
A Brief Review on Multivalent Intercalation Batteries with Aqueous Electrolytes

Abstract
:1. Introduction
2. Overview on Multivalent Intercalation Batteries
3. Literature Review on Multivalent Aqueous Batteries
3.1. Al–Ion Batteries
- TiO2
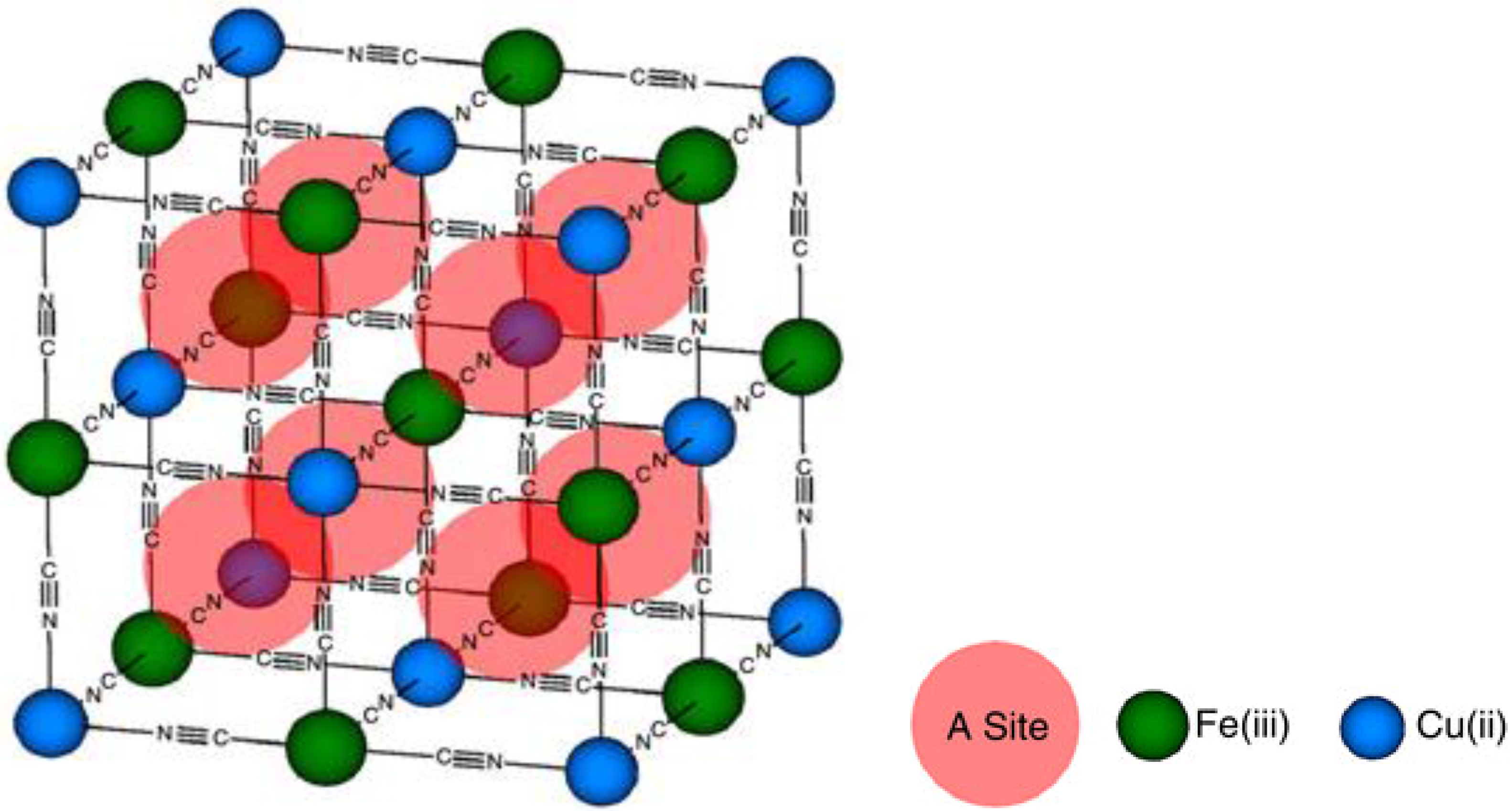
- Copper Hexacyanoferrate (CuHCF)
3.2. Zn–Ion Batteries
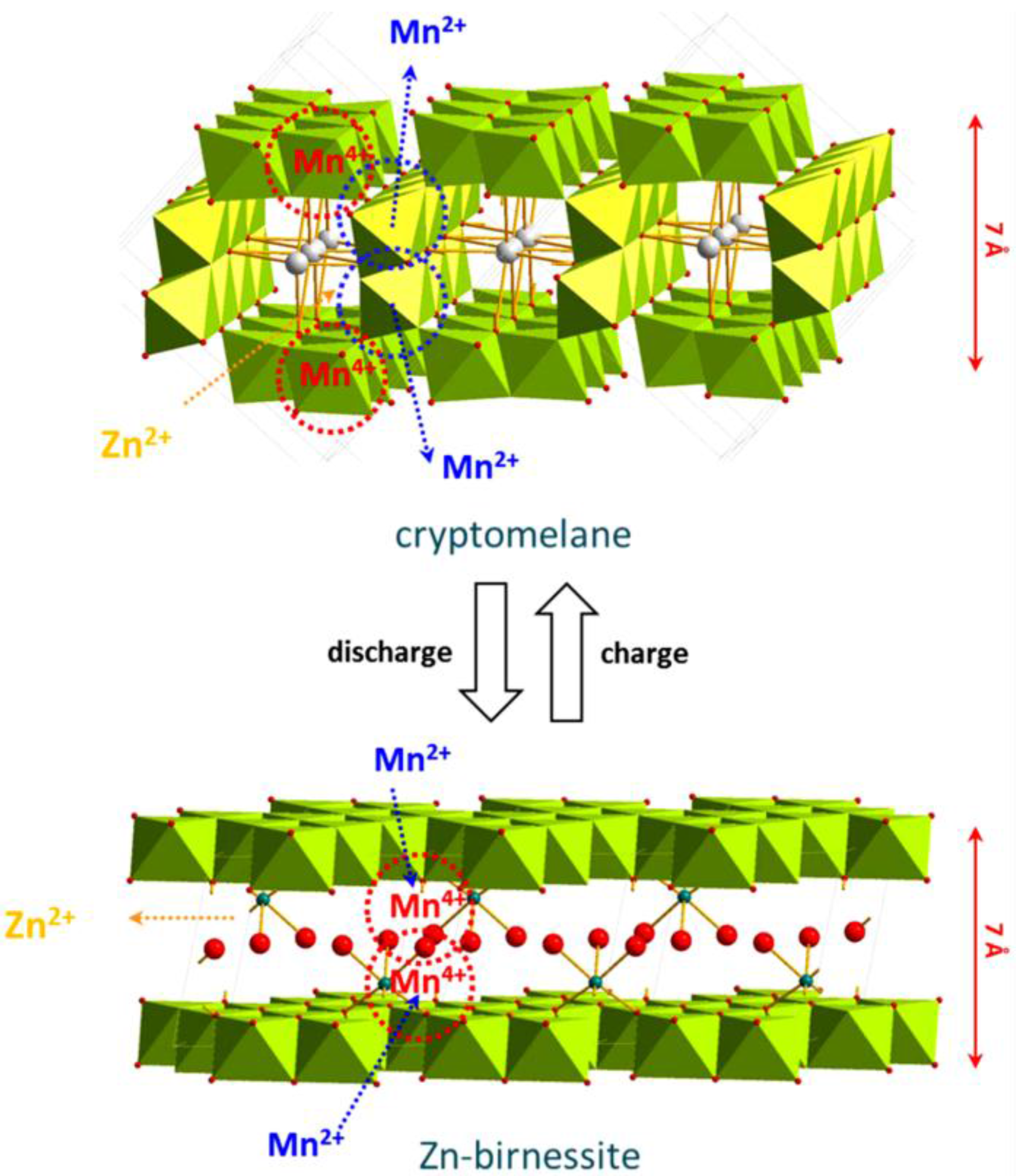
- α-MnO2
- λ–MnO2
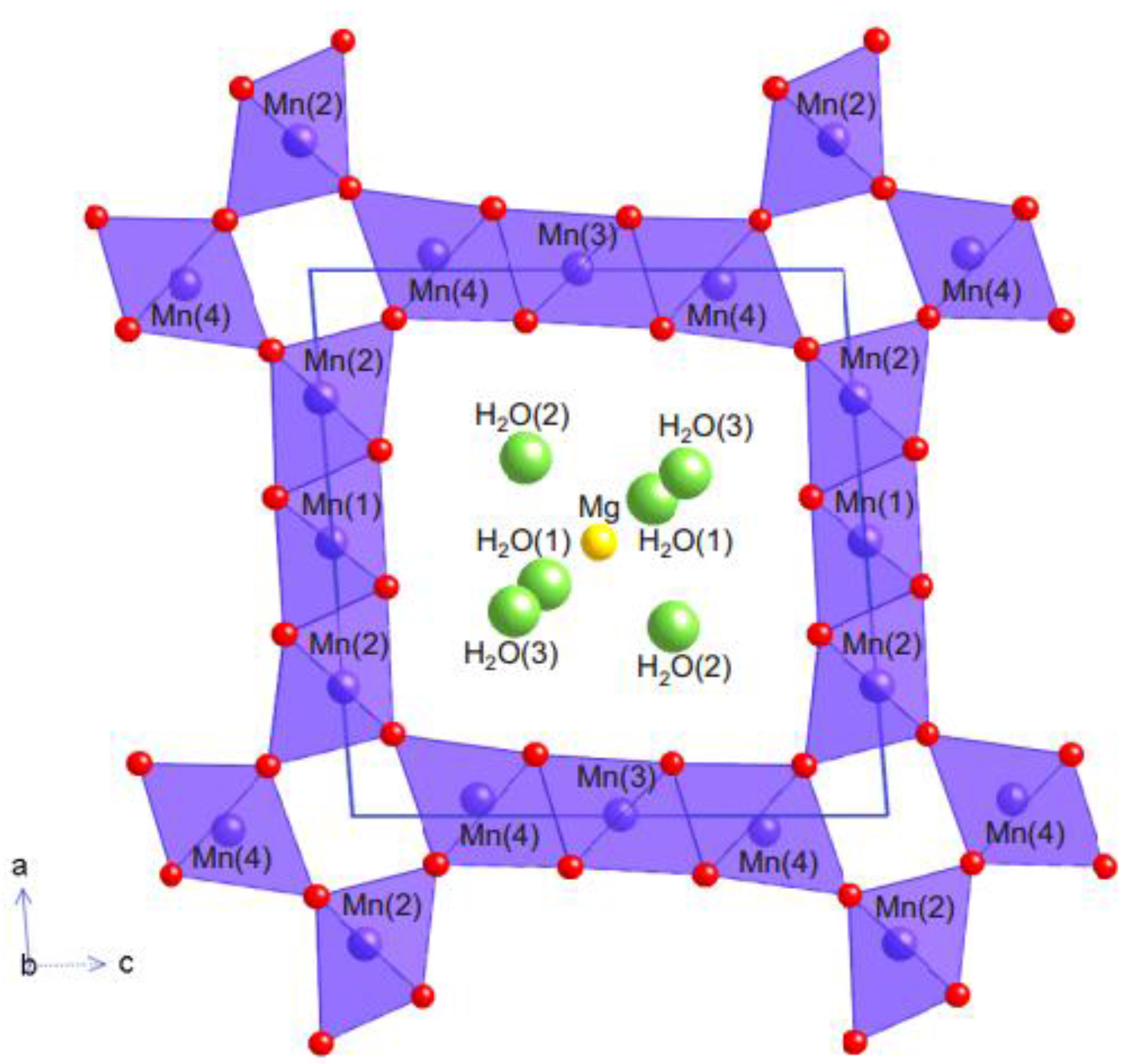
- Todorokite
- Zinc Hexacyanoferrate (ZnHCF)
- Copper Hexacyanoferrate (CuHCF)
3.3. Ni-Ion Batteries
- α-MnO2
- Copper Hexacyanoferrate (CuHCF)
3.4. Mg–Ion Batteries
- λ-MnO2 /Spinel Mn2O4
- MnO2 Birnessite
- Nickel Hexacyanoferrate (NiHCF)
- V2O5 Xerogels
3.5. Other Ions (Ca, Ba, Cu, Sr, Nd, La, Y, Sm, Pb and Ce)
- Nickel Hexacyanoferrate (NiHCF)
- Copper Hexacyanoferrate (CuHCF)
4. Future Directions
- Discover and/or develop new cathode materials with high voltages and high capacities.
- Develop new cathode materials with good reversibility for intercalation and deintercalation of multivalent ions.
- Improve the performance of currently discovered cathode materials with means of alterations in the electrode chemistries (e.g., doping) and structures, electrolytes, etc.
- Develop mechanisms to increase over-potentials for hydrogen and oxygen evolution by tailoring the chemistry of water, electrolytes and possibly electrode structures.
- Develop mechanisms to reduce the solvation energies of multivalent ions.
- Extend the research to nanostructured and porous cathode materials.
- Investigate the composite electrode materials in combination with the electrode materials used for supercapacitors.
- Explore the mechanisms and opportunities to enhance the interfacial capacitance at cathode/electrolyte interfaces.
- Reduce the polarization issues.
