Templated Synthesis of Magnetic Nanoparticles through the Self-Assembly of Polymers and Surfactants



Abstract
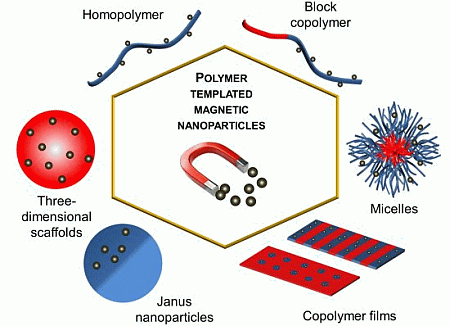
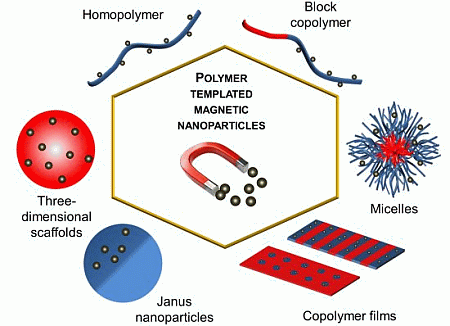
:
1. Introduction
2. In Situ Synthesis in Non-Polymeric Templates
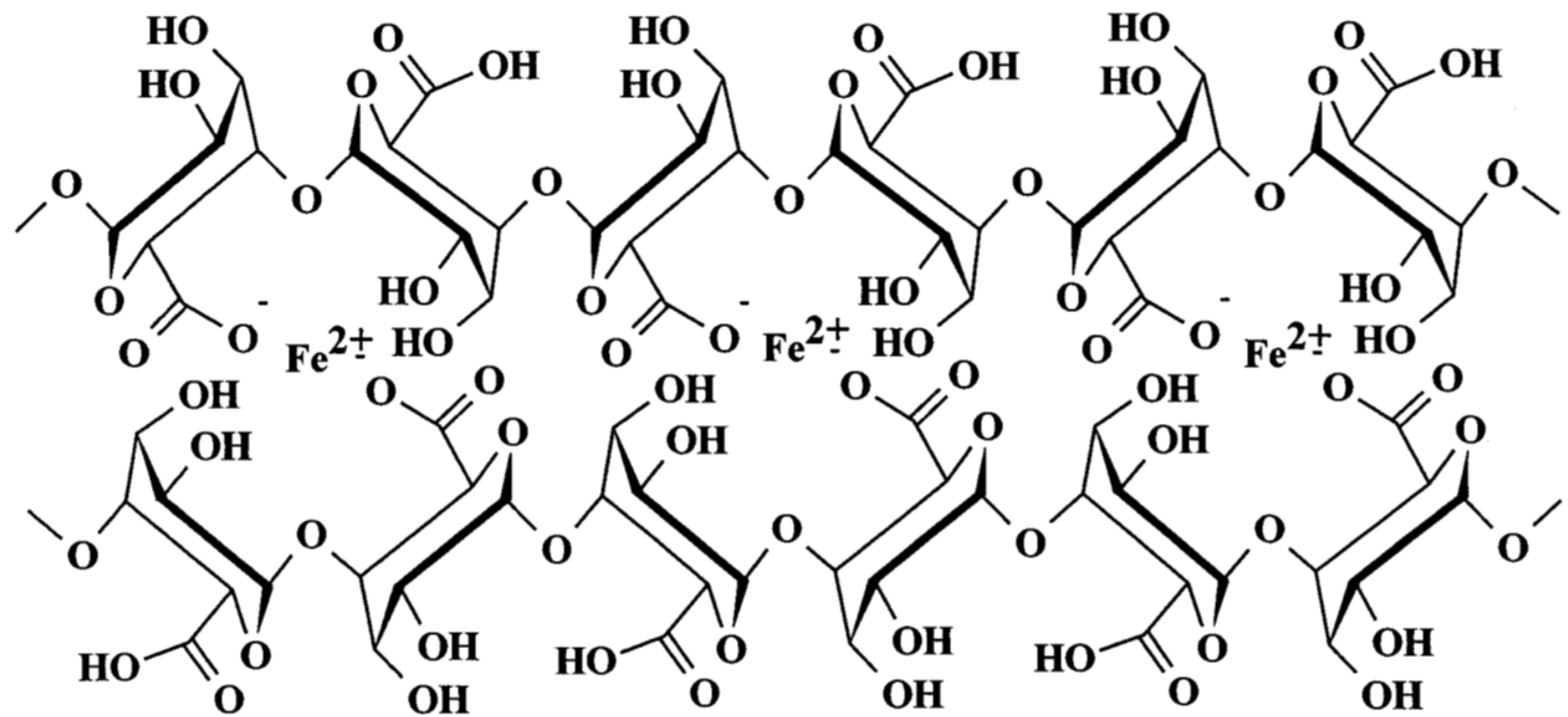
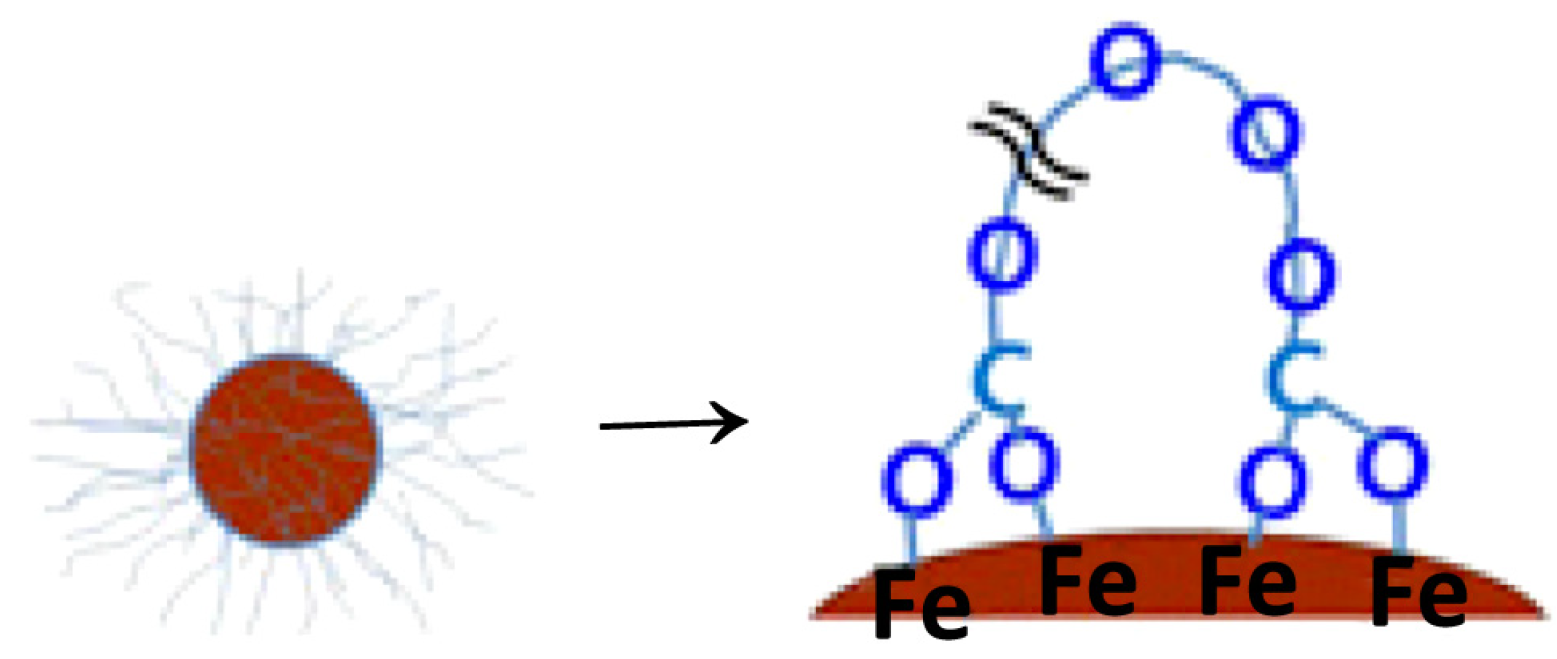
2.1. Carboxylates
Oleic Acid Coating in Combination with Reduction

2.2. Sulfonates and Sulfates
2.2.1. Sodium Dodecyl Sulfate (SDS) Microemulsions
2.2.2. Sodium Dodecylbenzenesulfonate (NaDBS) Microemulsions
3. In Situ Synthesis with Polymers in Solution or in the Bulk State
3.1. Dextran (DEX) and Polysaccharide Derivatives
3.1.1. Influence of DEX End-Group Reduction
3.1.2. DEX Sulfate
3.1.3. DEX-b-poly(methacrylic acid)(Fe3O4)-b-Poly(N-Isopropylacrylamide) with Thiol End Groups
3.1.4. Alginate (Alg) Beads

3.1.5. Precipitation in Aqueous Alginate Networks
3.2. Synthetic Linear Homopolymers
3.2.1. Thermal Decomposition of Poly(Vinyl Alcohol)-Fe(OH)3
3.2.2. Fe2+/3+ Coprecipitation in PVA Membranes
3.2.3. Conversion of Fe3+ in PVA into Hematite in Supercritical H2O
3.2.4. Fe(acac)3 Decomposition/Diol Reduction in Octyl Ether (OE) with Polyvinylpyrrolidone (PVP)
3.2.5. Controlled Precipitation in the Presence of Poly(4-vinylpyridine) (P4VP)
3.2.6. Poly(ethylene oxide) PEO in the “One-Pot” Synthesis of Pegylated Oxide NPs
3.2.7. PEO in Alkaline Hydrothermal Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PEG:H2O (in volume) | Phases | Crystallite size (nm) |
|---|---|---|
| 1:3 | Fe3O4, α-Fe2O3 | 21.8 |
| 1:1 | Fe3O4, α-Fe2O3, γ-Fe2O3 | 24.7 |
| 3:1 | Fe3O4, α-Fe2O3, γ-Fe2O3 | 31.6 |
| 0:4 | Fe3O4, α-Fe2O3, γ-Fe2O3 | 42.2 |
| 4:0 | NaFeO2 | 64.5 |
| Conditions: temperature (°C)/time (h) | Phases | Crystallite size (nm) |
|---|---|---|
| 100/24 | Fe3O4 + NaFeS2(H2O)2 | 10.6 |
| 125/24 | Fe3O4 + α-Fe2O3 + NaFeS2(H2O)2 | 13.8 |
| 150/24 | Fe3O4 | 30.3 |
| 150/48 | Fe3O4 + α-Fe2O3 | 24.1 |
| 150/72 | Fe3O4 + α-Fe2O3 + γ-Fe2O3 | 28.4 |
3.2.8. Poly(acrylic acid) (PAA) Chains for Fe3O4 Growth Inhibition
3.2.9. FeCl3 Thermal Decomposition with PEG in Triethylene Glycol (Polyol)

