
Dimerization Effects and Negative Strain Energy in Silicon Monosulfide Nanotubes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
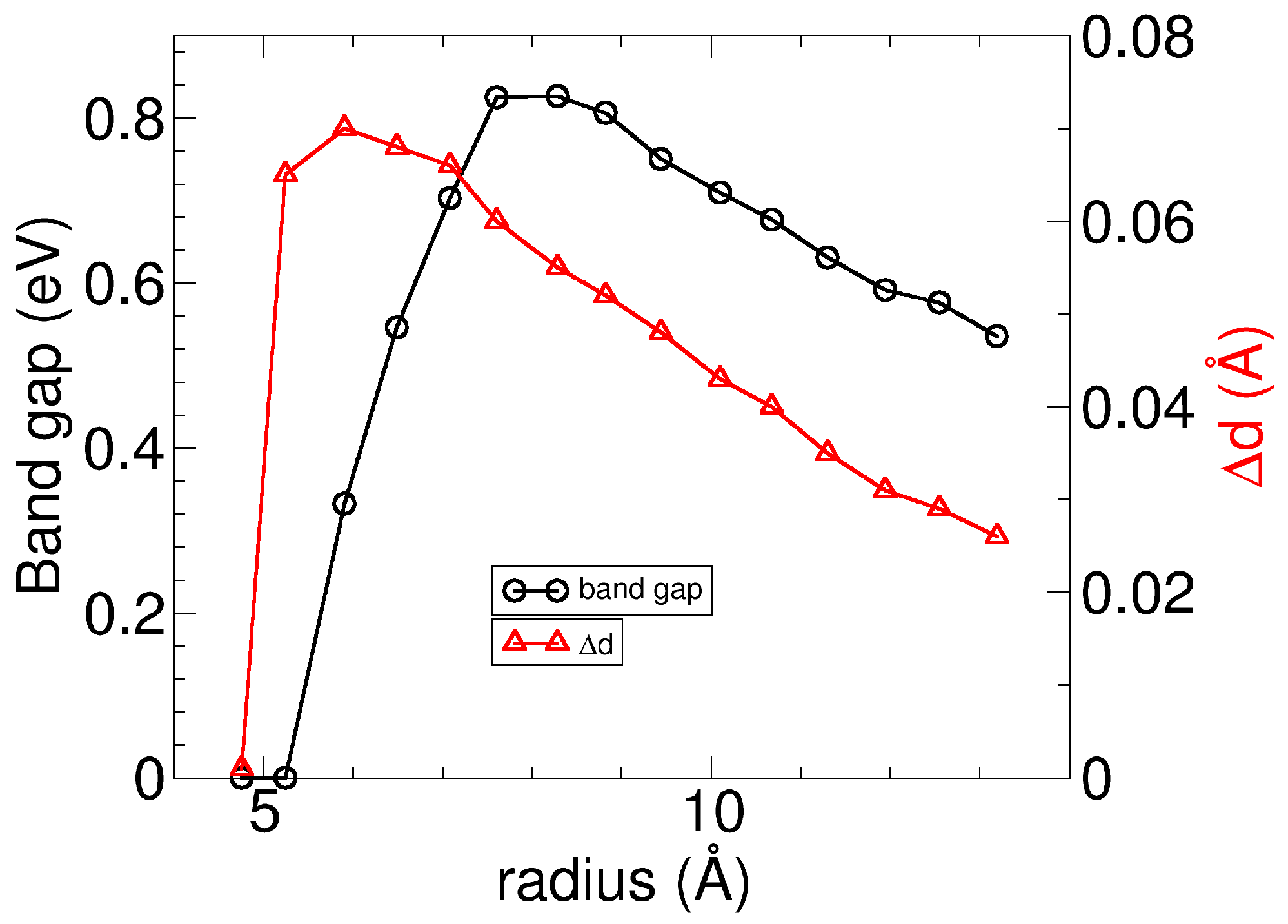
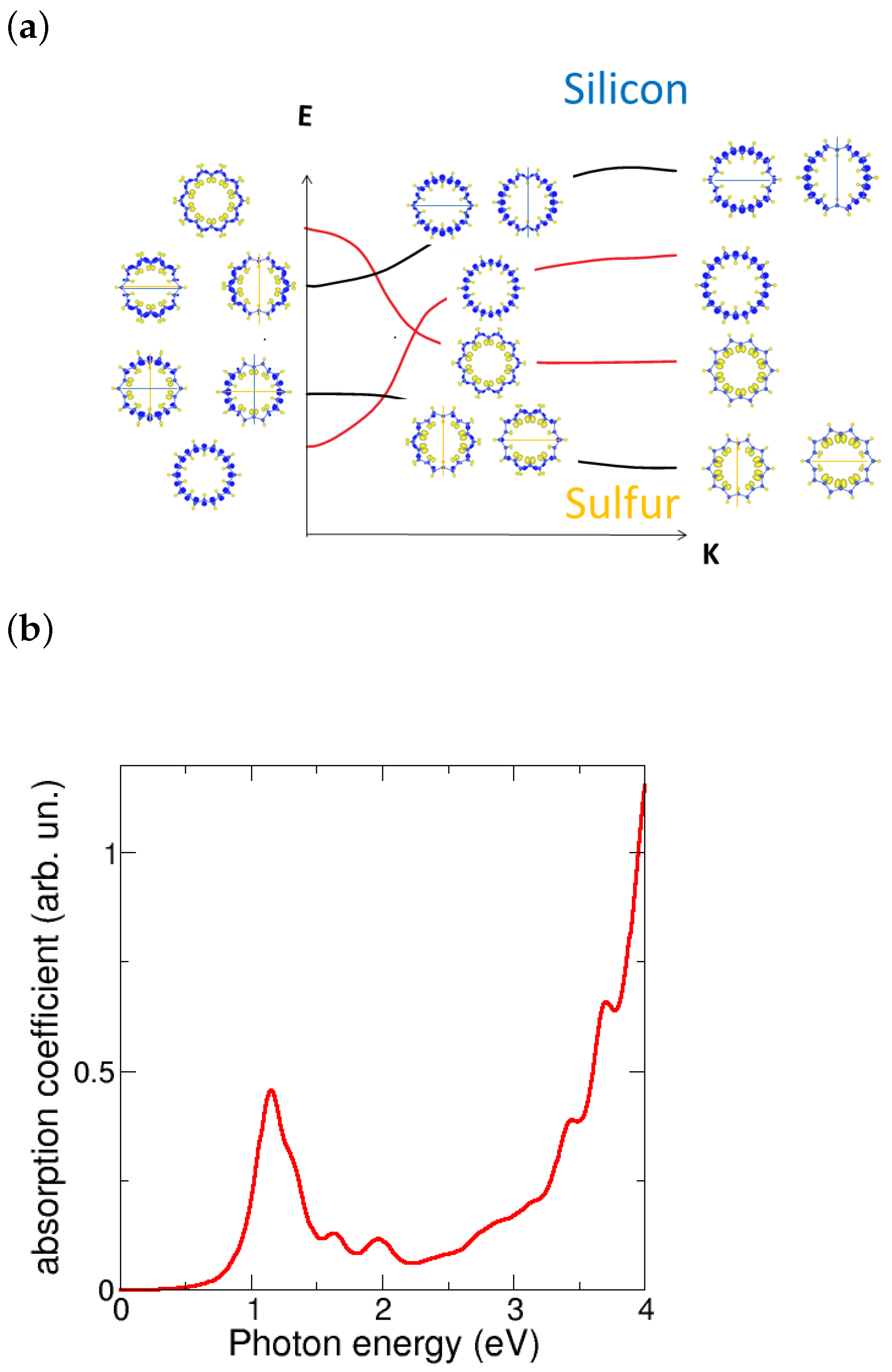
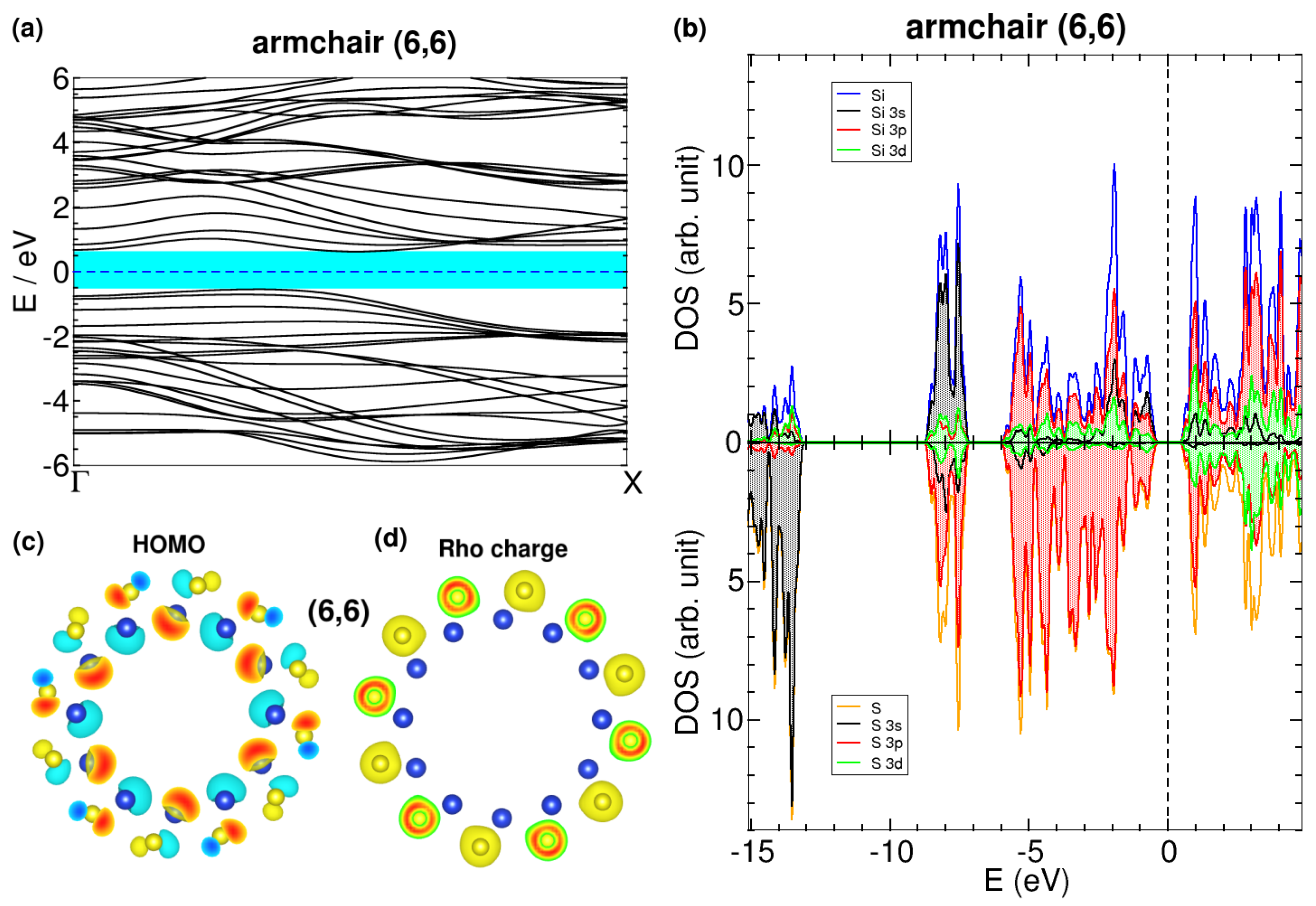
3.1. Pmma SiS Nanotubes: Dimerization Effects
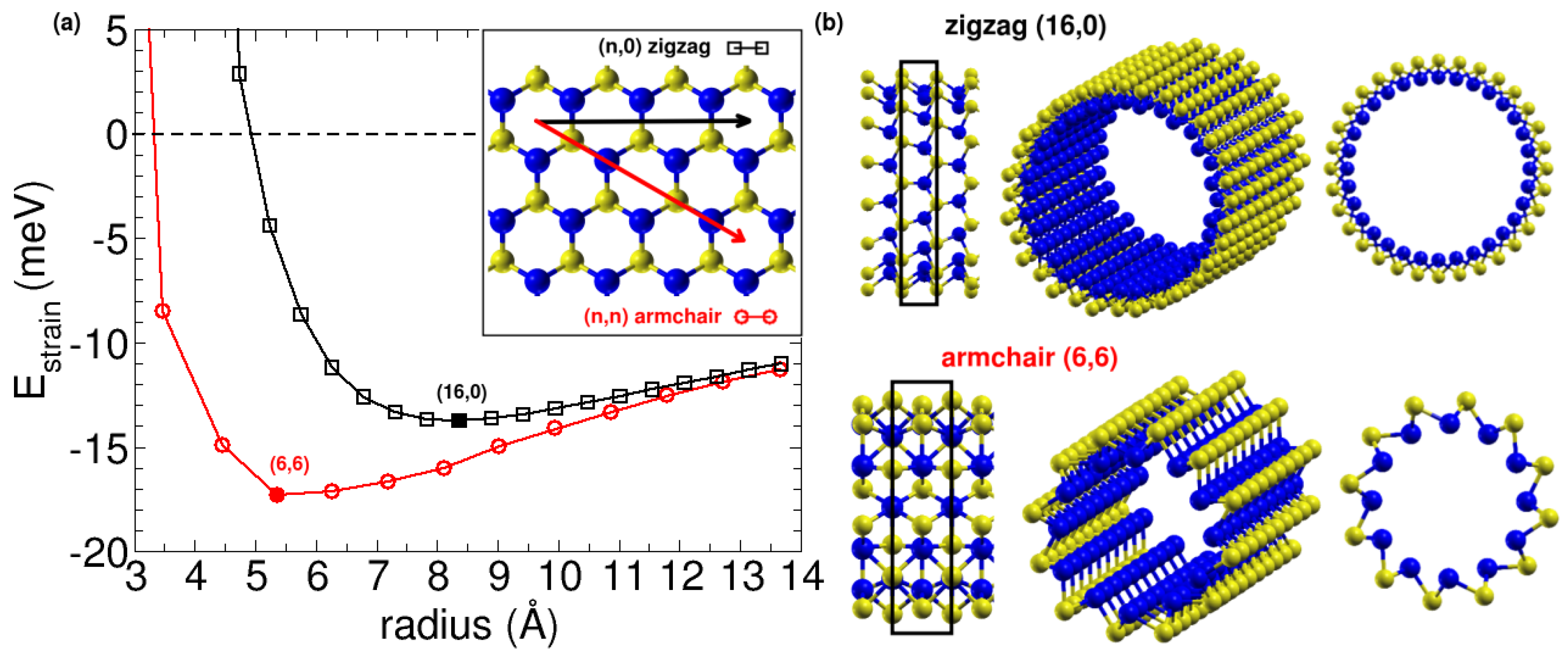
3.2. Nanotubes Rolling up SiS Monolayers: Negative Strain Energy
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TMDCs | transition metal dichalcogenides |
| DFT | density functional theory |
| GGA | generalized gradient approximation |
| NT | nanotube |
Appendix A. Pmma SiS Nanotubes
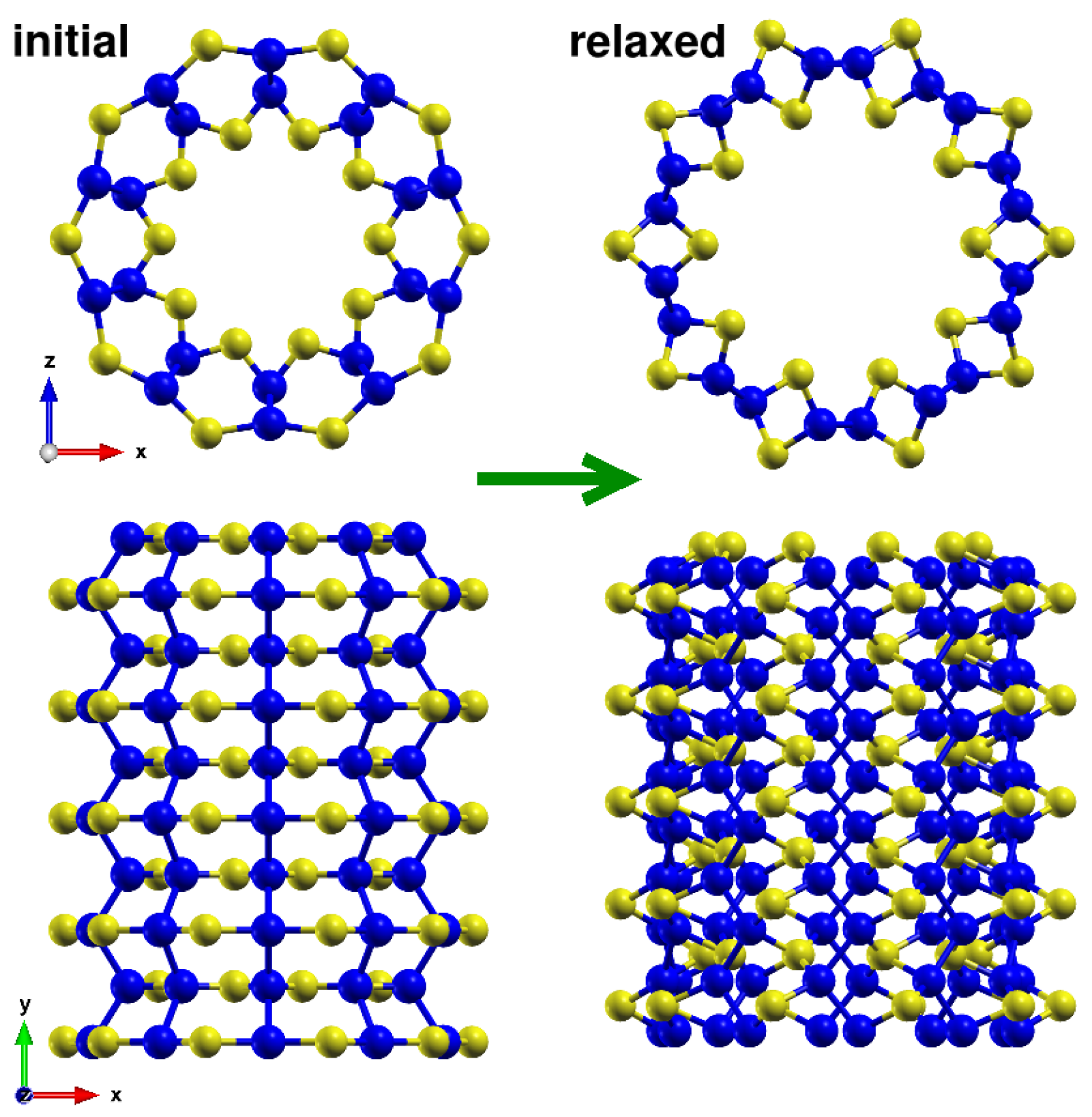
Appendix A.1. Reconstruction in (n,0) Nanotubes

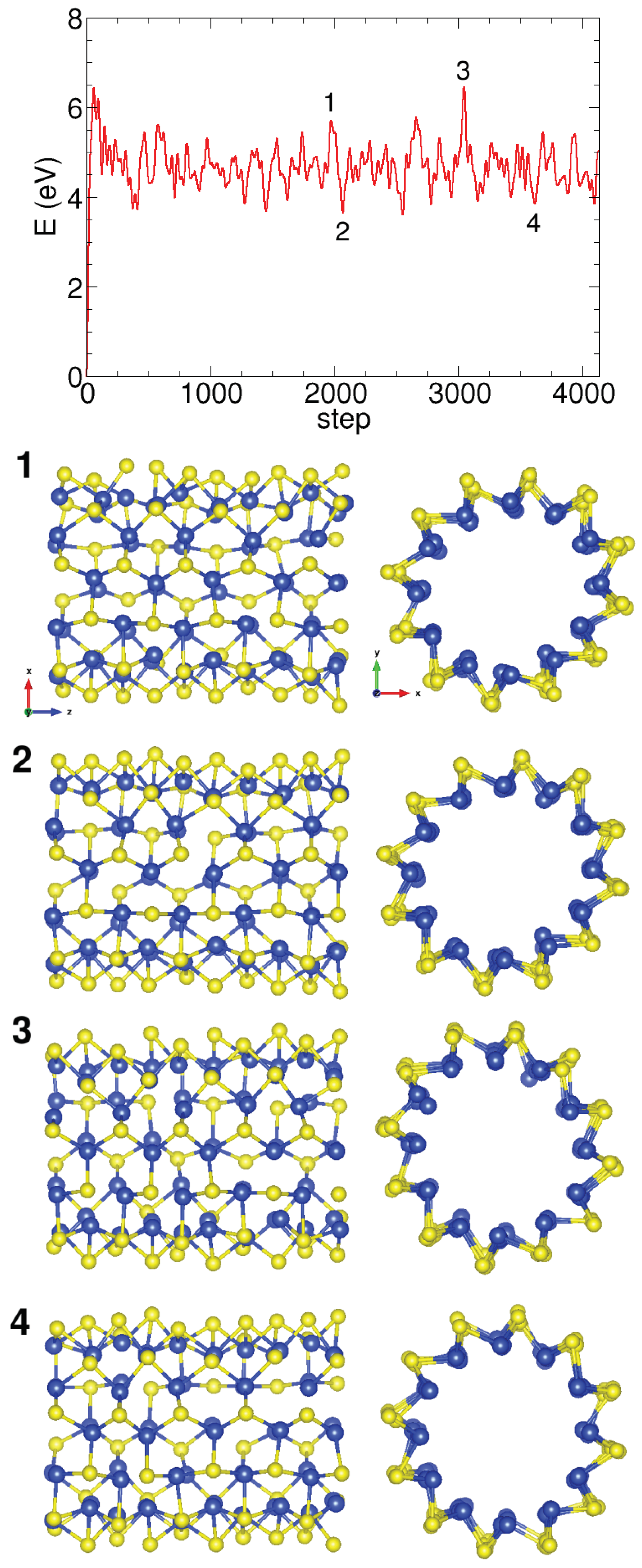
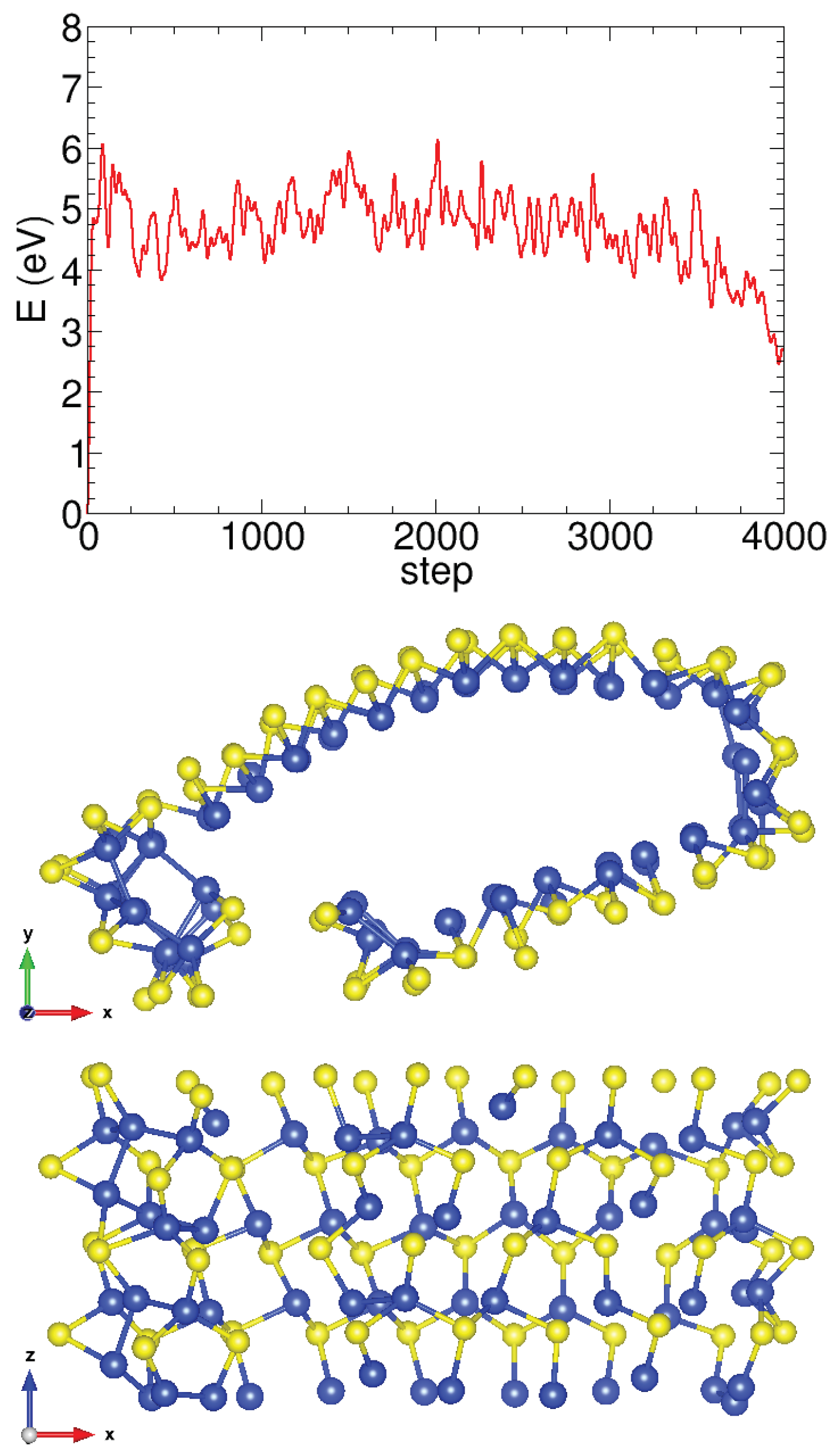
Appendix A.2. Molecular Dynamics

Appendix A.3. DOS of Dimerized Nanotube

Appendix B. Tests Using Hybrid Functionals

Appendix C. β SiS Nanotubes



References
