7178. [Google Scholar] [CrossRef]
- Kerr, L.L.; Li, S.S.; Johnston, S.W.; Anderson, T.J.; Crisalle, O.D.; Kim, W.K.; Abushama, J.; Noufi, R.N. Investigation of defect properties in Cu(In,Ga)Se-2 solar cells by deep-level transient spectroscopy. Solid-State Electron. 2004, 48, 1579–1586. [Google Scholar] [CrossRef]
- Goldmann, C.; Krellner, C.; Pernstich, K.P.; Haas, S.; Gundlach, D.J.; Batlogg, B. Determination of the interface trap density of rubrene single-crystal field-effect transistors and comparison to the bulk trap density. J. Appl. Phys. 2006, 99, 034507. [Google Scholar] [CrossRef]
- Shaw, P.E.; Ruseckas, A.; Samuel, I.D.W. Exciton diffusion measurements in poly(3-hexylthiophene). Adv. Mater. 2008, 20, 3516–3520. [Google Scholar] [CrossRef]
- Kroeze, J.E.; Savenije, T.J.; Vermeulen, M.J.W.; Warman, J.M. Contactless determination of the photoconductivity action spectrum, exciton diffusion length, and charge separation efficiency in polythiophene-sensitized TiO2 bilayers. J. Phys. Chem. B 2003, 107, 7696–7705. [Google Scholar] [CrossRef]
- Sheng, R.; Ho-Baillie, A.; Huang, S.J.; Chen, S.; Wen, X.M.; Hao, X.J.; Green, M.A. Methylammonium Lead Bromide Perovskite-Based Solar Cells by Vapor-Assisted Deposition. J. Phys. Chem. C 2015, 119, 3545–3549. [Google Scholar] [CrossRef]
- Gonzalez-Pedro, V.; Juarez-Perez, E.J.; Arsyad, W.S.; Barea, E.M.; Fabregat-Santiago, F.; Mora-Sero, I.; Bisquert, J. General Working Principles of CH3NH3PbX3 Perovskite Solar Cells. Nano Lett. 2014, 14, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, V.W.; Weber, S.A.L.; Ramos, F.J.; Nazeeruddin, M.K.; Gratzel, M.; Li, D.; Domanski, A.L.; Lieberwirth, I.; Ahmad, S.; Berger, R. Real-space observation of unbalanced charge distribution inside a perovskite-sensitized solar cell. Nat. Commun. 2014, 5, 5001. [Google Scholar] [CrossRef]
- Tian, W.M.; Zhao, C.Y.; Leng, J.; Gui, R.R.; Jin, S.G. Visualizing Carrier Diffusion in Individual Single-Crystal Organolead Halide Perovskite Nanowires and Nanoplates. J. Am. Chem. Soc. 2015, 137, 12458–12461. [Google Scholar] [CrossRef]
- Ishihara, T. Optical-Properties Of Pbi-Based Perovskite Structures. J. Lumin. 1994, 60–61, 269–274. [Google Scholar] [CrossRef]
- Tanaka, K.; Takahashi, T.; Ban, T.; Kondo, T.; Uchida, K.; Miura, N. Comparative study on the excitons in lead-halide-based perovskite-type crystals CH3NH3PbBr3CH3NH3PbI3. Solid State Commun. 2003, 127, 619–623. [Google Scholar] [CrossRef]
- Miyata, A.; Mitioglu, A.; Plochocka, P.; Portugall, O.; Wang, J.T.W.; Stranks, S.D.; Snaith, H.J.; Nicholas, R.J. Direct measurement of the exciton binding energy and effective masses for charge carriers in organic-inorganic tri-halide perovskites. Nat. Phys. 2015, 11, 582–587. [Google Scholar] [CrossRef]
- Wu, K.W.; Bera, A.; Ma, C.; Du, Y.M.; Yang, Y.; Li, L.; Wu, T. Temperature-dependent excitonic photoluminescence of hybrid organometal halide perovskite films. Phys. Chem. Chem. Phys. 2014, 16, 22476–22481. [Google Scholar] [CrossRef]