3.2.10. Coprecipitation in the Presence of Trithiol-Terminated Poly(methacrylic Acid) (PMAA-PTTM)
3.2.11. Poly(Methyl Glutarimide) (PMGI) Templated Iron NPs by Spin-Coating
3.3. Synthetic Linear Copolymers
3.3.1. Aqueous Coprecipitation with Double-Hydrophilic Block Copolymers (DHBCs)
3.3.2. Aqueous Coprecipitation with Triple-Hydrophilic Block Copolymers (THBCs)

3.3.3. Fe2+/Fe3+ Coprecipitation with Silicon-Containing Random Copolymers
3.3.4. Coprecipitation in Water in the Presence of Poly(ethylene oxide)-Block-Poly(Methacrylic Acid) (PEO-b-PMAA) DHBCs

3.3.5. Poly(Ethylene Glycol)-Block-Poly(Aspartic Acid) (PEG-b-PAsp) Leading to Akaganeite Rods

3.3.6. Coprecipitation in Water with Poly(Ethylene Oxide)-Block-Poly(Acrylic Acid) (PEO-b-PAA)

3.3.7. Deuterated Poly(Norbornene) (PNOR) Block Copolymers


3.3.8. Poly(Styrene Sulfonate-Alt-maleic Acid) (PSS-Alt-MA) Shell with Subsequent Cross-Linking
| Cross-linking (%) | Hydrodynamic diameter (DLS) (nm) | Zeta potential (mV) |
|---|---|---|
| PSS-alt-MA before cross-linking | 44 ± 9 | −48.6 ± 4.7 |
| PAA before cross-linking | 83 ± 2 | −39.7 ± 1.5 |
| PAA, 12.5% cross-linking | 69 ± 17 | −48.6 ± 1.1 |
| PAA, 50% cross-linking | 126 ± 9 | −44.3 ± 2.2 |
| PAA, 12.5% cross-linking | 77 ± 16 | −47.1 ± 2.2 |
| PAA, 100% cross-linking | 91 ± 2 | −38.3 ± 3.0 |
3.3.9. Brush Linear Poly(oligo(ethylene Glycol) Methacrylate-co-methacrylic Acid) P(OEGMA-co-MAA) as Nucleating and Stabilizing Ligand
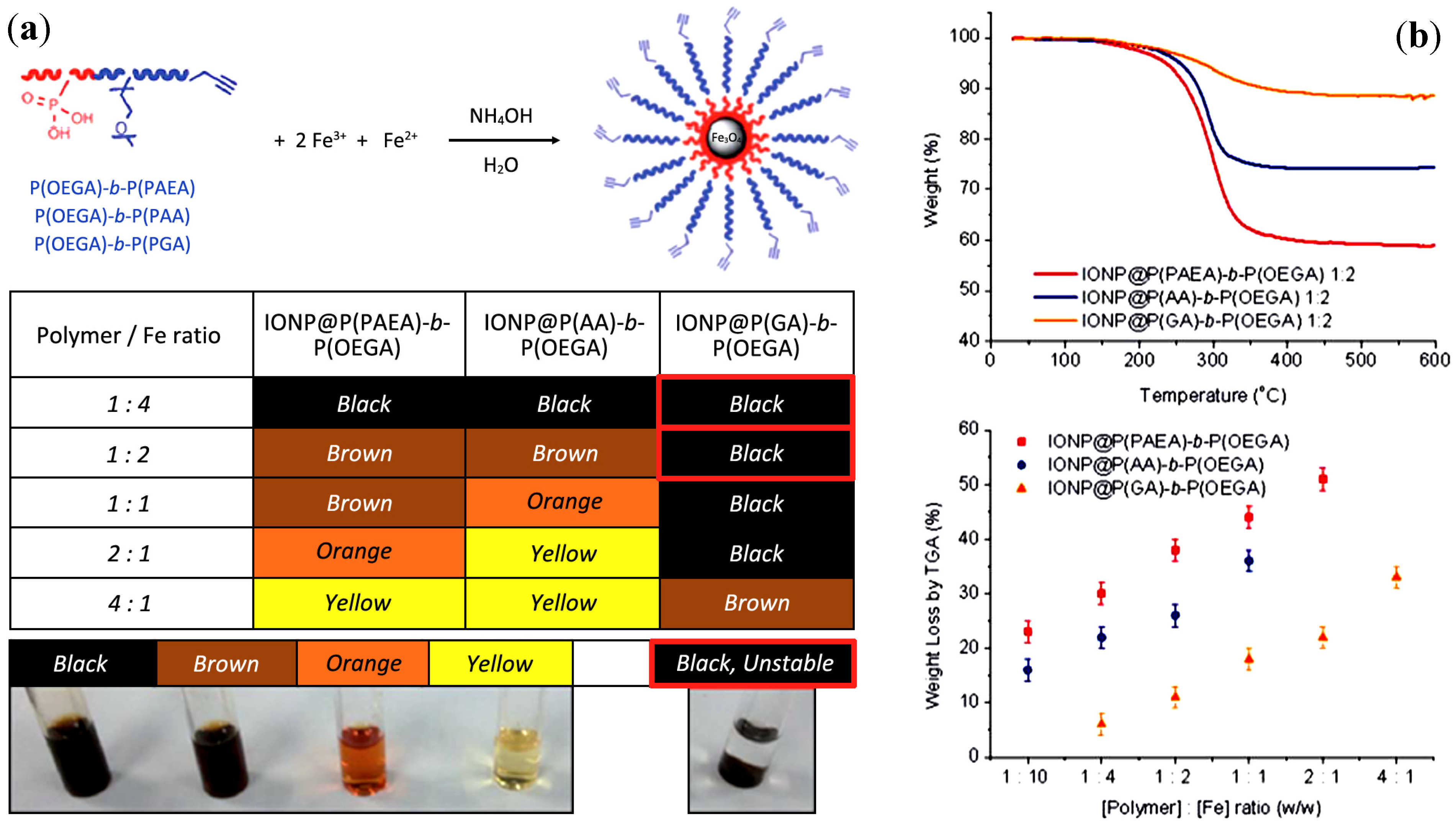
3.3.10. Coprecipitation with DHBCs of Poly(oligoethylene Glycol Acrylate) (POEGA) and Different Binding Blocks

| Hybrid magnetic core-shells | [Polymer]:[Fe] weight ratio | Wt loss TGA | dTEM (nm) | dXRD (nm) | Grafting density (nm−2) |
|---|---|---|---|---|---|
| IONP@P(PAEA)-b-P(OEGA) | 1:10 | 23% | 10.45 | 9.83 | 0.083 ± 0.005 |
| 1:4 | 30% | 9.80 | 9.10 | 0.124 ±0.003 | |
| 1:2 | 38% | 9.40 | 8.52 | 0.168 ± 0.008 | |
| 1:1 | 44% | 8.64 | 7.32 | 0.192 ± 0.01 | |
| 2:1 | 51% | - | - | - | |
| IONP@P(AA)-b-P(OEGA) | 1:10 | 16% | 10.25 | 10.09 | 0.065 ± 0.001 |
| 1:4 | 22% | 8.91 | 8.75 | 0.084 ± 0.003 | |
| 1:2 | 26% | 7.83 | 7.44 | 0.091 ± 0.003 | |
| 1:1 | 36% | - | - | - | |
| IONP@P(GA)-b-P(OEGA) | 1:4 | 6% | 12.39 | 11.47 | 0.023 ± 0.001 |
| 1:2 | 11% | 10.24 | 10.62 | 0.039 ± 0.001 | |
| 1:1 | 18% | 9.14 | 9.78 | 0.063 ± 0.002 | |
| 2:1 | 22% | 8.65 | 8.41 | 0.073 ± 0.001 | |
| 4:1 | 33% | - | - | - |
3.3.11. Spherical Micelles Loaded with IONPs

4. Synthesis Templated by Preformed Structures
4.1. Microemulsions in an Organic Solvent
4.1.1. Chitosan Shells in Fe2+ Microemulsions with Triton®-X

4.1.2. Quaternary Microemulsions to Produce Aligned Spinel CoFe2O4 Nanorods


4.2. Spherical Micelles in Water
4.2.1. Triblock Polyisoprene-Block-poly(2-cinnamoylethyl Methacrylate)-Block-Poly(Tert-Butyl Acrylate) (PI-b-PCEMA-b-PtBA) Copolymer Hollow Nanospheres


