

Silicate Dissolution Mechanism from Metakaolinite Using Density Functional Theory




Abstract
:1. Introduction
2. Materials and Computational Approach
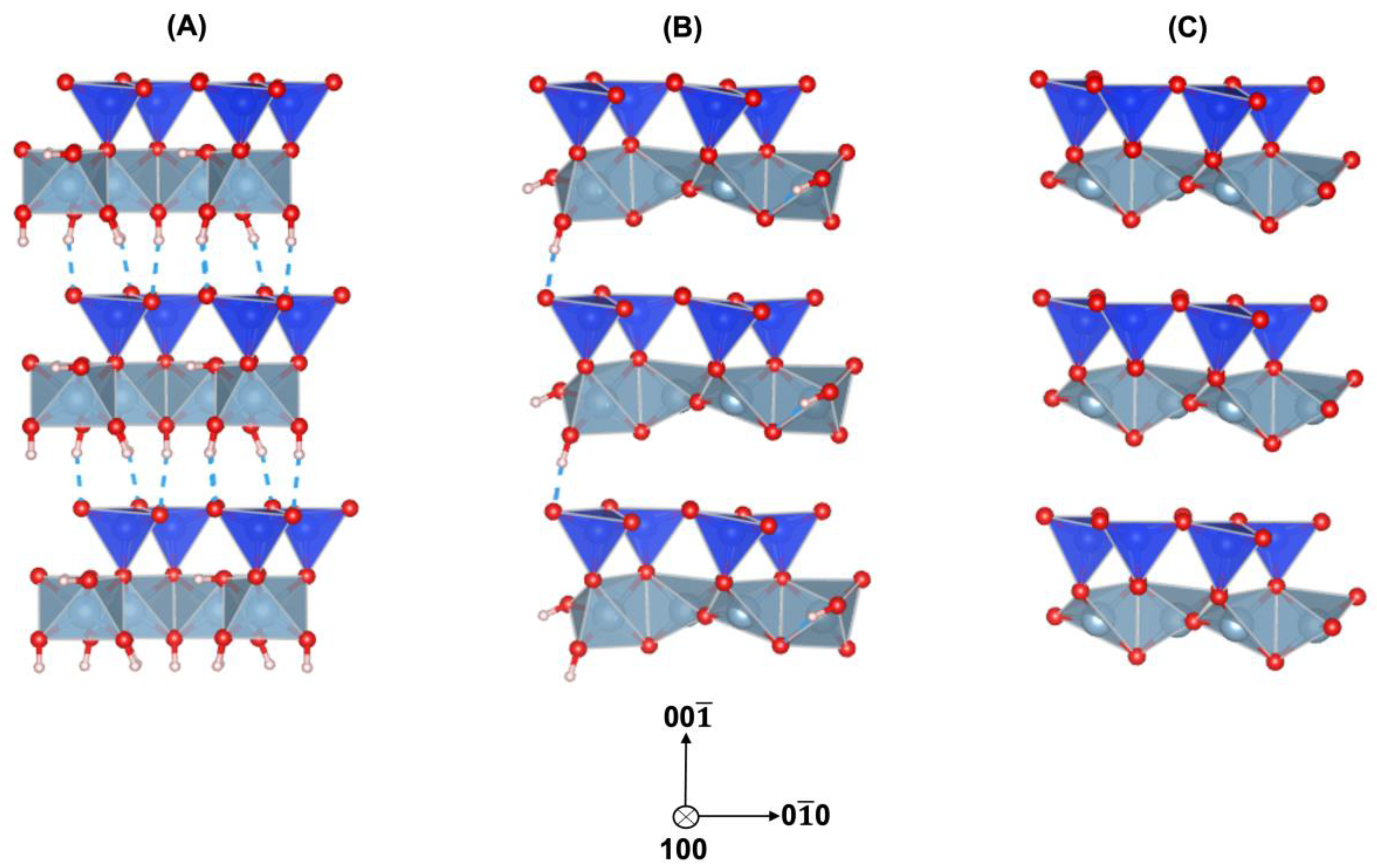
2.1. Structural Preparation
2.2. Density Functional Theory (DFT) Calculation
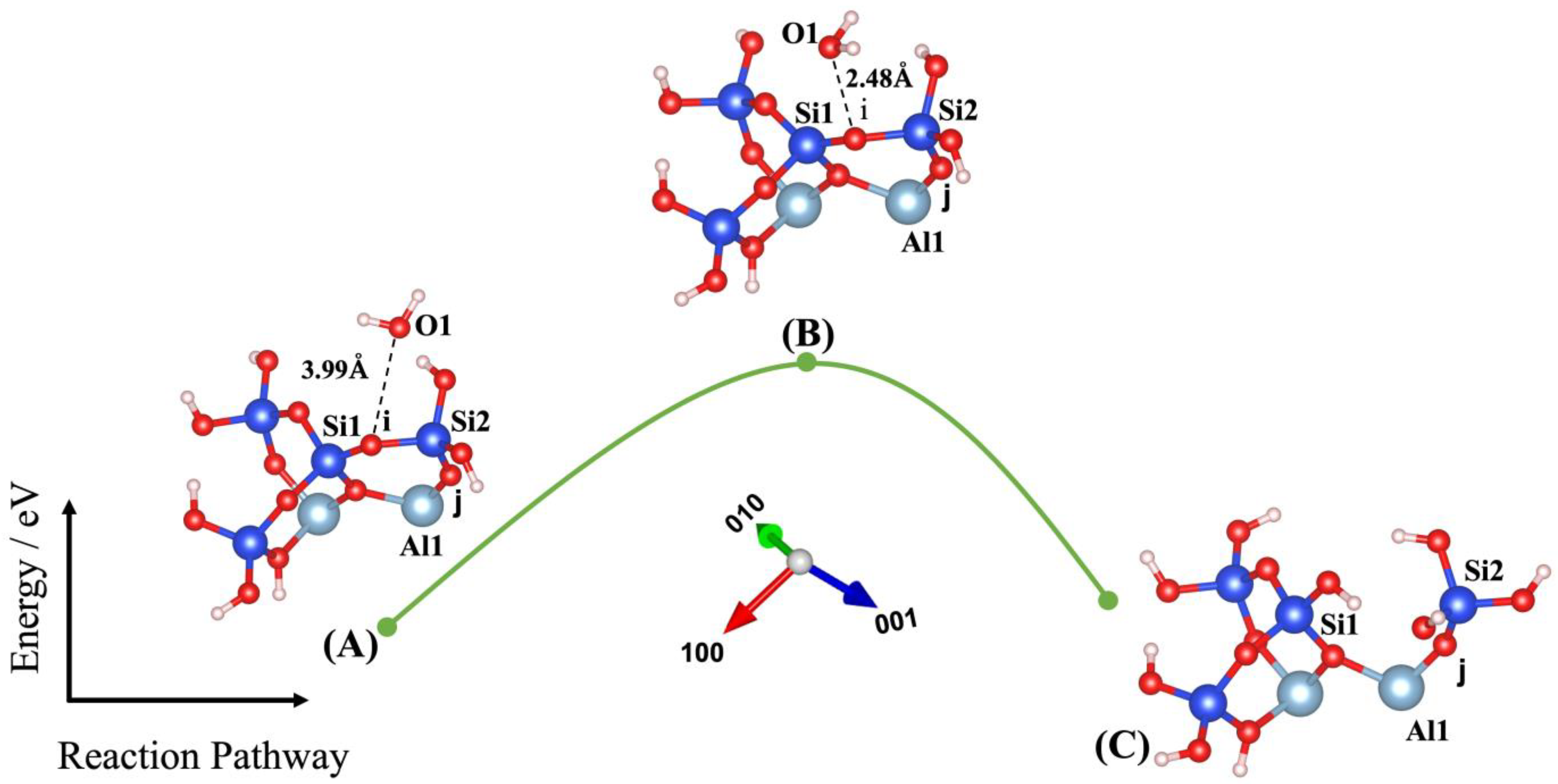
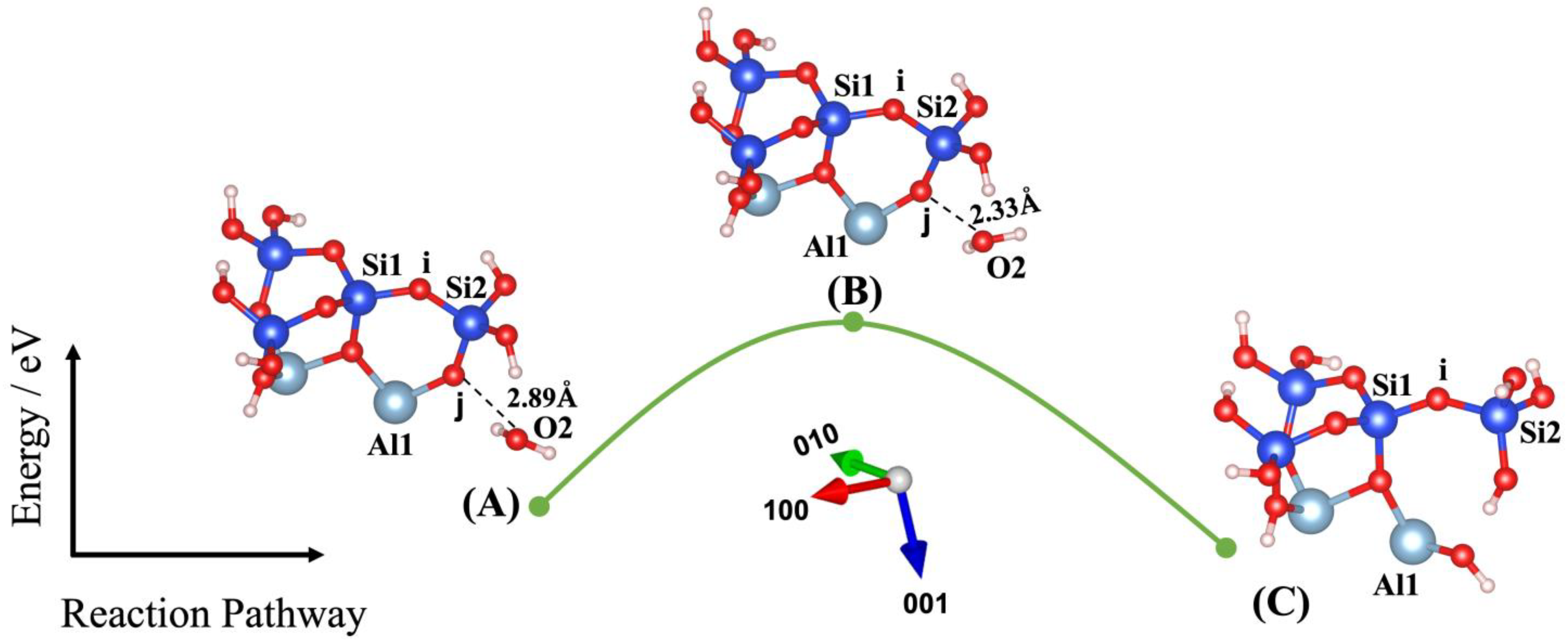
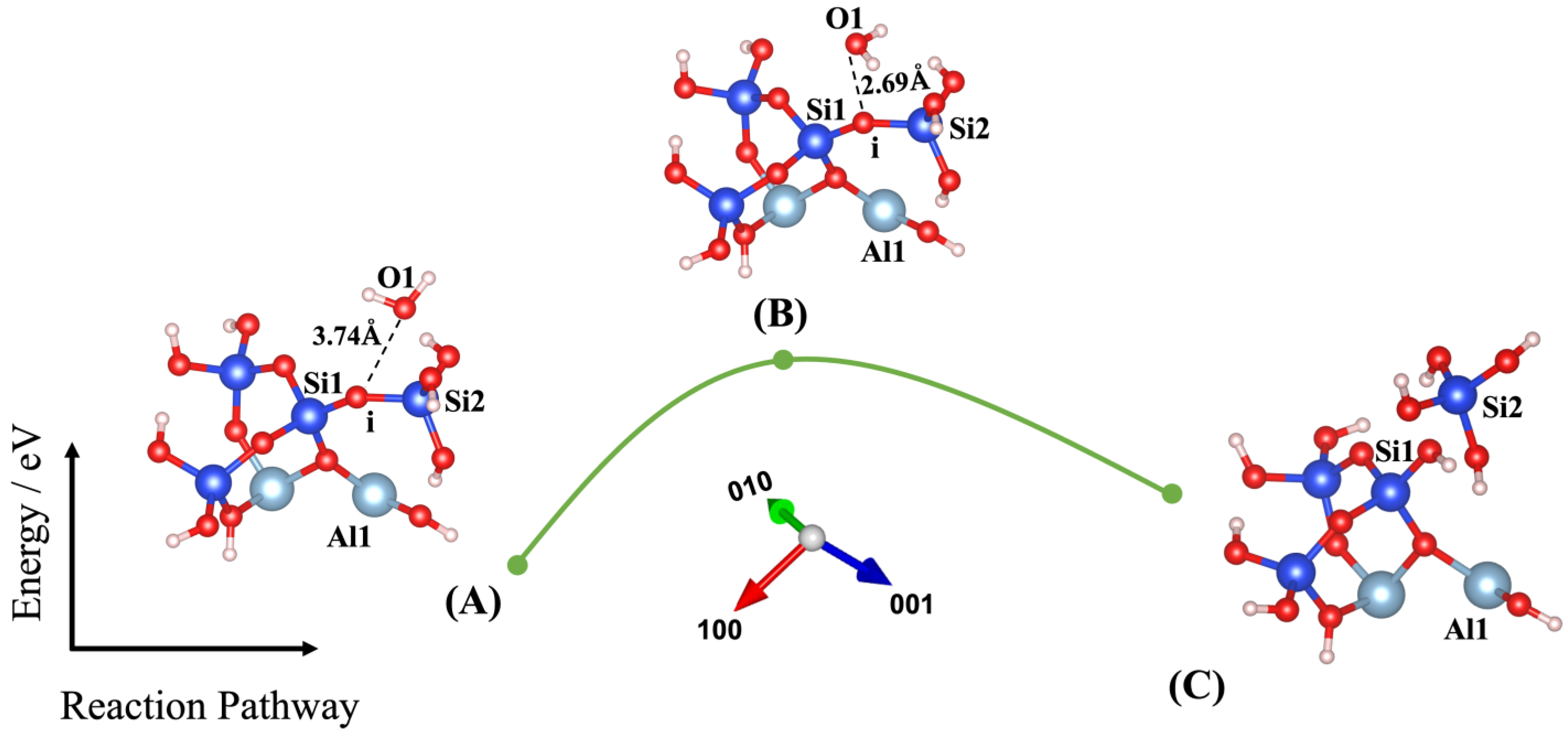
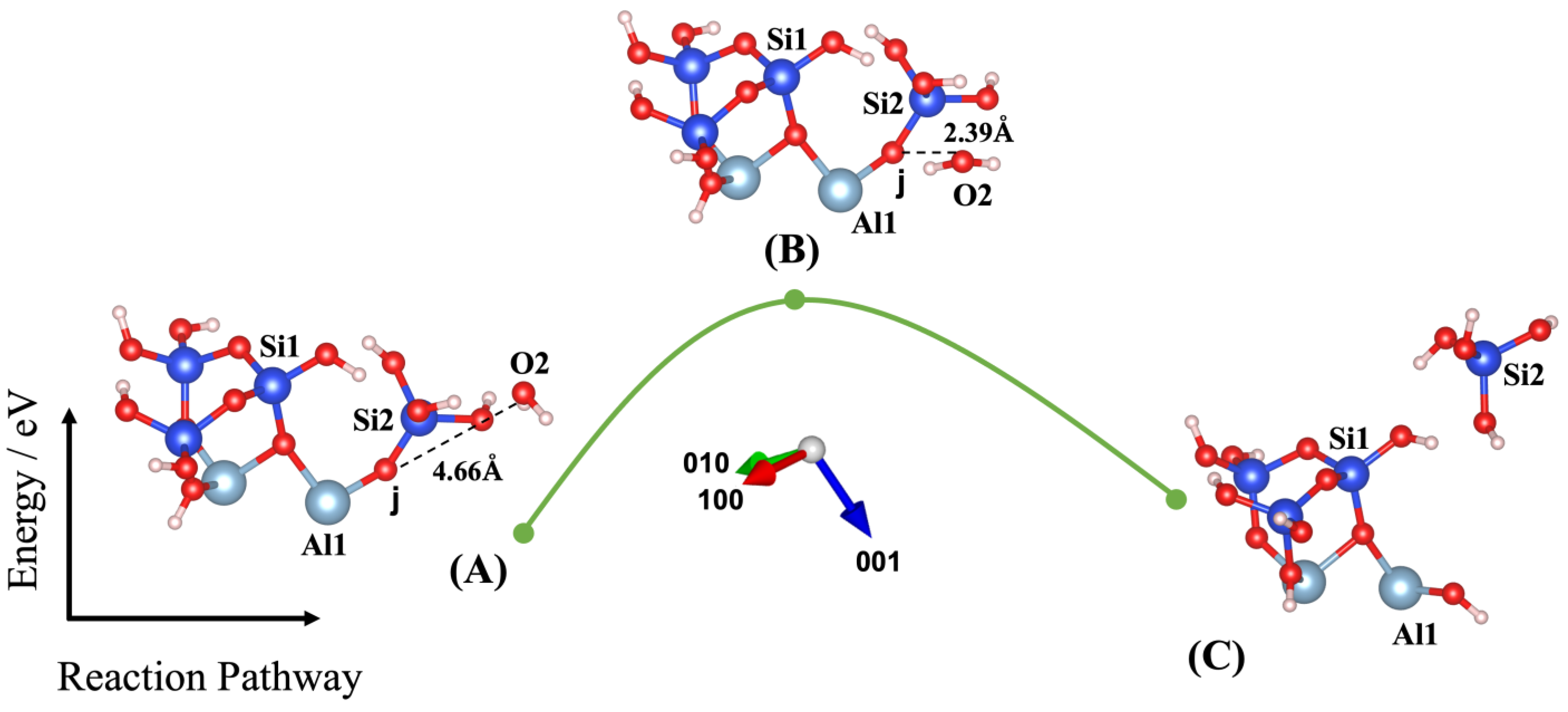
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worrell, E.; Price, L.; Martin, N.; Hendriks, C.; Meida, L.O. Carbon dioxide emissions from the global cement industry. Annu. Rev. Energy Environ. 2001, 26, 303–329. [Google Scholar] [CrossRef]
- Davidovits, J. Geopolymers: Inorganic polymeric new materials. J. Therm. Anal. 1991, 37, 1633–1656. [Google Scholar] [CrossRef]
- Skibsted, J.; Snellings, R. Reactivity of supplementary cementitious materials (SCMs) in cement blends. Cem. Concr. Res. 2019, 124, 105799. [Google Scholar] [CrossRef]
- El-Diadamony, H.; Amer, A.A.; Sokkary, T.M.; El-Hoseny, S. Hydration and characteristics of metakaolin pozzolanic cement pastes. HBRC J. 2018, 14, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Izadifar, M.; Thissen, P.; Steudel, A.; Kleeberg, R.; Kaufhold, S.; Kaltenbach, J.; Schuhmann, R.; Dehn, F.; Emmerich, K. Comprehensive examination of dehydroxylation of kaolinite, disordered kaolinite, and dickite: Experimental studies and density functional theory. Clays Clay Miner. 