- Juarez-Perez, E.J.; Sanchez, R.S.; Badia, L.; Garcia-Belmonte, G.; Kang, Y.S.; Mora-Sero, I.; Bisquert, J. Photoinduced Giant Dielectric Constant in Lead Halide Perovskite Solar Cells. J. Phys. Chem. Lett. 2014, 5, 2390–2394. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Whittaker-Brooks, L.; Fleming, G.R. Exciton and Free Charge Dynamics of Methylammonium Lead Iodide Perovskites Are Different in the Tetragonal and Orthorhombic Phases. J. Phys. Chem. C 2015, 119, 19590–19595. [Google Scholar] [CrossRef]
- Motta, C.; El-Mellouhi, F.; Kais, S.; Tabet, N.; Alharbi, F.; Sanvito, S. Revealing the role of organic cations in hybrid halide perovskite CH3NH3PbI3. Nat. Commun. 2015, 6, 7026. [Google Scholar] [CrossRef] [PubMed]
- Green, M.A.; Jiang, Y.; Soufiani, A.M.; Ho-Baillie, A. Optical Properties of Photovoltaic Organic–Inorganic Lead Halide Perovskites. J. Phys. Chem. Lett. 2015, 6, 4774–4785. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Liu, Y.; Zou, X.; Johansson, E.M.J.; Sá, J. Can photoluminescence quenching be a predictor for perovskite solar cell efficiencies? Phys. Chem. Chem. Phys. 2023, 25, 22607–22613. [Google Scholar] [CrossRef] [PubMed]
- Scholes, G.D.; Rumbles, G. Excitons in nanoscale systems. Nat. Mater. 2006, 5, 683–696. [Google Scholar] [CrossRef]
- Tyagi, P.; Arveson, S.M.; Tisdale, W.A. Colloidal Organohalide Perovskite Nanoplatelets Exhibiting Quantum Confinement. J. Phys. Chem. Lett. 2015, 6, 1911–1916. [Google Scholar] [CrossRef]
- Sichert, J.A.; Tong, Y.; Mutz, N.; Vollmer, M.; Fischer, S.; Milowska, K.Z.; Cortadella, R.G.; Nickel, B.; Cardenas-Daw, C.; Stolarczyk, J.K.; et al. Quantum Size Effect in Organometal Halide Perovskite Nanoplatelets. Nano Lett. 2015, 15, 6521–6527. [Google Scholar] [CrossRef]
- Huang, H.; Susha, A.S.; Kershaw, S.V.; Hung, T.F.; Rogach, A.L. Control of Emission Color of High Quantum Yield CH3NH3PbBr3 Perovskite Quantum Dots by Precipitation Temperature. Adv. Sci. 2015, 2, 1500194. [Google Scholar] [CrossRef]
- Di, D.W.; Musselman, K.P.; Li, G.R.; Sadhanala, A.; Ievskaya, Y.; Song, Q.L.; Tan, Z.K.; Lai, M.L.; MacManus-Driscoll, J.L.; Greenham, N.C.; et al. Size-Dependent Photon Emission from Organometal Halide Perovskite Nanocrystals Embedded in an Organic Matrix. J. Phys. Chem. Lett. 2015, 6, 446–450. [Google Scholar] [CrossRef]
- Zhu, F.; Men, L.; Guo, Y.J.; Zhu, Q.C.; Bhattacharjee, U.; Goodwin, P.M.; Petrich, J.W.; Smith, E.A.; Vela, J. Shape Evolution and Single Particle Luminescence of Organometal Halide Perovskite Nanocrystals. ACS Nano 2015, 9, 2948–2959. [Google Scholar] [CrossRef]
- Yang, S.; Zheng, Y.C.; Hou, Y.; Chen, X.; Chen, Y.; Wang, Y.; Zhao, H.J.; Yang, H.G. Formation Mechanism of Freestanding CH3NH3PbI3 Functional Crystals: In Situ Transformation vs Dissolution-Crystallization. Chem. Mater. 2014, 26, 6705–6710. [Google Scholar] [CrossRef]
- Schmidt, L.C.; Pertegas, A.; Gonzalez-Carrero, S.; Malinkiewicz, O.; Agouram, S.; Espallargas, G.M.; Bolink, H.J.; Galian, R.E.; Perez-Prieto, J. Nontemplate Synthesis of CH3NH3PbBr3 Perovskite Nanoparticles. J. Am. Chem. Soc. 2014, 136, 850–853. [Google Scholar] [CrossRef]
- Gonzalez-Carrero, S.; Galian, R.E.; Perez-Prieto, J. Maximizing the emissive properties of CH3NH3PbBr3 perovskite nanoparticles. J. Mater. Chem. A 2015, 3, 9187–9193. [Google Scholar] [CrossRef]