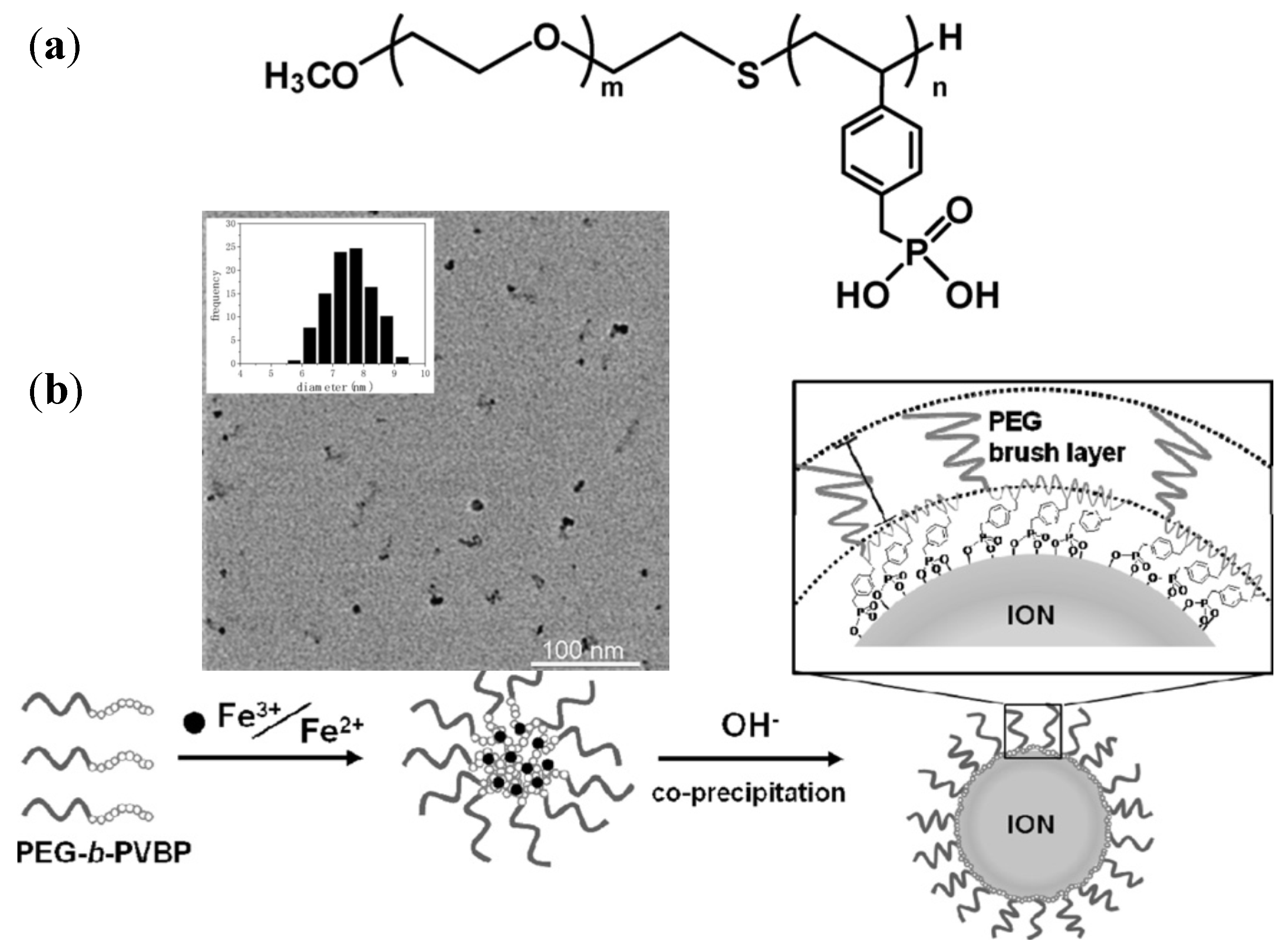
4.2.2. PEG-block-Poly(4-Vinylbenzylphosphonate) (PEG-b-PVBP) Micelles

4.2.3. Multiarm Star-Like Amphiphilic or DHBCs


4.3. Cylindrical Multimolecular Micelles

4.3.1. Fe3+ Loading in Poly(Acrylic Acid)-Graft-Poly(n-Butyl Acrylate) Brush

4.3.2. Fe3+/2+ Loading and Precipitation in Poly[Poly(Ethylene Glycol) Methylether Acrylate]-Graft-Poly(Methacrylic Acid) (PPEGMEA-g-PMAA) Brush

4.3.3. Poly(Glycerol Monoacrylate)-Graft-Poly(PEG Methyl Ether Acrylate) (PGA-g-PEG) Copolymers
4.3.4. Poly(Ethylene Oxide)-Graft-Poly(Acrylic Acid) (PEO-g-PAA) Graft Copolymer
4.4. Lamellar Films
Polystyrene-Block-Poly(2-Vinylpyridine) (PS-b-P2VP) Lamellae Hosting Pristine Iron Nanoparticles

4.5. Hexagonal Ordered Films
4.5.1. Monolayer Films of Polystyrene-block-Poly(4-Vinylpyridine) (PS-b-P4VP) Copolymer Micelles

4.5.2. Monolayer Films of Polystyrene-block-Poly(Ethylene Oxide) (PS-b-PEO) Copolymers


4.6. Holey Membranes
4.6.1. PVA-Fe3+/Citric Acid (CA)/Ethylene Glycol (EG) Calcination
4.6.2. Loading of the Pores of a Nafion® Membrane Followed by Reduction
4.7. Tridimensional Scaffolds (Macroscopic Samples)
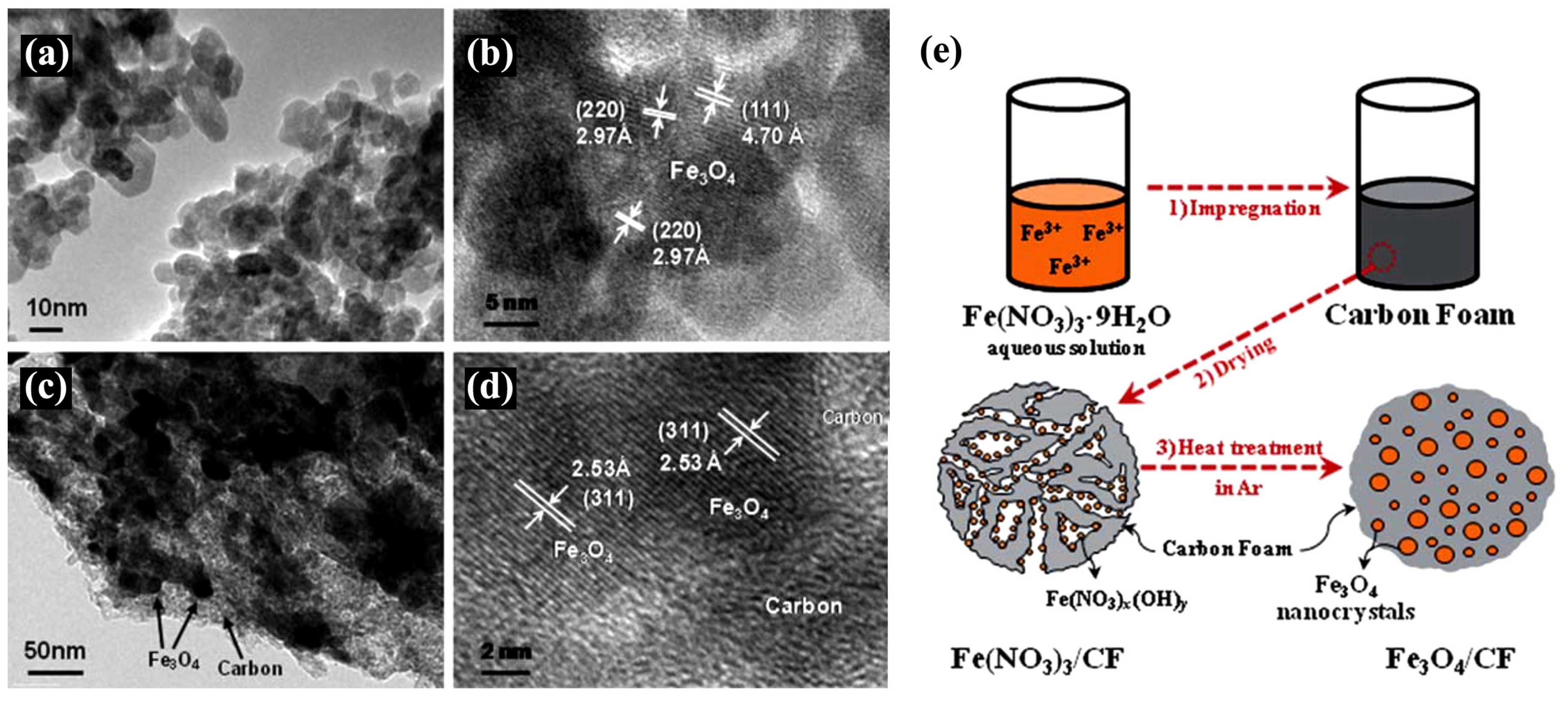
4.7.1. Porous Preformed Carbon Foams (CF)

4.7.2. Sponge-Like Polystyrene (PS)/Polyacrylate Copolymer Gel

4.7.3. Cross-Linked Poly[Styrene-co-(N-4-Carboxybutylmaleimide)] Copolymer
4.7.4. Semi-Interpenetrating (Semi-IPN) Polymer Networks of Alginate and Poly(N-Isopropylacryl Amide) (PNiPAAm)
4.7.5. Cross-Linked Polyacrylamide (PAAm) Hydrogels


4.7.6. Hydrogels of Poly(2-Acrylamido-2-Methyl-1-Propansulfonic Acid) (PAMPS) and P4VP
4.7.7. PAAm Hydrogels

4.8. Dispersed Colloids (Microscopic)
4.8.1. Poly(N-Isopropyl Acrylamide-co-Acrylic acid-co-2-Hydroxyethyl Acrylate) (Poly(NIPAM-co-AA-co-HEA)) Microgels Cross-Linked with N,N′-methylene Bisacrylamide (BIS)