Acknowledgments
Conflicts of Interest
References
- Chen, J. Recent Progress in Advanced Materials for Lithium Ion Batteries. Materials 2013, 6, 156–183. [Google Scholar] [CrossRef]
- Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D. Challenges in the development of advanced Li-ion batteries: A review. Energy Environ. Sci. 2011, 4, 3243–3243. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Park, K.-S. The Li-Ion Rechargeable Battery: A Perspective. J. Am. Chem. Soc. 2013, 135, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Srivastava, S.K. Nanostructured anode materials for lithium ion batteries. J. Mater. Chem. A 2015, 3, 2454–2484. [Google Scholar] [CrossRef]
- Hayner, C.; Zhao, X.; Kung, H. Materials for Rechargeable Lithium-Ion Batteries. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 445–471. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Lin, Z.; Alcoutlabi, M.; Zhang, X. Recent developments in nanostructured anode materials for rechargeable lithium-ion batteries. Energy Environ. Sci. 2011, 4, 2682–2699. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, G. Developments in Nanostructured Cathode Materials for High-Performance Lithium-Ion Batteries. Adv. Mater. 2008, 20, 2251–2269. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hong, J.; Park, K.-Y.; Kim, H.; Kim, S.-W.; Kang, K. Aqueous Rechargeable Li and Na Ion Batteries. Chem. Rev. 2014, 114, 11788–11827. [Google Scholar] [CrossRef] [PubMed]
- Nithya, C.; Gopukumar, S. Sodium ion batteries: A newer electrochemical storage. WIREs Energy Environ. 2014, 4, 253–278. [Google Scholar] [CrossRef]
- Han, M.H.; Gonzalo, E.; Singh, G.; Rojo, T. A comprehensive review of sodium layered oxides: Powerful cathodes for Na-ion batteries. Energy Environ. Sci. 2015, 8, 81–102. [Google Scholar] [CrossRef]
- Yoo, H.D.; Shterenberg, I.; Gofer, Y.; Gershinsky, G.; Pour, N.; Aurbach, D. Mg rechargeable batteries: An on-going challenge. Energy Environ. Sci. 2013, 6, 2265–2279. [Google Scholar] [CrossRef]
- Mohtadi, R.; Mizuno, F. Magnesium batteries: Current state of the art, issues and future perspectives. Beilstein J. Nanotechnol. 2014, 5, 1291–1311. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Wessells, C.D.; Huggins, R.A.; Cui, Y. Highly Reversible Open Framework Nanoscale Electrodes for Divalent Ion Batteries. Nano Lett. 2013, 13, 5748–5752. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, N.; Das, S.K.; Archer, L.A. The rechargeable aluminum-ion battery. Chem. Commun. 2011, 47, 12610–12612. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Yoon, C.S.; Lee, H.R.; Chung, K.Y.; Cho, B.W.; Oh, S.H. Electrochemically-induced reversible transition from the tunneled to layered polymorphs of manganese dioxide. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, S.H.; Rajput, N.N.; Qu, X.; Chapman, K.W.; Persson, K.A.; Chupas, P.J. Solvation structure and energetics of electrolytes for multivalent energy storage. Phys. Chem. 2014, 16, 21941–21945. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ju, J.B.; Cho, W.I.; Cho, B.W.; Oh, S.H. Todorokite-type MnO2 as a zinc-ion intercalating material. Electrochim. Acta 2013, 112, 138–143. [Google Scholar] [CrossRef]
- Yuan, C.; Zhang, Y.; Pan, Y.; Liu, X.; Wang, G.; Cao, D. Investigation of the intercalation of polyvalent cations (Mg2+, Zn2+) into λ-MnO2 for rechargeable aqueous battery. Electrochim. Acta 2014, 116, 404–412. [Google Scholar] [CrossRef]
- Liu, M.; Rong, Z.; Malik, R.; Canepa, P.; Jain, A.; Ceder, G.; Persson, K.A. Spinel compounds as multivalent battery cathodes: A systematic evaluation based on ab initio calculations. Energy Environ. Sci. 2015, 8, 964–974. [Google Scholar] [CrossRef]
- Gregory, T.D. Nonaqueous Electrochemistry of Magnesium. J. Electrochem. Soc. 1990, 137, 775–780. [Google Scholar] [CrossRef]
- Novák, P.; Desilvestro, J. Electrochemical Insertion of Magnesium in Metal Oxides and Sulfides from Aprotic Electrolytes. J. Electrochem. Soc. 1993, 140, 140–144. [Google Scholar] [CrossRef]