- Tenne, R.; Margulis, L.; Genut, M.E.; Hodes, G. Polyhedral and cylindrical structures of tungsten disulphide. Nature 1992, 360, 444. [Google Scholar] [CrossRef]
- Margulis, L.; Salitra, G.; Tenne, R.; Talianker, M. Nested fullerene-like structures. Nature 1993, 365, 113. [Google Scholar] [CrossRef]
- Guimarães, L.; Enyashin, A.N.; Frenzel, J.; Heine, T.; Duarte, H.A.; Seifert, G. Imogolite nanotubes: Stability, electronic, and mechanical properties. ACS Nano 2007, 1, 362–368. [Google Scholar] [CrossRef]
- Chopra, N.G.; Luyken, R.; Cherrey, K.; Crespi, V.H.; Cohen, M.L.; Louie, S.G.; Zettl, A. Boron nitride nanotubes. Science 1995, 269, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Golberg, D.; Bando, Y.; Huang, Y.; Terao, T.; Mitome, M.; Tang, C.; Zhi, C. Boron nitride nanotubes and nanosheets. ACS Nano 2010, 4, 2979–2993. [Google Scholar] [CrossRef] [PubMed]
- Musfeldt, J.L.; Iwasa, Y.; Tenne, R. Nanotubes from layered transition metal dichalcogenides. Phys. Today 2020, 73, 42–48. [Google Scholar] [CrossRef]