4.8.2. Acetoacetoxyethyl Methacrylate (AAEM)-N-Vinylcaprolactam (VCL) Microgels

4.8.3. Sulfonated Copolymer Beads
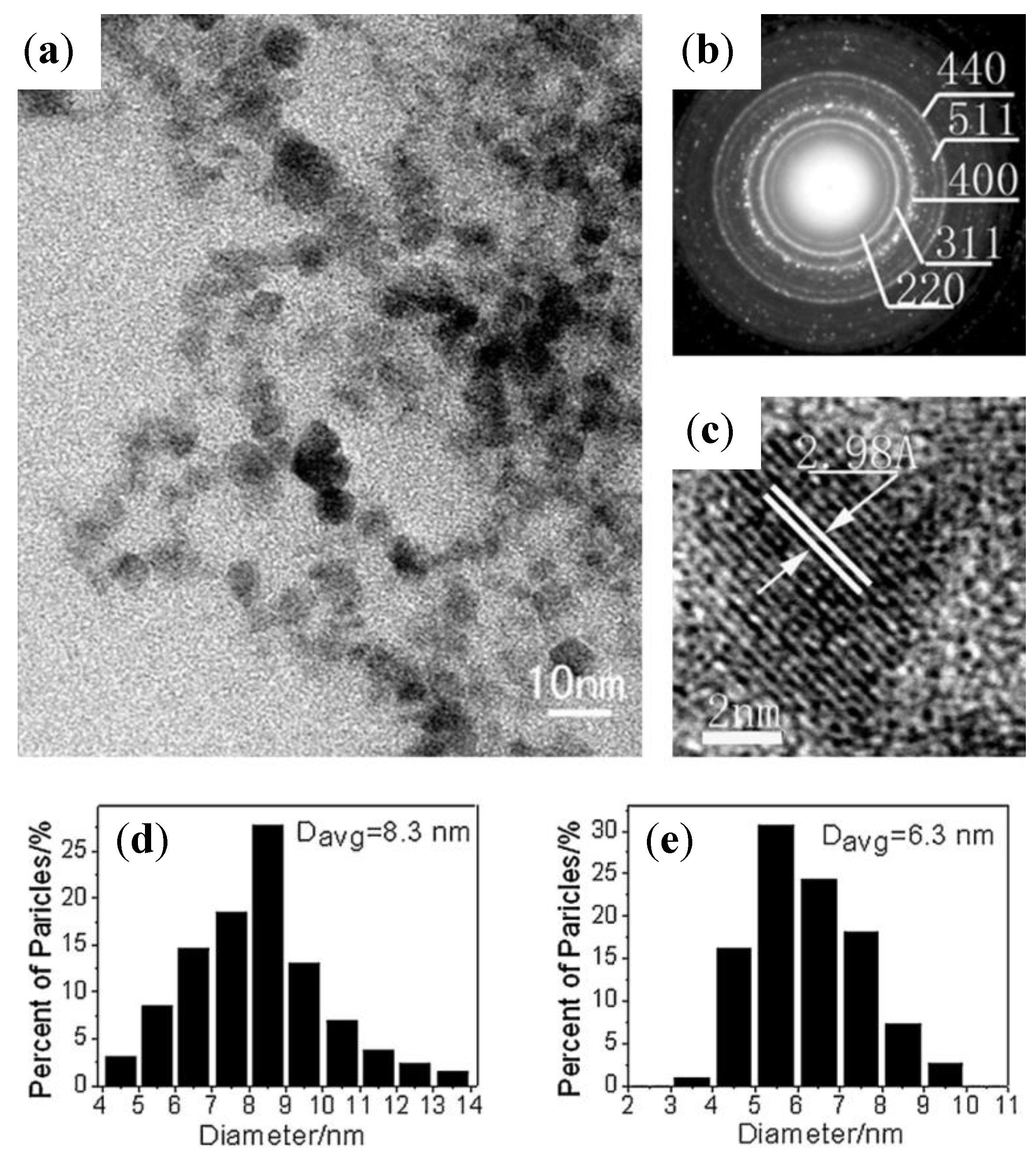
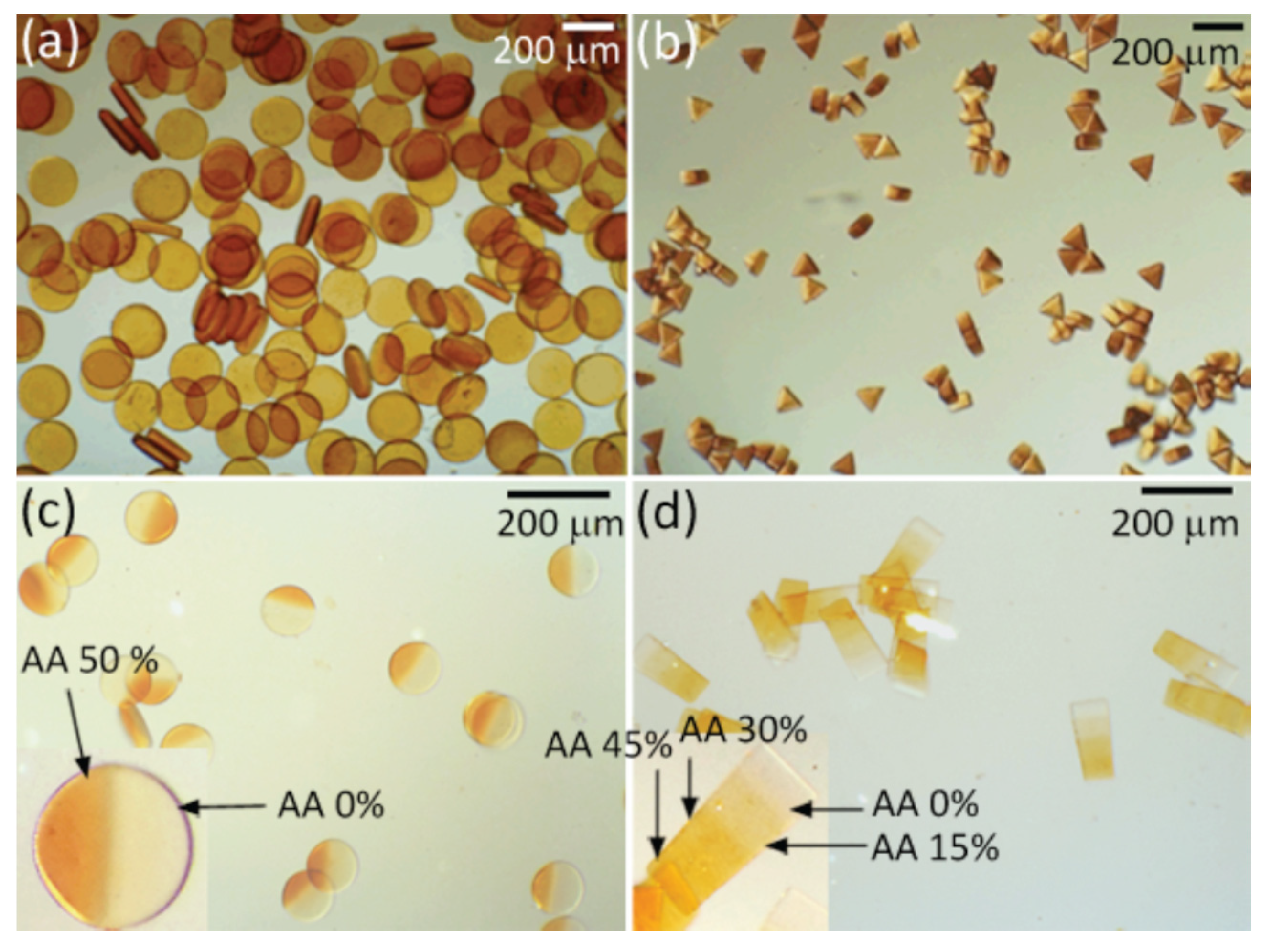
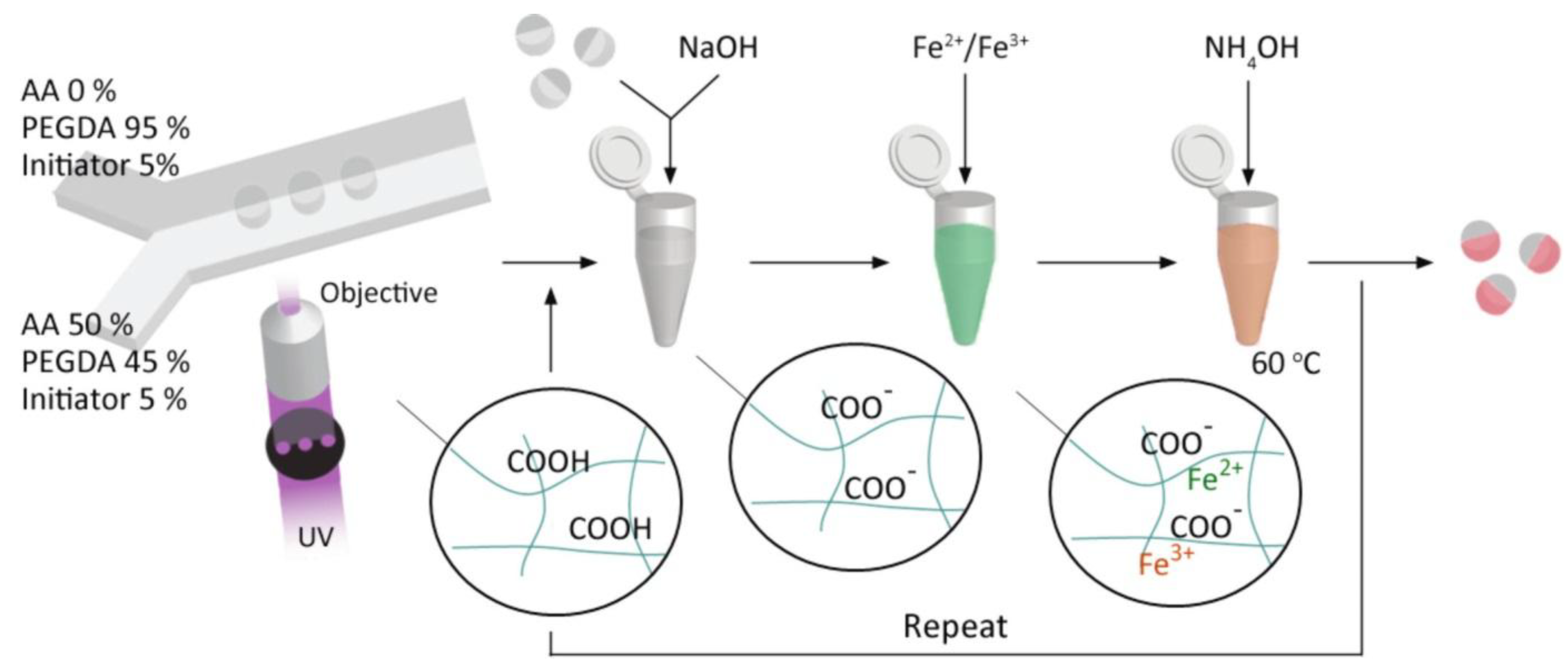
4.8.4. Poly[Poly(Ethylene Glycol Diacrylate-co-Acrylic acid) (PEGDA-co-PAA) Copolymer Hydrogels Prepared by Microfluidics