2020, 68, 319–333. [Google Scholar] [CrossRef]
- Garg, N.; Skibsted, J. Dissolution kinetics of calcined kaolinite and montmorillonite in alkaline conditions: Evidence for reactive Al(V) sites. J. Am. Ceram. Soc. 2019, 102, 7720–7734. [Google Scholar] [CrossRef]
- Briki, Y.; Zajac, M.; Ben Haha, M.; Scrivener, K. Factors affecting the reactivity of slag at early and late ages. Cem. Concr. Res. 2021, 150, 106604. [Google Scholar] [CrossRef]
- Werling, N.; Dehn, F.; Krause, F.; Steudel, A.; Schuhmann, R.; Emmerich, K. Solubility of precursors and carbonation of waterglass-free geopolymers. Clays Clay Miner. 2020, 68, 524–531. [Google Scholar] [CrossRef]
- Uddin, K.M.S.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of Portlandite in Pure Water: Part 1 Molecular Dynamics (MD) Approach. Materials 2022, 15, 1404. [Google Scholar] [CrossRef]
- Uddin, K.M.S.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets. Materials 2022, 15, 6388. [Google Scholar] [CrossRef]
- Izadifar, M.; Ukrainczyk, N.; Uddin, K.S.; Middendorf, B.; Koenders, E. Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials 2022, 15, 1442. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Ukrainczyk, N.; Uddin, K.M.S.; Middendorf, B.; Koenders, E. Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach. Materials 2022, 15, 6716. [Google Scholar] [CrossRef]
- Gong, K.; White, C.E. Predicting CaO-(MgO)-Al2O3-SiO2 glass reactivity in alkaline environments from force field molecular dynamics simulations. Cem. Concr. Res. 2021, 150, 106588. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Abadi, R.; Jam, A.N.; Gohari, S.; Burvill, C.; Rabczuk, T. Fracture toughness of various percentage of doping of boron atoms on the mechanical properties of polycrystalline graphene: A molecular dynamics study. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 114, 113614. [Google Scholar] [CrossRef]
- Izadifar, M.; Abadi, R.; Shirazi, A.H.N.; Alajlan, N.; Rabczuk, T. Nanopores creation in boron and nitrogen doped polycrystalline graphene: A molecular dynamics study. Phys. E Low-Dimens. Syst. Nanostruct. 2018, 99, 24–36. [Google Scholar] [CrossRef]
- Izadifar, M.; Abadi, R.; Jam, A.N.; Rabczuk, T. Investigation into the effect of doping of boron and nitrogen atoms in the mechanical properties of single-layer polycrystalline graphene. Comput. Mater. Sci. 2017, 138, 435–447. [Google Scholar] [CrossRef]