- Spahr, M.E.; Novak, P.; Schnyder, B.; Haas, O.; Nesper, R. Characterization of Layered Lithium Nickel Manganese Oxides Synthesized by a Novel Oxidative Coprecipitation Method and Their Electrochemical Performance as Lithium Insertion Electrode Materials. J. Electrochem. Soc. 1998, 145, 1113–1121. [Google Scholar] [CrossRef]
- Novák, P.; Imhof, R.; Haas, O. Magnesium insertion electrodes for rechargeable nonaqueous batteries—A competitive alternative to lithium? Electrochim. Acta 1999, 45, 351–367. [Google Scholar] [CrossRef]
- Novák, P.; Scheifele, W.; Haas, O. Magnesium insertion batteries—An alternative to lithium? J. Power Sources 1995, 54, 479–482. [Google Scholar] [CrossRef]
- Novák, P.; Shklover, V.; Nesper, R. Magnesium Insertion in Vanadium Oxides: A Structural Study. Z. Phys. Chem. 1994, 185, 51–68. [Google Scholar] [CrossRef]
- Sánchez, L.; Pereira-Ramos, J.-P. Electrochemical insertion of magnesium in a mixed manganese–cobalt oxide. J. Mater. Chem. 1997, 7, 471–473. [Google Scholar] [CrossRef]
- Le, D.B.; Passerini, S.; Coustier, F.; Guo, J.; Soderstrom, T.; Owens, B.B.; Smyrl, W.H. Intercalation of Polyvalent Cations into V2O5 Aerogels. Chem. Mater. 1998, 10, 682–684. [Google Scholar] [CrossRef]
- Imamura, D.; Hibino, M.; Miyayama, M.; Judo, T. High Rate Magnesium Intercalation into V2O5/Carbon Composites. Trans. Mater. Res. Soc. Jpn. 2002, 27, 675–678. [Google Scholar]
- Imamura, D. Characterization of magnesium-intercalated V2O5/carbon composites. Solid State Ion. 2003, 161, 173–180. [Google Scholar] [CrossRef]
- Stojković, I.; Cvjetićanin, N.; Markovic, S.; Mitrić, M.; Mentus, S. Electrochemical behavior of V2O5 Xerogel and V2O5 Xerogel/C composite in an aqueous LiNO3 and Mg(NO3)2 solutions. Acta Phys. Polonica A 2010, 117, 837–840. [Google Scholar] [CrossRef]
- Vujković, M.; Pašti, I.; Simatović, I.; Šljukić, B.; Milenković, M.; Mentus, S. The Influence Of Intercalated Ions On Cyclic Stability Of V2O5/graphite Composite In Aqueous Electrolytic Solutions: Experimental And Theoretical Approach. Electrochim. Acta 2015, 176, 130–140. [Google Scholar] [CrossRef]
- Imamura, D.; Miyayama, M.; Hibino, M.; Kudo, T. Mg Intercalation Properties into V2O5 gel/Carbon Composites under High-Rate Condition. J. Electrochem. Soc. 2003, 150, A753–A758. [Google Scholar] [CrossRef]
- Yang, S.; Gong, Y.; Liu, Z.; Zhan, L.; Hashim, D.P.; Ma, L.; Vajtai, R.; Ajayan, P.M. Bottom-up Approach toward Single-Crystalline VO2-Graphene Ribbons as Cathodes for Ultrafast Lithium Storage. Nano Lett. 2013, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Katayama, Y.; Miura, T.; Kishi, T. Preparation and electrochemical magnesium insertion behaviors of Mg0.5+y(MeyTi1−y)2(PO4)3 (Me = Cr, Fe). J. Power Sources 2002, 112, 85–89. [Google Scholar] [CrossRef]
- Yuan, H.; Jiao, L.; Cao, J.; Liu, X.; Zhao, M.; Wang, Y. Development of Magnesium-Insertion Positive Electrode for Rechargeable Magnesium Batteries. J. Mater. Sci. Technol. 2004, 20, 41–45. [Google Scholar]
- Aurbach, D.; Weissman, I.; Gofer, Y.; Levi, E. Nonaqueous magnesium electrochemistry and its application in secondary batteries. Chem. Rec. 2003, 3, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.-F.; Yuan, H.-T.; Si, Y.-C.; Wang, Y.-J.; Wang, Y.-M. Synthesis of Cu0.1-doped vanadium oxide nanotubes and their application as cathode materials for rechargeable magnesium batteries. Electrochem. Commun. 2006, 8, 1041–1044. [Google Scholar] [CrossRef]
- Giraudet, J.; Claves, D.; Guérin, K.; Dubois, M.; Houdayer, A.; Masin, F.; Hamwi, A. Magnesium batteries: Towards a first use of graphite fluorides. J. Power Sources 2007, 173, 592–598. [Google Scholar] [CrossRef]
- Aurbach, D.; Lu, Z.; Schechter, A.; Gofer, Y.; Gizbar, H.; Turgeman, R.; Cohen, Y.; Moshkovich, M.; Levi, E. Prototype systems for rechargeable magnesium batteries. Nature 2000, 407, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Amatucci, G.G.; Badway, F.; Singhal, A.; Beaudoin, B.; Skandan, G.; Bowmer, T.; Plitz, I.; Pereira, N.; Chapman, T.; Jaworski, R. Investigation of Yttrium and Polyvalent Ion Intercalation into Nanocrystalline Vanadium Oxide. J. Electrochem. Soc. 2001, 148, A940–A950. [Google Scholar] [CrossRef]