- Radovsky, G.; Popovitz-Biro, R.; Staiger, M.; Gartsman, K.; Thomsen, C.; Lorenz, T.; Seifert, G.; Tenne, R. Synthesis of copious amounts of SnS2 and SnS2/SnS nanotubes with ordered superstructures. Angew. Chem. 2011, 123, 12524–12528. [Google Scholar] [CrossRef]
- Hettler, S.; Sreedhara, M.; Serra, M.; Sinha, S.S.; Popovitz-Biro, R.; Pinkas, I.; Enyashin, A.N.; Tenne, R.; Arenal, R. YS-TaS2 and YxLa1−xS-TaS2 (0≤ x≤ 1) Nanotubes: A Family of Misfit Layered Compounds. ACS Nano 2020, 14, 5445–5458. [Google Scholar] [CrossRef]
- Allec, S.I.; Wong, B.M. Inconsistencies in the electronic properties of phosphorene nanotubes: New insights from large-scale DFT calculations. J. Phys. Chem. Lett. 2016, 7, 4340–4345. [Google Scholar] [CrossRef]
- Guan, J.; Zhu, Z.; Tománek, D. High stability of faceted nanotubes and fullerenes of multiphase layered phosphorus: A computational study. Phys. Rev. Lett. 2014, 113, 226801. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, P.; Xue, J. Ti2CO2 nanotubes with negative strain energies and tunable band gaps predicted from first-principles calculations. J. Phys. Chem. Lett. 2016, 7, 5280–5284. [Google Scholar] [CrossRef] [PubMed]