4.8.5. Multi-Responsive Microgels Made from VCL, AAEM and Vinylimidazole (VIm)

4.9. Preformed Microspheres in Organic Solvents
PS Microsphere Swollen in CHCl3

5. Conclusions
| Surfactants: –COO−, –SO3−, –SO4− Natural polysaccharides: DEX, DEX sulfate, Alg, DEX-b-PMAA-b-PNiPAAm | Synthetic linear polymers: PVA, PVP, P4VP, PEO, PAA, PMAA, PMGI Linear DHBC or amphiphilic copolymers | Hydrophilic blocks: iron-complexing (PGMA, PGA, P(norbornene-methanol or di-carboxy), PMAA, PAA, PAsp, PAEA, PAEMA) or repulsive block (PEO, PEOGMA, PDMAEMA) | Hydrophobic blocks: PMMA, PNOR, PVBP Tri-blocks: Pluronics®, PI-b-PCEMA-b-PtBA |
Three-dimensional scaffold macroscopic matrices: Carbon foam, sponge-like PS-co-PAA, P(S-co-CBMi) networks, Alg/PNiPAAm semi-IPN, PAAm, PAMPS, P(4VP-co-HEMA) hydrogels |  | Star-like (PPO-b-PAA, PEO-b-PS) Comb-like (PPEGMEA-g-PMAA, PEO-g-PAA) Spherical micelles (PS-b-P2VP) Cylindrical multi-molecular/unimolecular micelles | |
| Dispersed colloids: microgels, hydrophobic PS microbeads | Janus, gradient or triangle: PEGDA, PVCL-co-AAEM core-VIm shell microgels | Lamellar or hexagonally ordered cylinders (PS-b-P2VP, PS-b-PEO) Membranes (PVA, Nafion®) | Microemulsions: hexane/Triton®-X/Chitosan, CTAB |
Abbreviations and Acronyms
| acac | acetylacetonate |
| AAEM | acetoacetoxyethyl methacrylate |
| ADR | antitumor drug adriamycin |
| AFM | atomic force microscopy |
| Alg | alginate |
| AuNR | gold nanorods |
| BIS | N,N′-methylene bisacrylamide |
| CA | citric acid |
| CBMi | N-4-carboxybutylmaleimide |
| CD | cyclodextrin |
| CF | carbon foams |
| CNT | carbon nanotube |
| CHP | continuous hydrothermal processing |
| CTAB | cetyl trimethylammonium bromide |
| DEX | dextran |
| DHBC | double-hydrophilic block copolymer |
| DLS | dynamic light scattering |
| EDS | energy-dispersive X-ray spectroscopy |
| EFTEM | energy filtering transmission electron microscopy |
| EG | ethylene glycol |
| FC | field cooled |
| FE-SEM | field emission-scanning electron microscopy |
| FTIR | Fourier transform infrared spectroscopy |
| G | 1,4-linked α-l-guluronic acid |
| GDA | glutaraldehyde |
| Gly | glycidyl |
| Hc | coercivity |
| HEA | 2-hydroxyethyl acrylate |
| HGMS | high-gradient magnetic separator |
| HRTEM | high resolution transmission electron microscopy |
| IONP | iron oxide nanoparticle |
| LCST | lower critical solution temperature |
| M | 1,4-linked β-d-mannuronic acid |
| MA | maleic acid |
| MNP | magnetic nanoparticle |
| mPEG | poly(ethylene glycol) monomethyl |
| MRI | magnetic resonance imaging |
| Ms | saturation magnetization |
| NIR | near infrared radiation |
| MW | molecular weight |
| NaDBS | sodium dodecylbenzenesulfonate |
| NP | nanoparticle |
| OE | octyl ether |
| P2VP | poly(2-vinylpyridine) |
| P4VP | poly(4-vinylpyridine) |
| PAA | poly(acrylic acid) |
| PAEA | phosphonic acid ethyl acrylate |
| PAEMA | poly[2-(acetoacetoxy) ethyl] methacrylate |
| PAAm | polyacrylamide |
| PAMPS | poly(2-acrylamido-2-methyl-1-propansulfonic acid) |
| PAsp | poly(aspartic acid) |
| PBO | poly(ethylene-co-butylene) |
| PCEMA | poly(2-cinnamoylethyl methacrylate) |
| PDMAEMA | poly[(N,N-dimethylamino)ethyl methacrylate] |
| PEG | poly(ethylene glycol) |
| PEGDA | poly(ethylene glycol) diacrylate |
| PEGMA | poly (ethylene glycol methyl ether methacrylate) |
| PEO | poly(ethylene oxide) |
| PGA | poly(glycerol acrylate) |
| PGMA | poly(glycerol methacrylate) |
| PI | polyisoprene |
| PION | pegylated iron oxide nanoparticle |
| PMAA | poly(methacrylic acid) |
| PMAA-PTTM | trithiol-terminated poly(methacrylic acid) |
| PMGI | poly(methyl glutarimide) |
| PnBA | poly(n-butyl acrylate) |
| PNiPAAm | poly(N-isopropylacrylamide) |
| PNOR | poly(norbornene) |
| PNORCOOH | poly(norbornene dicarboxylic acid) |
| PNORMEOH | poly(norbornene methanol) |
| POEGA | poly(oligoethylene glycol acrylate) |
| POEGMA | poly(oligo(ethylene glycol) methacrylate |
| PP | polymer precursor |
| PPEGMEA | poly(ethylene glycol) methylether acrylate |
| PPO | poly(propylene oxide) |
| PS | poly styrene |
| PSS | poly (styrene sulfonate) |
| PtBA | poly(tert-butyl acrylate) |
| PVA | poly(vinyl alcohol) |
| PVBP | poly(4-vinylbenzylphosphonate) |
| PVP | polyvinylpyrrolidone |
| PXRD | powder X-ray diffraction |
| RAFT/MADIX | xanthate reversible addition-fragmentation chain-transfer |
| Rh | hydrodynamic radius |
| ROMP | ring-opening metathesis polymerization |
| SAED | selected area electron diffraction |
| SANS | small-angle neutron scattering |
| SDS | sodium dodecyl sulfate |
| SEM | scanning electron microscope |
| semi-IPN | semi-interpenetrating |
| SFL | stop-flow lithography |
| SPIONs | superparamagnetic iron oxide nanoparticles |
| SQUID | superconducting quantum interference device |
| STEM | scanning transmission electron microscopy |
| TB | blocking temperature |
| TEM | transmission electron microscopy |
| TGA | thermogravimetric analysis |
| THBC | triple-hydrophilic block copolymer |
| TMSMA | (trimethoxysilyl)propyl methacrylate |
| triEG | triethylene glycol |
| USPIO | ultra-small superparamagnetic iron oxide |
| UV-Vis | ultraviolet-visible spectroscopy |
| VCL | N-vinylcaprolactam |
| VIm | vinylimidazole |
| VSM | vibrating sample magnetometer |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
| ZFC | zero field cooled |