- Wang, R.Y.; Shyam, B.; Stone, K.H.; Weker, J.N.; Pasta, M.; Lee, H.-W.; Toney, M.F.; Cui, Y. Reversible Multivalent (Monovalent, Divalent, Trivalent) Ion Insertion in Open Framework Materials. Adv. Energy Mater. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Yagi, S.; Ichitsubo, T.; Shirai, Y.; Yanai, S.; Doi, T.; Murase, K.; Matsubara, E. A concept of dual-salt polyvalent-metal storage battery. J. Mater. Chem. A 2014, 2, 1144–1149. [Google Scholar] [CrossRef] [Green Version]
- Lisbona, D.; Snee, T. A review of hazards associated with primary lithium and lithium-ion batteries. Process Saf. Environ. Prot. 2011, 89, 434–442. [Google Scholar] [CrossRef]
- Scrosati, B.; Hassoun, J.; Sun, Y.-K. Lithium-ion batteries. A look into the future. Energy Environ. Sci. 2011, 4, 3287–3295. [Google Scholar] [CrossRef]
- Wang, Q.; Ping, P.; Zhao, X.; Chu, G.; Sun, J.; Chen, C. Thermal runaway caused fire and explosion of lithium ion battery. J. Power Sources 2012, 208, 210–224. [Google Scholar] [CrossRef]
- Beck, F.; Rüetschi, P. Rechargeable batteries with aqueous electrolytes. Electrochim. Acta 2000, 45, 2467–2482. [Google Scholar] [CrossRef]
- Levi, E.; Levi, M.D.; Chasid, O.; Aurbach, D. A review on the problems of the solid state ions diffusion in cathodes for rechargeable Mg batteries. J. Electroceram 2007, 22, 13–19. [Google Scholar] [CrossRef]
- Li, W.; Dahn, J.R.; Wainwright, D.S. Rechargeable Lithium Batteries with Aqueous Electrolytes. Science 1994, 20, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hu, J.J.; Yan, N.F.; Pan, G.L.; Li, G.R.; Gao, X.P. Aluminum storage behavior of anatase TiO2 nanotube arrays in aqueous solution for aluminum ion batteries. Energy Environ. Sci. 2012, 5, 9743–9746. [Google Scholar] [CrossRef]
- Liu, S.; Pan, G.L.; Li, G.R.; Gao, X.P. Copper hexacyanoferrate nanoparticles as cathode material for aqueous Al-ion batteries. J. Mater. Chem. A 2015, 3, 959–962. [Google Scholar] [CrossRef]
- He, Y.J.; Peng, J.F.; Chu, W.; Li, Y.Z.; Tong, D.G. Black mesoporous anatase TiO2 nano leaves: A high capacity and high rate anode for aqueous Al-ion batteries. J. Mater. Chem. A 2014, 2, 1721–1731. [Google Scholar] [CrossRef]
- Liu, Y.; Sang, S.; Wu, Q.; Lu, Z.; Liu, K.; Liu, H. The electrochemical behavior of Cl− assisted Al3+ insertion into titanium dioxide nanotube arrays in aqueous solution for aluminum ion batteries. Electrochim. Acta 2014, 143, 340–346. [Google Scholar] [CrossRef]
- Xu, C.; Li, B.; Du, H.; Kang, F. Energetic Zinc Ion Chemistry: The Rechargeable Zinc Ion Battery. Angew. Chem. 2012, 51, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Manickam, M.; Singh, P.; Issa, T.B.; Thurgate, S. Electrochemical behavior of anatase TiO2 in aqueous lithium hydroxide electrolyte. J. Appl. Electrochem. 2006, 36, 599–602. [Google Scholar] [CrossRef]
- Wessells, C.D.; Huggins, R.A.; Cui, Y. Copper Hexacyanoferrate Battery Electrodes with Long Cycle Life and High Power. Nat. Commun. 2011, 2, 550. [Google Scholar] [CrossRef] [PubMed]
- Wessells, C.D.; Peddada, S.V.; Huggins, R.A.; Cui, Y. Nickel Hexacyanoferrate Nanoparticle Electrodes For Aqueous Sodium and Potassium Ion Batteries. Nano Lett. 2014, 11, 5421–5425. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, Y.; Shi, S.; Li, J.; Kang, F.; Su, D. Secondary batteries with multivalent ions for energy storage. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, L.; Zhou, X.; Liu, Z. Towards High-Voltage Aqueous Metal-Ion Batteries Beyond 1.5 V: The Zinc/Zinc Hexacyanoferrate System. Adv. Energy Mater. 2014, 5. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, B.; Wang, Y. Copper hexacyanoferrate with a well-defined open framework as a positive electrode for aqueous zinc ion batteries. Mater. Chem. Phys. 2015, 149–150, 601–606. [Google Scholar] [CrossRef]
- Trócoli, R.; Mantia, F.L. An Aqueous Zinc-Ion Battery Based on Copper Hexacyanoferrate. ChemSusChem 2014, 8, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Kim, A.; Phadke, S.; Biswas, S.; Luong, T.; Hertzberg, B.J.; Chamoun, M.; Evans-Lutterodt, K.; Steingart, D.A. Improving the cycle life of a high-rate, high-potential aqueous dual-ion battery using hyper-dendritic zinc and copper hexacyanoferrate. J. Power Sources 2016, 305, 22–29. [Google Scholar] [CrossRef]