- Monet, G.; Amara, M.S.; Rouzière, S.; Paineau, E.; Chai, Z.; Elliott, J.D.; Poli, E.; Liu, L.M.; Teobaldi, G.; Launois, P. Structural resolution of inorganic nanotubes with complex stoichiometry. Nat. Commun. 2018, 9, 2033. [Google Scholar] [CrossRef] [PubMed]
- Paineau, E.; Monet, G.; Peyre, V.; Goldmann, C.; Rouzière, S.; Launois, P. Colloidal Stability of Imogolite Nanotube Dispersions: A Phase Diagram Study. Langmuir 2019, 35, 12451–12459. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Arenal, R.; Tenne, R. An overview of the recent advances in inorganic nanotubes. Nanoscale 2019, 11, 8073–8090. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhu, H.; Eshun, K.; Arab, A.; Badwan, A.; Li, Q. A computational study of the electronic properties of one-dimensional armchair phosphorene nanotubes. J. App. Phys. 2015, 118, 164306. [Google Scholar] [CrossRef]
- Zhu, Z.; Guan, J.; Liu, D.; Tománek, D. Designing isoelectronic counterparts to layered group V semiconductors. ACS Nano 2015, 9, 8284–8290. [Google Scholar] [CrossRef]
- Yang, J.H.; Zhang, Y.; Yin, W.J.; Gong, X.; Yakobson, B.I.; Wei, S.H. Two-Dimensional SiS Layers with Promising Electronic and Optoelectronic Properties: Theoretical Prediction. Nano Lett. 2016, 16, 1110–1117. [Google Scholar] [CrossRef]
- Alonso-Lanza, T.; Ayuela, A.; Aguilera-Granja, F. An array of layers in silicon sulfides: Chainlike and monolayer. Phys. Rev. B 2016, 94, 245441. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, X.; Wang, C.; Peng, L.; Qian, Q.; Wang, S. Layer-dependent electronic properties of phosphorene-like materials and phosphorene-based van der Waals heterostructures. Nanoscale 2017, 9, 8616–8622. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for Ab Initio Total-Energy Calc. Using A Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Souza, A.M.; Rocha, A.R.; Fazzio, A.; da Silva, A.J.R. Ab-initio calculations for a realistic sensor: A study of CO sensors based on nitrogen-rich carbon nanotubes. AIP Adv. 2012, 2, 032115. [Google Scholar] [CrossRef]