| WAXS | wide-angle X-ray scattering |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ahn, T.; Kim, J.H.; Yang, H.-M.; Lee, J.W.; Kim, J.-D. Formation pathways of magnetite nanoparticles by coprecipitation method. J. Phys. Chem. C 2012, 116, 6069–6076. [Google Scholar] [CrossRef]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; vander Elst, L.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Shen, Z.; Feng, C.; Li, Y.; Lu, G.; Huang, X.; Wang, G.; Huang, J. Synthesis of PPEGMEA-g-PMAA densely grafted double hydrophilic copolymer and its use as a template for the preparation of size-controlled superparamagnetic Fe3O4/polymer nano-composites. J. Mater. Chem. 2008, 18, 4332–4340. [Google Scholar] [CrossRef]
- Yathindranath, V.; Rebbouh, L.; Moore, D.F.; Miller, D.W.; van Lierop, J.; Hegmann, T. A versatile method for the reductive, one-pot synthesis of bare, hydrophilic and hydrophobic magnetite nanoparticles. Adv. Funct. Mater. 2011, 21, 1457–1464. [Google Scholar] [CrossRef]
- Bee, A.; Massart, R.; Neveu, S. Synthesis of very fine maghemite particles. J. Magn. Magn. Mater. 1995, 149, 6–9. [Google Scholar] [CrossRef]
- Liu, C.; Rondinone, A.J.; Zhang, Z.J. Synthesis of magnetic spinel ferrite CoFe2O4 nanoparticles from ferric salt and characterization of the size-dependent superparamagnetic properties. Pure Appl. Chem. 2000, 72, 37–45. [Google Scholar]
- Lee, Y.; Lee, J.; Bae, C.J.; Park, J.G.; Noh, H.J.; Park, J.H.; Hyeon, T. Large-scale synthesis of uniform and crystalline magnetite nanoparticles using reverse micelles as nanoreactors under reflux conditions. Adv. Funct. Mater. 2005, 15, 503–509. [Google Scholar] [CrossRef]
- Patrinoiu, G.; Visinescu, D.; Tirsoaga, A.; Carp, O. Green synthetic strategy for oxide materials: Polysaccharides-assisted synthesis. Part III. Dextran-assisted synthesis of nanosized metal-oxides. Rev. Roum. Chim. 2011, 56, 145–150. [Google Scholar]
- Paul, K.G.; Frigo, T.B.; Groman, J.Y.; Groman, E.V. Synthesis of ultrasmall superparamagnetic iron oxides using reduced polysaccharides. Bioconjug. Chem. 2004, 15, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.J.; Michele, F.; Jacob, V.; Angelique, Y.L. Size-controlled synthesis of dextran sulfate coated iron oxide nanoparticles for magnetic resonance imaging. Nanotechnology 2007, 18. [Google Scholar] [CrossRef]
- Feng, W.; Lv, W.; Qi, J.; Zhang, G.; Zhang, F.; Fan, X. Quadruple-responsive nanocomposite based on dextran–PMAA–PNIPAM, iron oxide nanoparticles, and gold nanorods. Macromol. Rapid Commun. 2012, 33, 133–139. [Google Scholar] [CrossRef]
- Srivastava, M.; Singh, J.; Yashpal, M.; Gupta, D.K.; Mishra, R.K.; Tripathi, S.; Ojha, A.K. Synthesis of superparamagnetic bare Fe3O4 nanostructures and core/shell (Fe3O4/alginate) nanocomposites. Carbohydr. Polym. 2012, 89, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Kantoh, T. Thermal decomposition of the iron(III) hydroxide and magnetite composites of poly(vinyl alcohol). Preparation of magnetite and metallic iron particles. Bull. Chem. Soc. Jpn. 1993, 66, 1536–1541. [Google Scholar]
- Sairam, M.; Naidu, B.V.K.; Nataraj, S.K.; Sreedhar, B.; Aminabhavi, T.M. Poly(vinyl alcohol)-iron oxide nanocomposite membranes for pervaporation dehydration of isopropanol, 1,4-dioxane and tetrahydrofuran. J. Membr. Sci. 2006, 283, 65–73. [Google Scholar] [CrossRef]
- Xu, C.; Teja, A.S. Continuous hydrothermal synthesis of iron oxide and pva-protected iron oxide nanoparticles. J. Supercrit. Fluids 2008, 44, 85–91. [Google Scholar] [CrossRef]
- Liu, H.-L.; Ko, S.P.; Wu, J.-H.; Jung, M.-H.; Min, J.H.; Lee, J.H.; An, B.H.; Kim, Y.K. One-pot polyol synthesis of monosize PVP-coated sub-5 nm Fe3O4 nanoparticles for biomedical applications. J. Magn. Magn. Mater. 2007, 310, e815–e817. [Google Scholar] [CrossRef]
- Millan, A.; Urtizberea, A.; Natividad, E.; Luis, F.; Silva, N.J.O.; Palacio, F.; Mayoral, I.; Ruiz-González, M.L.; González-Calbet, J.M.; Lecante, P.; et al. Akaganeite polymer nanocomposites. Polymer 2009, 50, 1088–1094. [Google Scholar] [CrossRef]
- Karakoti, A.S.; Das, S.; Thevuthasan, S.; Seal, S. PEGylated inorganic nanoparticles. Angew. Chem. Int. Ed. 2011, 50, 1980–1994. [Google Scholar] [CrossRef]
- Harraz, F.A. Polyethylene glycol-assisted hydrothermal growth of magnetite nanowires: Synthesis and magnetic properties. Phys. E Low Dimens. Syst. Nanostruct. 2008, 40, 3131–3136. [Google Scholar] [CrossRef]
- Lin, C.-L.; Lee, C.-F.; Chiu, W.-Y. Preparation and properties of poly(acrylic acid) oligomer stabilized superparamagnetic ferrofluid. J. Colloid Interface Sci. 2005, 291, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Ja Young, P.; Patel, D.; Gang Ho, L.; Seungtae, W.; Yongmin, C. Highly water-dispersible PEG surface modified ultra small superparamagnetic iron oxide nanoparticles useful for target-specific biomedical applications. Nanotechnology 2008, 19. [Google Scholar] [CrossRef]
- Li, Z.; Tan, B.; Allix, M.; Cooper, A.I.; Rosseinsky, M.J. Direct coprecipitation route to monodisperse dual-functionalized magnetic iron oxide nanocrystals without size selection. Small 2008, 4, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Tural, B.; Özkan, N.; Volkan, M. Preparation andcharacterization of polymer coated superparamagnetic magnetite nanoparticle agglomerates. J. Phys. Chem. Solids 2009, 70, 860–866. [Google Scholar]
- Lu, J.Q.; Moll, N.; Fu, Q.; Liu, J. Iron nanoparticles derived from iron-complexed polymethylglutarimide to produce high-quality lithographically defined single-walled carbon nanotubes. Chem. Mater. 2005, 17, 2237–2240. [Google Scholar] [CrossRef]