- Sinha, N.N.; Munichandraiah, N. Electrochemical Conversion of LiMn2O4 to MgMn2O4 in Aqueous Electrolytes. Electrochem. Solid-State Lett. 2008, 11, F23–F26. [Google Scholar] [CrossRef]
- Nam, K.W.; Kim, S.; Lee, S.; Salama, M.; Shterenberg, I.; Gofer, Y.; Kim, J.-S.; Yang, E.; Park, C.S.; Kim, J.-S.; et al. The High Performance of Crystal Water Containing Manganese Birnessite Cathodes for Magnesium Batteries. Nano Lett. 2015, 15, 4071–4079. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Phillips, P.J.; Key, B.; Yi, T.; Nordlund, D.; Yu, Y.-S.; Bayliss, R.D.; Han, S.-D.; He, M.; Zhang, Z.; et al. Direct Observation of Reversible Magnesium Ion Intercalation into a Spinel Oxide Host. Adv. Mater. 2015, 27, 3377–3384. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, I.; Pasti, I.; Cvijeticanin, N.; Mitric, M.; Mentus, S. Electrochemical behavior of V2O5 xerogel in aqueous LiNO3 solution. Electrochem. Commun. 2009, 11, 1512–1514. [Google Scholar] [CrossRef]
- Choi, J.; Alvarez, E.; Arunkumar, T.A.; Manthiram, A. Proton Insertion into Oxide Cathodes during Chemical Delithiation. Electrochem. Solid-State Lett. 2006, 9, A241–A244. [Google Scholar] [CrossRef]
- Manthiram, A.; Choi, J. Chemical and structural instabilities of lithium ion battery cathodes. J. Power Sources 2006, 159, 249–253. [Google Scholar] [CrossRef]
- Wang, Y.-G.; Luo, J.-Y.; Wang, C.-X.; Xia, Y.-Y. Hybrid Aqueous Energy Storage Cells Using Activated Carbon and Lithium-Ion Intercalated Compounds II. Comparison of LiMn2O4, LiCo1/3Ni1/3Mn1/3O2, and LiCoO2 Positive Electrodes. J. Electrochem. Soc. 2006, 153, A1425–A1431. [Google Scholar] [CrossRef]
- Luo, J.-Y.; Cui, W.-J.; He, P.; Xia, Y.-Y. Raising the cycling stability of aqueous lithium-ion batteries by eliminating oxygen in the electrolyte. Nat. Chem. 2010, 2, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.W.; Donoue, K.; Kadohata, T.; Murata, T.; Matsuta, S.; Fujitani, S. Impurities in LiFePO4 and Their Influence on Material Characteristics. J. Electrochem. Soc. 2008, 155, A526–A530. [Google Scholar] [CrossRef]
- Li, G.; Yang, Z.; Jiang, Y.; Zhang, W.; Huang, Y. Hybrid aqueous battery based on Na3V2(PO4)3/C cathode and zinc anode for potential large-scale energy storage. J. Power Sources 2016, 308, 52–57. [Google Scholar] [CrossRef]
- Pasta, M.; Wessells, C.D.; Huggins, R.A.; Cui, Y. A high-rate and long cycle life aqueous electrolyte battery for grid-scale energy storage. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Tong, X.; Gan, L.; Zhang, S.; Zhang, X.; Liu, X. Effect of adding various carbon additives to porous zinc anode in rechargeable hybrid aqueous battery. J. Alloys Comp. 2016, 658, 119–124. [Google Scholar] [CrossRef]
- Vujković, M.; Nedić, Z.; Tančić, P.; Aleksić, O.S.; Nikolić, M.V.; Mioč, U.; Mentus, S. Electrochemical lithiation/delithiation kinetics and capacity of phosphate tungsten bronze and its chemically pre-lithiated derivatives in aqueous solutions. J. Mater. Sci. 2015, 51, 2481–2489. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Li, C.; He, Z.; Xiang, Y.; Xiong, L.; Chen, D.; Yu, Y.; Sun, K.; He, Z.; et al. The electrochemical performance improvement of LiMn2O4/Zn based on zinc foil as the current collector and thiourea as an electrolyte additive. J. Power Sources 2015, 300, 453–459. [Google Scholar] [CrossRef]
- Neff, V.D. Electrochemical Oxidation and Reduction of Thin Films of Prussian Blue. J. Electrochem. Soc. 1978, 125, 886–887. [Google Scholar] [CrossRef]
- Itaya, K.; Uchida, I.; Neff, V.D. Electrochemistry of polynuclear transition metal cyanides: Prussian blue and its analogues. Acc. Chem. Res. 1986, 19, 162–168. [Google Scholar] [CrossRef]
- Stilwell, D.E.; Park, K.H.; Miles, M.H. Electrochemical studies of the factors influencing the cycle stability of Prussian Blue films. J. Appl. Electrochem. 1992, 22, 325–331. [Google Scholar] [CrossRef]
- De Tacconi, N.R.; Rajeshwar, K.; Lezna, R.O. Metal Hexacyanoferrates: Electro Synthesis, in situ Characterization and Applications. Chem. Inform 2003, 15, 3046–3062. [Google Scholar]