- Zanolli, Z.; Charlier, J.C. Defective carbon nanotubes for single-molecule sensing. Phys. Rev. B 2009, 80, 155447. [Google Scholar] [CrossRef]
- Hobi, E., Jr.; Pontes, R.B.; Fazzio, A.; da Silva, A.J. Formation of atomic carbon chains from graphene nanoribbons. Phys. Rev. B 2010, 81, 201406. [Google Scholar] [CrossRef]
- Adams, G.B.; Sankey, O.F.; Page, J.B.; O’Keeffe, M.; Drabold, D.A. Energetics of large fullerenes: Balls, tubes, and capsules. Science 1992, 256, 1792–1795. [Google Scholar] [CrossRef]
- Robertson, D.H.; Brenner, D.W.; Mintmire, J.W. Energetics of nanoscale graphitic tubules. Phys. Rev. B 1992, 45, 12592–12595. [Google Scholar] [CrossRef]
- Seifert, G.; Terrones, H.; Terrones, M.; Jungnickel, G.; Frauenheim, T. Structure and Electronic Properties of MoS2 Nanotubes. Phys. Rev. Lett. 2000, 85, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Barik, G.; Pal, S. Structural, Electronic, and Mechanical Properties of Nitrogen Nanotubes: The Effect of Size and Strain. J. Phys. Chem. C 2023, 127, 21704–21712. [Google Scholar] [CrossRef]