- Mortazavi, B.; Cuniberti, G.; Rabczuk, T. Mechanical properties and thermal conductivity of graphitic carbon nitride: A molecular dynamics study. Comput. Mater. Sci. 2015, 99, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Mortazavi, B.; Aldakheel, F. Molecular Dynamics Modeling of Mechanical Properties of Polymer Nanocomposites Reinforced by C7N6 Nanosheet. Surfaces 2021, 4, 240–254. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. A comprehensive stochastic model of phyllosilicate dissolution: Structure and kinematics of etch pits formed on muscovite basal face. Geochim. Cosmochim. Acta 2013, 120, 545–560. [Google Scholar] [CrossRef]
- Izadifar, M.; Valencia, N.C.; Xiao, P.; Ukrainczyk, N.; Koenders, E. 3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels. Materials 2023, 16, 1863. [Google Scholar] [CrossRef]
- White, C.E.; Provis, J.L.; Proffen, T.; van Deventer, J.S.J. Molecular mechanisms responsible for the structural changes occurring during geopolymerization: Multiscale simulation. AIChE J. 2012, 58, 2241–2253. [Google Scholar] [CrossRef]
- Morrow, C.P.; Nangia, S.; Garrison, B.J. Ab Initio Investigation of Dissolution Mechanisms in Aluminosilicate Minerals. J. Phys. Chem. A 2009, 113, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Schliemann, R.; Churakov, S.V. Pyrophyllite dissolution at elevated pressure conditions: An ab initio study. Geochim. Cosmochim. Acta 2021, 307, 42–55. [Google Scholar] [CrossRef]