- Wan, S.; Zheng, Y.; Liu, Y.; Yan, H.; Liu, K. Fe3O4 nanoparticles coated with homopolymers of glycerol mono(meth)acrylate and their block copolymers. J. Mater. Chem. 2005, 15, 3424–3430. [Google Scholar] [CrossRef]
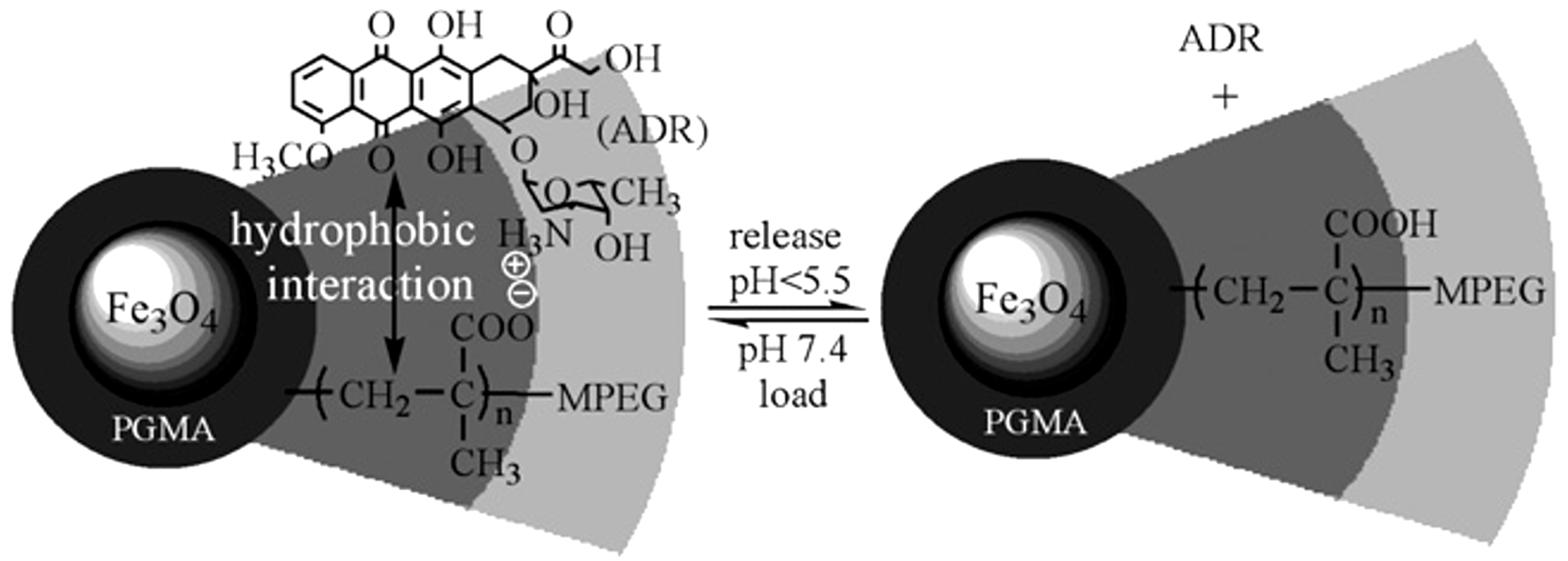
- Guo, M.; Yan, Y.; Zhang, H.; Yan, H.; Cao, Y.; Liu, K.; Wan, S.; Huang, J.; Yue, W. Magnetic and pH-responsive nanocarriers with multilayer core-shell architecture for anticancer drug delivery. J. Mater. Chem. 2008, 18, 5104–5112. [Google Scholar] [CrossRef]
- Lee, H.; Lee, E.; Kim, D.K.; Jang, N.K.; Jeong, Y.Y.; Jon, S. Antibiofouling polymer-coated superparamagnetic iron oxide nanoparticles as potential magnetic resonance contrast agents for in vivo cancer imaging. J. Am. Chem. Soc. 2006, 128, 7383–7389. [Google Scholar] [CrossRef] [PubMed]
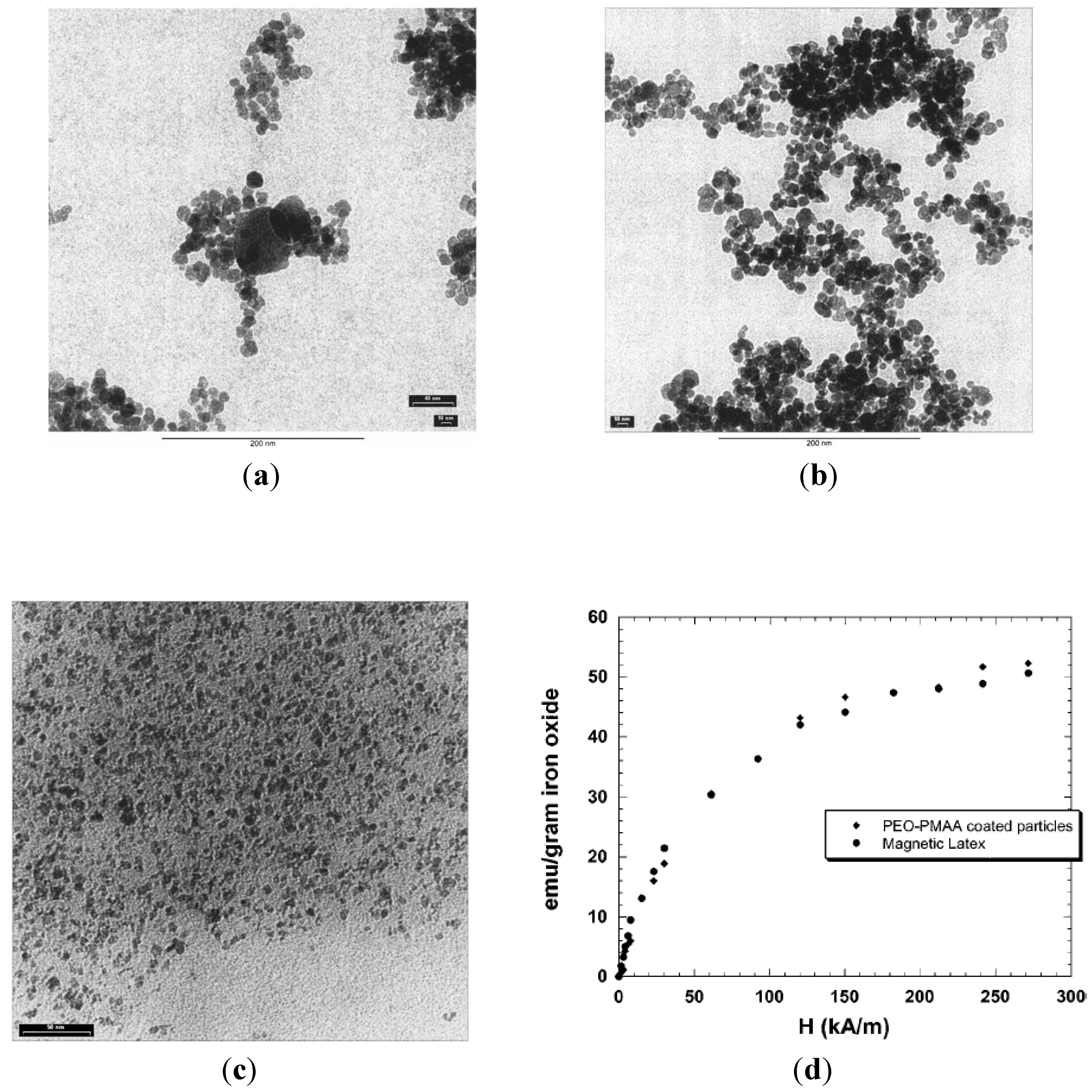
- Wormuth, K. Superparamagnetic latex via inverse emulsion polymerization. J. Colloid Interface Sci. 2001, 241, 366–377. [Google Scholar] [CrossRef]
- Kumagai, M.; Imai, Y.; Nakamura, T.; Yamasaki, Y.; Sekino, M.; Ueno, S.; Hanaoka, K.; Kikuchi, K.; Nagano, T.; Kaneko, E.; et al. Iron hydroxide nanoparticles coated with poly(ethylene glycol)-poly(aspartic acid) block copolymer as novel magnetic resonance contrast agents for in vivo cancer imaging. Colloids Surfaces B 2007, 56, 174–181. [Google Scholar] [CrossRef]
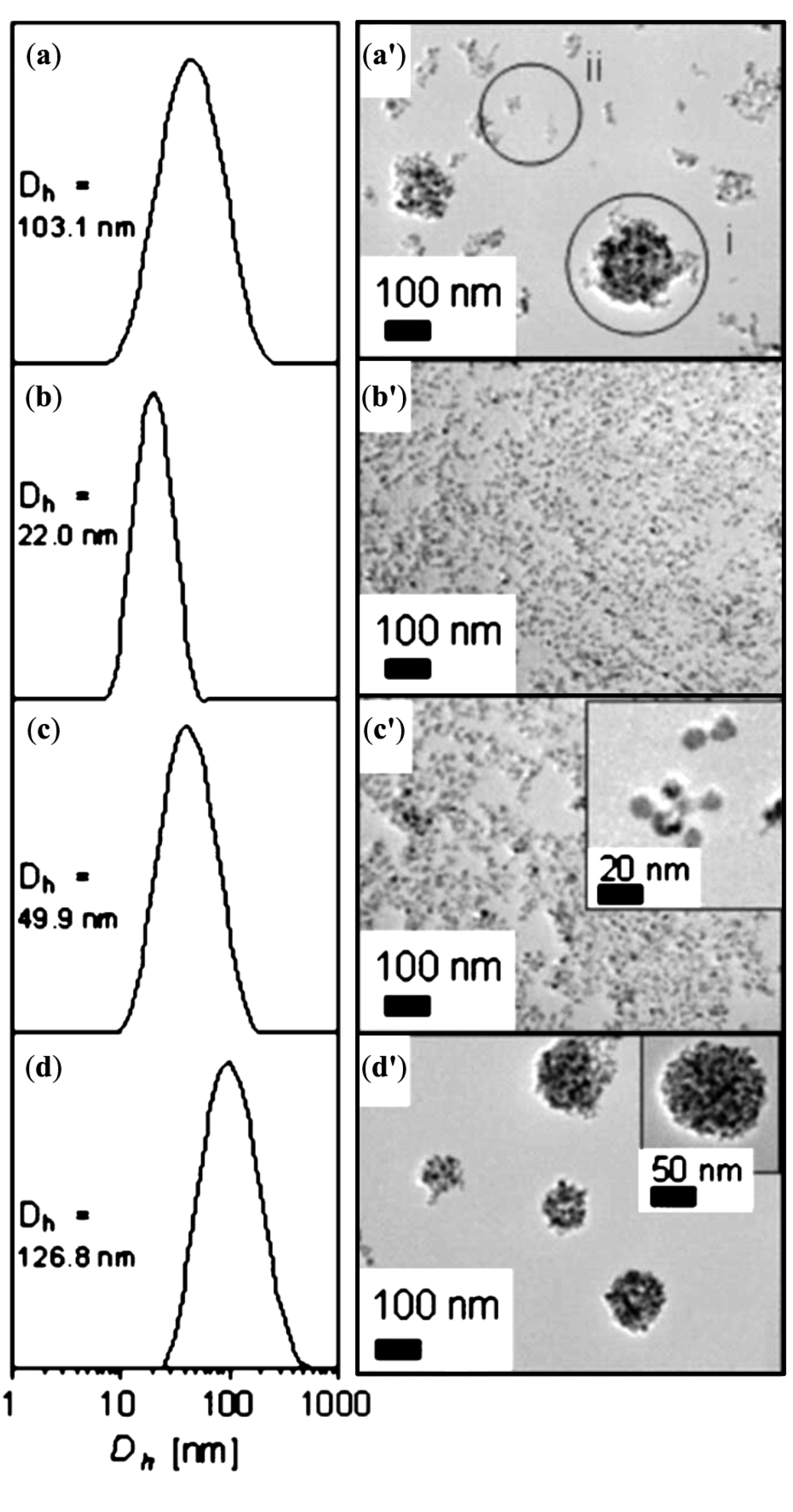
- Sondjaja, R.; Alan Hatton, T.; Tam, M.K.C. Clustering of magnetic nanoparticles using a double hydrophilic block copolymer, poly(ethylene oxide)-b-poly(acrylic acid). J. Magn. Magn. Mater. 2009, 321, 2393–2397. [Google Scholar] [CrossRef]
- Akcora, P.; Zhang, X.; Varughese, B.; Briber, R.M.; Kofinas, P. Structural and magnetic characterization of norbornene–deuterated norbornene dicarboxylic acid diblock copolymers doped with iron oxide nanoparticles. Polymer 2005, 46, 5194–5201. [Google Scholar] [CrossRef]
- Akcora, P.; Briber, R.M.; Kofinas, P. TEM characterization of diblock copolymer templated iron oxide nanoparticles: Bulk solution and thin film surface doping approach. Polymer 2006, 47, 2018–2022. [Google Scholar] [CrossRef]
- Yoon, K.Y.; Kotsmar, C.; Ingram, D.R.; Huh, C.; Bryant, S.L.; Milner, T.E.; Johnston, K.P. Stabilization of superparamagnetic iron oxide nanoclusters in concentrated brine with cross-linked polymer shells. Langmuir 2011, 27, 10962–10969. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.-F.; Stiller, S.; Hoth, A.; Kaufner, L.; Pison, U.; Cartier, R. One-pot synthesis of PEGylated ultrasmall iron-oxide nanoparticles and their in vivo evaluation as magnetic resonance imaging contrast agents. Biomacromolecules 2006, 7, 3132–3138. [Google Scholar] [CrossRef] [PubMed]
- Basuki, J.S.; Jacquemin, A.; Esser, L.; Li, Y.; Boyer, C.; Davis, T.P. A block copolymer-stabilized co-precipitation approach to magnetic iron oxide nanoparticles for potential use as MRI contrast agents. Polym. Chem. 2014, 5, 2611–2620. [Google Scholar] [CrossRef]
- Papaphilippou, P.; Loizou, L.; Popa, N.C.; Han, A.; Vekas, L.; Odysseos, A.; Krasia-Christoforou, T. Superparamagnetic hybrid micelles, based on iron oxide nanoparticles and well-defined diblock copolymers possessing β-ketoester functionalities. Biomacromolecules 2009, 10, 2662–2671. [Google Scholar] [CrossRef] [PubMed]
- Moeser, G.D.; Green, W.H.; Laibinis, P.E.; Linse, P.; Hatton, T.A. Structure of polymer-stabilized magnetic fluids: Small-angle neutron scattering and mean-field lattice modeling. Langmuir 2004, 20, 5223–5234. [Google Scholar] [CrossRef] [PubMed]
- Zhi, J.; Wang, Y.; Lu, Y.; Ma, J.; Luo, G. In situ preparation of magnetic chitosan/Fe3O4 composite nanoparticles in tiny pools of water-in-oil microemulsion. React. Funct. Polym. 2006, 66, 1552–1558. [Google Scholar] [CrossRef]
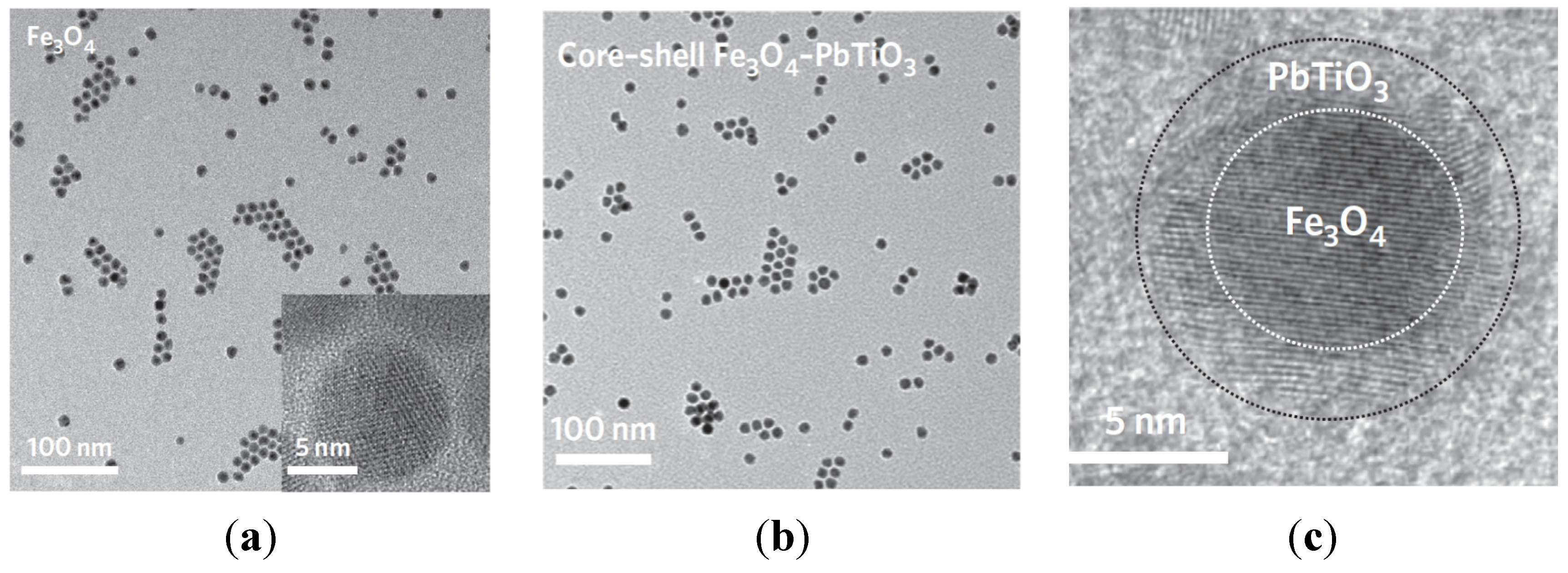
- Zhang, Z.; Rondinone, A.J.; Ma, J.X.; Shen, J.; Dai, S. Morphologically templated growth of aligned spinel CoFe2O4 nanorods. Adv. Mater. 2005, 17, 1415–1419. [Google Scholar] [CrossRef]
- Underhill, R.S.; Liu, G. Triblock nanospheres and their use as templates for inorganic nanoparticle preparation. Chem. Mater. 2000, 12, 2082–2091. [Google Scholar] [CrossRef]
- Ujiie, K.; Kanayama, N.; Asai, K.; Kishimoto, M.; Ohara, Y.; Akashi, Y.; Yamada, K.; Hashimoto, S.; Oda, T.; Ohkohchi, N.; et al. Preparation of highly dispersible and tumor-accumulative, iron oxide nanoparticles: Multi-point anchoring of PEG-b-poly(4-vinylbenzylphosphonate) improves performance significantly. Colloids Surfaces B Biointerfaces 2011, 88, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Zhao, L.; Han, W.; Xin, X.; Lin, Z. A general and robust strategy for the synthesis of nearly monodisperse colloidal nanocrystals. Nat. Nanotechnol. 2013, 8, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, J.-S. Gradual phase and morphology transformation of Fe3O4 nanoparticles to α-FeOOH nanorods in alcohol/water media in the presence of surfactant F127. J. Mater. Sci. 2008, 43, 4340–4343. [Google Scholar] [CrossRef]
- Zhang, M.; Müller, A.H.E.; Teissier, P.; Cabuil, V.; Krekhova, M. Polychelates of Amphiphilic Cylindrical Core–Shell Polymer Brushes with Iron Cations. In Trends in Colloid and Interface Science XVII; Springer: Berlin/Heidelberg, Germany, 2004; Volume 126, pp. 35–39. [Google Scholar]
- Wan, S.; Huang, J.; Yan, H.; Liu, K. Size-controlled preparation of magnetite nanoparticles in the presence of graft copolymers. J. Mater. Chem. 2006, 16, 298–303. [Google Scholar] [CrossRef]