- Matsuda, Y.; Tahir-Kheli, J.; Goddard III, W.A. Definitive band gaps for single-wall carbon nanotubes. J. Phys. Chem. Lett. 2010, 1, 2946–2950. [Google Scholar] [CrossRef]
- Dolado, J.S.; Goracci, G.; Arrese-Igor, S.; Ayuela, A.; Torres, A.; Liberal, I.; Beruete, M.; Gaitero, J.J.; Cagnoni, M.; Cappelluti, F. Radiative Cooling Properties of Portlandite and Tobermorite: Two Cementitious Minerals of Great Relevance in Concrete Science and Technology. ACS Appl. Optic. Mater. 2023. [Google Scholar] [CrossRef]
- Poli, E.; Elliott, J.; Ratcliff, L.E.; Andrinopoulos, L.; Dziedzic, J.; Hine, N.; Mostofi, A.A.; Skylaris, C.K.; Haynes, P.D.; Teobaldi, G. The potential of imogolite nanotubes as (co-) photocatalysts: A linear-scaling density functional theory study. J. Phys. Condens. Matter 2016, 28, 074003. [Google Scholar] [CrossRef]
- Zhang, J.L.; Zhao, S.; Han, C.; Wang, Z.; Zhong, S.; Sun, S.; Guo, R.; Zhou, X.; Gu, C.D.; Yuan, K.D.; et al. Epitaxial growth of single layer blue phosphorus: A new phase of two-dimensional phosphorus. Nano Lett. 2016, 16, 4903–4908. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Lanza, T.; Aguilera-Granja, F.; Ayuela, A. Dimerization Effects and Negative Strain Energy in Silicon Monosulfide Nanotubes. Nanomaterials 2023, 13, 3033. https://doi.org/10.3390/nano13233033
Alonso-Lanza T, Aguilera-Granja F, Ayuela A. Dimerization Effects and Negative Strain Energy in Silicon Monosulfide Nanotubes. Nanomaterials. 2023; 13(23):3033. https://doi.org/10.3390/nano13233033
Chicago/Turabian StyleAlonso-Lanza, Tomás, Faustino Aguilera-Granja, and Andrés Ayuela. 2023. "Dimerization Effects and Negative Strain Energy in Silicon Monosulfide Nanotubes" Nanomaterials 13, no. 23: 3033. https://doi.org/10.3390/nano13233033
APA StyleAlonso-Lanza, T., Aguilera-Granja, F., & Ayuela, A. (2023). Dimerization Effects and Negative Strain Energy in Silicon Monosulfide Nanotubes. Nanomaterials, 13(23), 3033. https://doi.org/10.3390/nano13233033