- Schliemann, R.; Churakov, S.V. Atomic scale mechanism of clay minerals dissolution revealed by ab initio simulations. Geochim. Cosmochim. Acta 2021, 293, 438–460. [Google Scholar] [CrossRef]
- Tian, K.V.; Mahmoud, M.Z.; Cozza, P.; Licoccia, S.; Fang, D.C.; Di Tommaso, D.; Chass, G.A.; Greaves, G.N. Periodic vs. molecular cluster approaches to resolving glass structure and properties: Anorthite a case study. J. Non-Cryst. Solids 2016, 451, 138–145. [Google Scholar] [CrossRef]
- Salha, M.S.; Yada, R.Y.; Farrar, D.H.; Chass, G.A.; Tian, K.V.; Bodo, E. Aluminium catalysed oligomerisation in cement-forming silicate systems. Phys. Chem. Chem. Phys. 2023, 25, 455–461. [Google Scholar] [CrossRef]
- Pelmenschikov, A.; Strandh, H.; Pettersson, L.G.M.; Leszczynski, J. Lattice Resistance to Hydrolysis of Si−O−Si Bonds of Silicate Minerals: Ab Initio Calculations of a Single Water Attack onto the (001) and (111) β-Cristobalite Surfaces. J. Phys. Chem. B 2000, 104, 5779–5783. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Izadifar, M.; Königer, F.; Gerdes, A.; Wöll, C.; Thissen, P. Correlation between Composition and Mechanical Properties of Calcium Silicate Hydrates Identified by Infrared Spectroscopy and Density Functional Theory. J. Phys. Chem. C 2019, 123, 10868–10873. [Google Scholar] [CrossRef]