- Li, P.; Huang, J. Preparation of poly (ethylene oxide)-graft-poly (acrylic acid) copolymer stabilized iron oxide nanoparticles via an in situ templated process. J. Appl. Polym. Sci. 2008, 109, 501–507. [Google Scholar] [CrossRef]
- Abes, J.I.; Cohen, R.E.; Ross, C.A. Block-copolymer-templated synthesis of iron, iron-cobalt, and cobalt-nickel alloy nanoparticles. Mater. Sci. Eng. C 2003, 23, 641–650. [Google Scholar] [CrossRef]
- Yun, S.-H.; Sohn, B.-H.; Jung, J.C.; Zin, W.-C.; Lee, J.-K.; Song, O. Tunable magnetic arrangement of iron oxide nanoparticles in situ synthesized on the solid substrate from diblock copolymer micelles. Langmuir 2005, 21, 6548–6552. [Google Scholar] [CrossRef] [PubMed]
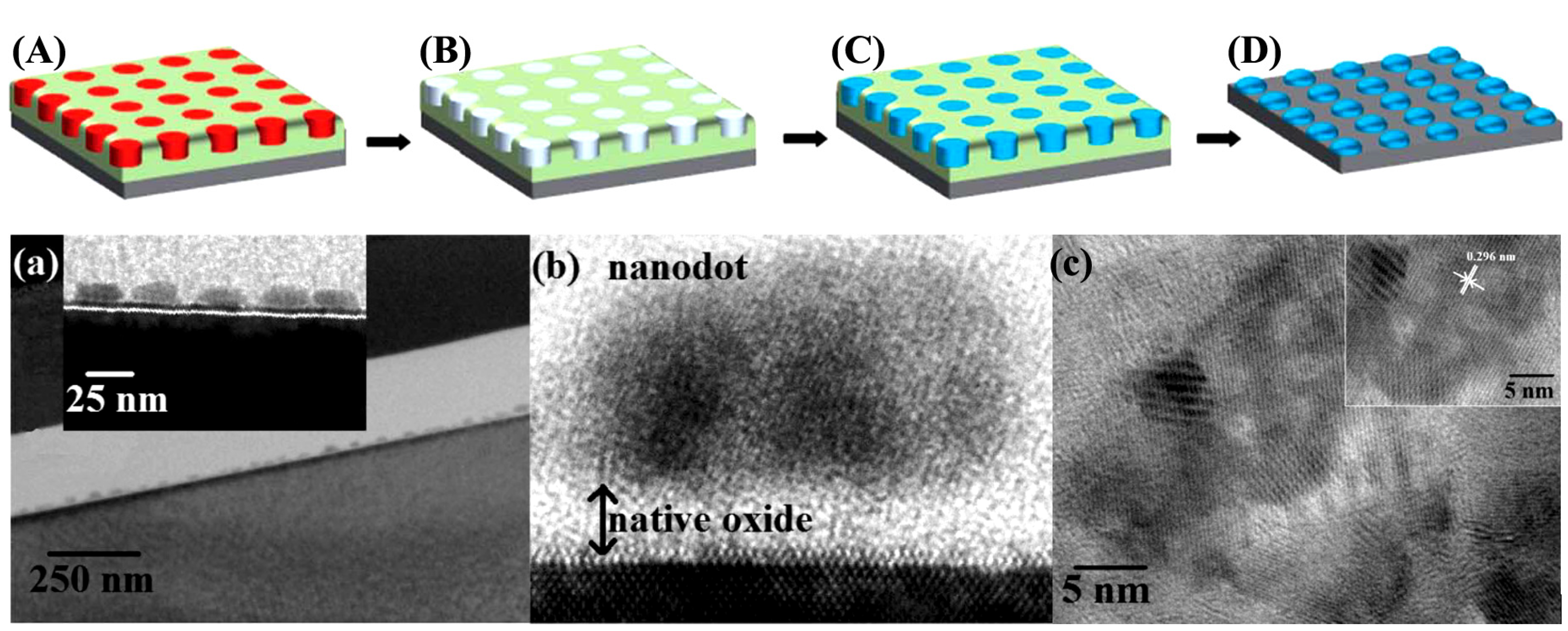
- Ghoshal, T.; Shaw, M.T.; Bolger, C.T.; Holmes, J.D.; Morris, M.A. A general method for controlled nanopatterning of oxide dots: A microphase separated block copolymer platform. J. Mater. Chem. 2012, 22, 12083–12089. [Google Scholar] [CrossRef]
- Uekawa, N.; Kaneko, K. Non stoichiometric properties of nanoporous iron oxide films. J. Phys. Chem. B 1998, 102, 8719–8724. [Google Scholar] [CrossRef]
- Yoon, M.; Kim, Y.M.; Kim, Y.; Volkov, V.; Song, H.J.; Park, Y.J.; Vasilyak, S.L.; Park, I.W. Magnetic properties of iron nanoparticles in a polymer film. J. Magn. Magn. Mater. 2003, 265, 357–362. [Google Scholar] [CrossRef]
- Yoon, T.; Chae, C.; Sun, Y.-K.; Zhao, X.; Kung, H.H.; Lee, J.K. Bottom-up in situ formation of Fe3O4 nanocrystals in a porous carbon foam for lithium-ion battery anodes. J. Mater. Chem. 2011, 21, 17325–17330. [Google Scholar] [CrossRef]
- Breulmann, M.; Cölfen, H.; Hentze, H.-P.; Antonietti, M.; Walsh, D.; Mann, S. Elastic magnets: Template-controlled mineralization of iron oxide colloids in a sponge-like gel matrix. Adv. Mater. 1998, 10, 237–241. [Google Scholar] [CrossRef]
- Sepúlveda-Guzmán, S.; Lara, L.; Pérez-Camacho, O.; Rodríguez-Fernández, O.; Olivas, A.; Escudero, R. Synthesis and characterization of an iron oxide poly(styrene-co-carboxybutylmaleimide) ferrimagnetic composite. Polymer 2007, 48, 720–727. [Google Scholar] [CrossRef]
- Hernández, R.; Sacristán, J.; Nogales, A.; Ezquerra, T.A.; Mijangos, C. Structural organization of iron oxide nanoparticles synthesized inside hybrid polymer gels derived from alginate studied with small-angle X-ray scattering. Langmuir 2009, 25, 13212–13218. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Sun, Z.-B.; Zheng, M.-L.; Cao, Y.-Y.; Jin, F.; Chen, W.-Q.; Zhao, Z.-S.; Duan, X.-M. A facile method for the room-temperature synthesis of water-soluble magnetic Fe3O4 nanoparticles: Combination of in situ synthesis and decomposition of polymer hydrogel. Mater. Chem. Phys. 2011, 130, 72–78. [Google Scholar] [CrossRef]
- Ozay, O.; Ekici, S.; Baran, Y.; Aktas, N.; Sahiner, N. Removal of toxic metal ions with magnetic hydrogels. Water Res. 2009, 43, 4403–4411. [Google Scholar] [CrossRef] [PubMed]
- Ozay, O.; Ekici, S.; Aktas, N.; Sahiner, N. P(4-vinyl pyridine) hydrogel use for the removal of UO22+ and Th4+ from aqueous environments. J. Environ. Manag. 2011, 92, 3121–3129. [Google Scholar] [CrossRef]
- Sivudu, K.S.; Rhee, K.Y. Preparation and characterization of pH-responsive hydrogel magnetite nanocomposite. Colloids Surfaces A Physicochem. Eng. Asp. 2009, 349, 29–34. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, S.; Kumacheva, E. Polymer microgels: Reactors for semiconductor, metal, and magnetic nanoparticles. J. Am. Chem. Soc. 2004, 126, 7908–7914. [Google Scholar] [CrossRef] [PubMed]
- Pich, A.; Bhattacharya, S.; Lu, Y.; Boyko, V.; Adler, H.-J.P. Temperature-sensitive hybrid microgels with magnetic properties. Langmuir 2004, 20, 10706–10711. [Google Scholar] [CrossRef] [PubMed]
- García-Cerda, L.A.; Chapa-Rodríguez, R.; Bonilla-Ríos, J. In situ synthesis of iron oxide nanoparticles in a styrene-divinylbenzene copolymer. Polym. Bull. 2007, 58, 989–994. [Google Scholar] [CrossRef]
- Suh, S.K.; Yuet, K.; Hwang, D.K.; Bong, K.W.; Doyle, P.S.; Hatton, T.A. Synthesis of nonspherical superparamagnetic particles: In situ coprecipitation of magnetic nanoparticles in microgels prepared by stop-flow lithography. J. Am. Chem. Soc. 2012, 134, 7337–7343. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Eckert, F.; Boyko, V.; Pich, A. Temperature-, pH-, and magnetic-field-sensitive hybrid microgels. Small 2007, 3, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Shao, Q.; He, J.; Jiang, B. Preparation of monodisperse magnetic polymer microspheres by swelling and thermolysis technique. Langmuir 2009, 26, 5179–5183. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nguyen, V.T.A.; Gauthier, M.; Sandre, O. Templated Synthesis of Magnetic Nanoparticles through the Self-Assembly of Polymers and Surfactants. Nanomaterials 2014, 4, 628-685. https://doi.org/10.3390/nano4030628
Nguyen VTA, Gauthier M, Sandre O. Templated Synthesis of Magnetic Nanoparticles through the Self-Assembly of Polymers and Surfactants. Nanomaterials. 2014; 4(3):628-685. https://doi.org/10.3390/nano4030628
Chicago/Turabian StyleNguyen, Vo Thu An, Mario Gauthier, and Olivier Sandre. 2014. "Templated Synthesis of Magnetic Nanoparticles through the Self-Assembly of Polymers and Surfactants" Nanomaterials 4, no. 3: 628-685. https://doi.org/10.3390/nano4030628
APA StyleNguyen, V. T. A., Gauthier, M., & Sandre, O. (2014). Templated Synthesis of Magnetic Nanoparticles through the Self-Assembly of Polymers and Surfactants. Nanomaterials, 4(3), 628-685. https://doi.org/10.3390/nano4030628