- Rosi, N.L.; Eckert, J.; Eddaudi, M.; Vodak, D.T.; Kim, J.; O’keeffe, M.; Yaghi, O.M. Hydrogen storage in microporous metal-organic frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Post, J.E.; Heaney, P.J.; Hanson, J. Synchrotron X-ray diffraction study of the structure and dehydration behavior of todorokite. Am. Mineral. 2003, 88, 142–150. [Google Scholar] [CrossRef]
- Lodovico, L.; Torresi, R.M.; Martins, V.L.; Benedetti, T.M. Electrochemical Behavior of Iron and Magnesium in Ionic Liquids. J. Braz. Chem. Soc. 2013, 25, 460–468. [Google Scholar] [CrossRef]
- Lu, Z.; Schechter, A.; Moshkovich, M.; Aurbach, D. On the electrochemical behavior of magnesium electrodes in polar aprotic electrolyte solutions. J. Electroanal. Chem. 1999, 466, 203–217. [Google Scholar] [CrossRef]
- Aurbach, D.; Suresh, G.S.; Levi, E.; Mitelman, A.; Mizrahi, O.; Chusid, O.; Brunelli, M. Progress in Rechargeable Magnesium Battery Technology. Adv. Mater. 2007, 19, 4260–4267. [Google Scholar] [CrossRef]
- Liang, Y.; Feng, R.; Yang, S.; Ma, H.; Liang, J.; Chen, J. Rechargeable Mg Batteries with Graphene-like MoS2 Cathode and Ultrasmall Mg Nanoparticle Anode. Adv. Mater. 2010, 23, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Luo, T.; Mu, G.; Wang, X.; Chen, D.; Shen, G. Rechargeable Mg-Ion Batteries Based on WSe2 Nanowire Cathodes. ACS Nano 2013, 7, 8051–8058. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yoo, H.D.; Li, Y.; Shuai, J.; Calderon, H.A.; Hernandez, F.C.R.; Grabow, L.C.; Yao, Y. Interlayer-Expanded Molybdenum Disulfide Nanocomposites for Electrochemical Magnesium Storage. Nano Lett. 2015, 15, 2194–2202. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, J.; Bucur, C.B.; Gregory, T. Quest for Nonaqueous Multivalent Secondary Batteries: Magnesium and Beyond. Chem. Rev. 2014, 114, 11683–11720. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, M.; David, W.; Bruce, P.; Goodenough, J. Lithium insertion into manganese spinels. Mater. Res. Bull. 1983, 18, 461–472. [Google Scholar] [CrossRef]
- Arthur, T.S.; Zhang, R.; Ling, C.; Glans, P.-A.; Fan, X.; Guo, J.; Mizuno, F. Understanding the Electrochemical Mechanism of K-αMnO2 for Magnesium Battery Cathodes. ACS Appl. Mater. Interfaces 2014, 6, 7004–7008. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Zhang, R.; Arthur, T.S.; Mizuno, F. How General is the Conversion Reaction in Mg Battery Cathode: A Case Study of the Magnesiation of α-MnO2. Chem. Mater. 2015, 27, 5799–5807. [Google Scholar] [CrossRef]
- Garche, J.; Dyer, C.K.; Moseley, P.; Ogumi, Z.; Rand, D.; Newnes, B. Encyclopedia of Electrochemical Power Sources; Academic Press: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Gautam, G.S.; Canepa, P.; Abdellahi, A.; Urban, A.; Malik, R.; Ceder, G. The Intercalation Phase Diagram of Mg in V2O5 from First-Principles. Chem. Mater. 2015, 27, 3733–3742. [Google Scholar] [CrossRef]
- Gershinsky, G.; Yoo, H.D.; Gofer, Y.; Aurbach, D. Electrochemical and Spectroscopic Analysis of Mg2+ Intercalation into Thin Film Electrodes of Layered Oxides: V2O5 and MoO3. Langmuir 2013, 29, 10964–10972. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Ion | Standard Electrode Potential (V) | Theoretical Capacity | |
|---|---|---|---|
| Specific Capacity (mAh/g) | Volumetric Capacity (mAh/cm3) | ||
| Li+ | −3.05 | 3829 | 2044 |
| Na+ | −2.71 | 1165 | 1128 |
| Mg2+ | −2.36 | 2234 | 3882 |
| Ca2+ | −2.87 | 1337 | 2073 |
| Ni2+ | −0.257 | 913 | 8133 |
| Zn2+ | −0.76 | 820 | 5854 |
| Al3+ | −1.66 | 2980 | 8046 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guduru, R.K.; Icaza, J.C. A Brief Review on Multivalent Intercalation Batteries with Aqueous Electrolytes. Nanomaterials 2016, 6, 41. https://doi.org/10.3390/nano6030041
Guduru RK, Icaza JC. A Brief Review on Multivalent Intercalation Batteries with Aqueous Electrolytes. Nanomaterials. 2016; 6(3):41. https://doi.org/10.3390/nano6030041
Chicago/Turabian StyleGuduru, Ramesh K., and Juan C. Icaza. 2016. "A Brief Review on Multivalent Intercalation Batteries with Aqueous Electrolytes" Nanomaterials 6, no. 3: 41. https://doi.org/10.3390/nano6030041
APA StyleGuduru, R. K., & Icaza, J. C. (2016). A Brief Review on Multivalent Intercalation Batteries with Aqueous Electrolytes. Nanomaterials, 6(3), 41. https://doi.org/10.3390/nano6030041