- Izadifar, M.; Dolado, J.S.; Thissen, P.; Ayuela, A. Interactions between Reduced Graphene Oxide with Monomers of (Calcium) Silicate Hydrates: A First-Principles Study. Nanomaterials 2021, 11, 2248. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Natzeck, C.; Emmerich, K.; Weidler, P.G.; Gohari, S.; Burvill, C.; Thissen, P. Unexpected Chemical Activity of a Mineral Surface: The Role of Crystal Water in Tobermorite. J. Phys. Chem. C 2022, 126, 12405–12412. [Google Scholar] [CrossRef]
- Blöchl, B.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Heyden, A.; Bell, A.T.; Keil, F.J. Efficient methods for finding transition states in chemical reactions: Comparison of improved dimer method and partitioned rational function optimization method. J. Chem. Phys. 2005, 123, 224101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Liu, C.; Meng, C. Oligomerization of Silicic Acids in Neutral Aqueous Solution: A First-Principles Investigation. IJMS 2019, 20, 3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Brantley, S.L. Temperature- and pH-dependence of albite dissolution rate at acid pH. Chem. Geol. 1997, 135, 275–290. [Google Scholar] [CrossRef]
- Nangia, S.; Garrison, B.J. Reaction Rates and Dissolution Mechanisms of Quartz as a Function of pH. J. Phys. Chem. A 2008, 112, 2027–2033. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | ΔEa (eV) | ΔEa (kJ/mol) | E_Reactant (eV) | E_Transition State (eV) | E_Product (eV) | ΔEe (eV) | Owater–Ooxo Distance (Transition State) (Å) | Imaginary Frequencies (THz) |
|---|---|---|---|---|---|---|---|---|
| (1) | 1.753 | 169.138 | −178.155 | −176.402 | −177.983 | 0.172 | 2.480 | −74.480 |
| (2) | 0.891 | 85.968 | −177.920 | −177.029 | −178.044 | −0.124 | 2.330 | −74.106 |
| (3) | 1.420 | 137.009 | −192.843 | −191.423 | −191.900 | 0.943 | 2.690 | −76.330 |
| (4) | 1.641 | 158.330 | −192.664 | −191.023 | −191.900 | 0.764 | 2.390 | −81.593 |
| Models | O1-i (Å) | O1-Si1 (Å) | O1-Si2 (Å) | Si1-i (Å) | Si2-i (Å) | O2-j (Å) | O2-Si1 (Å) | O2-Si2 (Å) | O2-Al1 (Å) | Al1-j (Å) | Si2-j (Å) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (1) | 3.99 | 4.23 | 3.79 | 1.61 | 1.67 | - | - | - | - | 1.76 | 1.62 |
| (2) | - | - | - | 1.62 | 1.66 | 2.89 | 5.58 | 3.39 | 4.02 | 1.78 | 1.64 |
| (3) | 3.74 | 4.09 | 3.86 | 1.60 | 1.66 | - | - | - | - | - | - |
| (4) | - | - | - | - | - | 4.66 | 6.35 | 3.37 | 5.97 | 1.75 | 1.61 |