- Smith, M.D.; Connor, B.A.; Karunadasa, H.I. Tuning the Luminescence of Layered Halide Perovskites. Chem. Rev. 2019, 119, 3104–3139. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.; Filip, M.R.; Qiu, D.Y. Minimal Molecular Building Blocks for Screening in Quasi-Two-Dimensional Organic–Inorganic Lead Halide Perovskites. Nano Lett. 2023, 23, 3796–3802. [Google Scholar] [CrossRef]
- Yu, H.; Wang, F.; Xie, F.Y.; Li, W.W.; Chen, J.; Zhao, N. The Role of Chlorine in the Formation Process of “CH3NH3PbI3-xCl(x)” Perovskite. Adv. Funct. Mater. 2014, 24, 7102–7108. [Google Scholar] [CrossRef]
- Pan, H. Quantum-confinement effects on binding energies and optical properties of excitons in quantum dots. Chin. Phys. Lett. 2004, 21, 160–163. [Google Scholar]
- Tanaka, K.; Takahashi, T.; Kondo, T.; Umeda, K.; Ema, K.; Umebayashi, T.; Asai, K.; Uchida, K.; Miura, N. Electronic and excitonic structures of inorganic-organic perovskite-type quantum-well crystal (C4H9NH3)(2)PbBr4. Jpn. J. Appl. Phys. 2005, 44, 5923–5932. [Google Scholar] [CrossRef]
- Shi, E.; Gao, Y.; Finkenauer, B.P.; Akriti; Coffey, A.H.; Dou, L. Two-dimensional halide perovskite nanomaterials and heterostructures. Chem. Soc. Rev. 2018, 47, 6046–6072. [Google Scholar] [CrossRef]
- Mao, L.; Wu, Y.; Stoumpos, C.C.; Wasielewski, M.R.; Kanatzidis, M.G. White-Light Emission and Structural Distortion in New Corrugated Two-Dimensional Lead Bromide Perovskites. J. Am. Chem. Soc. 2017, 139, 5210–5215. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.J.; Shi, T.T.; Yan, Y.F. Unusual defect physics in CH3NH3PbI3 perovskite solar cell absorber. Appl. Phys. Lett. 2014, 104, 063903. [Google Scholar] [CrossRef]
- Yamada, Y.; Nakamura, T.; Endo, M.; Wakamiya, A.; Kanemitsu, Y. Photocarrier Recombination Dynamics in Perovskite CH3NH3PbI3 for Solar Cell Applications. J. Am. Chem. Soc. 2014, 136, 11610–11613. [Google Scholar] [CrossRef] [PubMed]
- Cassette, E.; Mirkovic, T.; Scholes, G.D. Toward the Control of Nonradiative Processes in Semiconductor Nanocrystals. J. Phys. Chem. Lett. 2013, 4, 2091–2093. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Zidek, K.; Abdellah, M.; Messing, M.E.; Al-Marri, M.J.; Pullerits, T. Trap States and Their Dynamics in Organometal Halide Perovskite Nanoparticles and Bulk Crystals. J. Phys. Chem. C 2016, 120, 3077–3084. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhao, Z.; Liu, Z.; Tang, A. The Scale Effects of Organometal Halide Perovskites. Nanomaterials 2023, 13, 2935. https://doi.org/10.3390/nano13222935
Zhang Y, Zhao Z, Liu Z, Tang A. The Scale Effects of Organometal Halide Perovskites. Nanomaterials. 2023; 13(22):2935. https://doi.org/10.3390/nano13222935
Chicago/Turabian StyleZhang, Yibo, Zhenze Zhao, Zhe Liu, and Aiwei Tang. 2023. "The Scale Effects of Organometal Halide Perovskites" Nanomaterials 13, no. 22: 2935. https://doi.org/10.3390/nano13222935
APA StyleZhang, Y., Zhao, Z., Liu, Z., & Tang, A. (2023). The Scale Effects of Organometal Halide Perovskites. Nanomaterials, 13(22), 2935. https://doi.org/10.3390/nano13222935