| Models | O1-i (Å) | O1-Si1 (Å) | O1-Si2 (Å) | Si1-i (Å) | Si2-i (Å) | O2-j (Å) | O2-Si1 (Å) | O2-Si2 (Å) | O2-Al1 (Å) | Al1-j (Å) | Si2-j (Å) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (1) | 2.48 | 2.54 | 3.15 | 1.65 | 1.67 | - | - | - | - | 1.78 | 1.62 |
| (2) | - | - | - | 1.61 | 1.66 | 2.33 | 5.09 | 2.79 | 3.44 | 1.74 | 1.63 |
| (3) | 2.69 | 2.89 | 3.49 | 1.64 | 1.67 | - | - | - | - | - | - |
| (4) | - | - | - | - | - | 2.39 | 5.11 | 2.16 | 3.33 | 1.79 | 1.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izadifar, M.; Ukrainczyk, N.; Koenders, E. Silicate Dissolution Mechanism from Metakaolinite Using Density Functional Theory. Nanomaterials 2023, 13, 1196. https://doi.org/10.3390/nano13071196
Izadifar M, Ukrainczyk N, Koenders E. Silicate Dissolution Mechanism from Metakaolinite Using Density Functional Theory. Nanomaterials. 2023; 13(7):1196. https://doi.org/10.3390/nano13071196
Chicago/Turabian StyleIzadifar, Mohammadreza, Neven Ukrainczyk, and Eduardus Koenders. 2023. "Silicate Dissolution Mechanism from Metakaolinite Using Density Functional Theory" Nanomaterials 13, no. 7: 1196. https://doi.org/10.3390/nano13071196
APA StyleIzadifar, M., Ukrainczyk, N., & Koenders, E. (2023). Silicate Dissolution Mechanism from Metakaolinite Using Density Functional Theory. Nanomaterials, 13(7), 1196. https://doi.org/10.3390/nano13071196