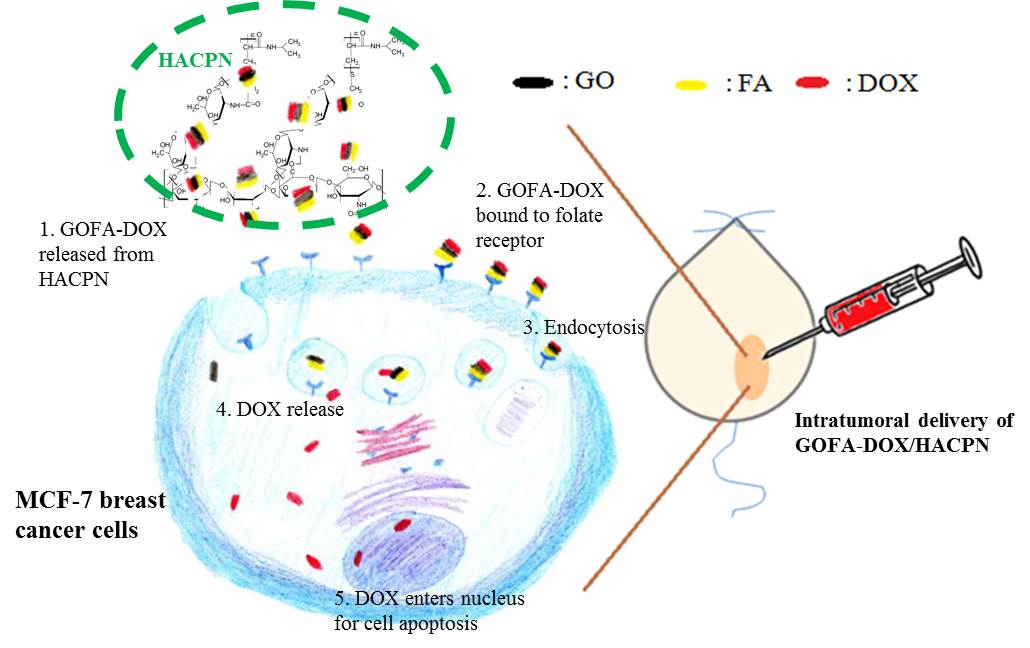
Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of GO and GOFA
2.2. Synthesis and Characterization of HACPN
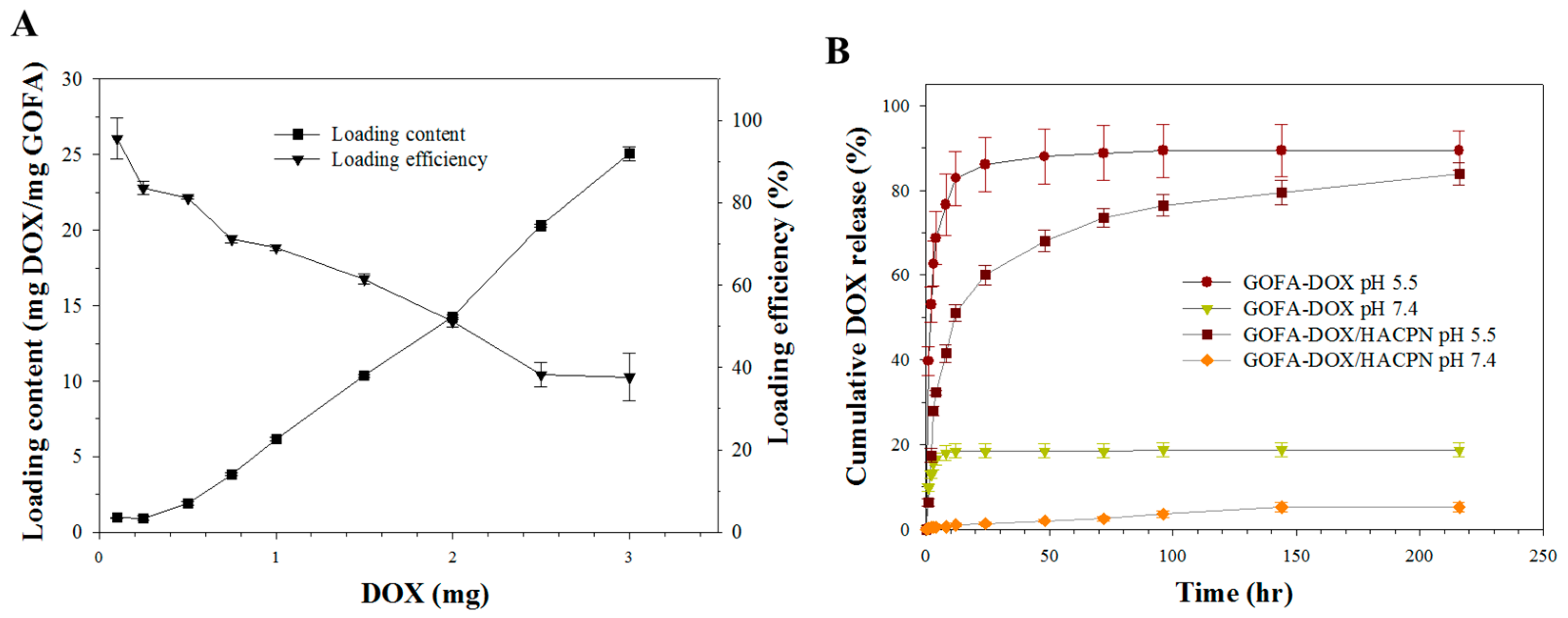
2.3. DOX Loading and Release
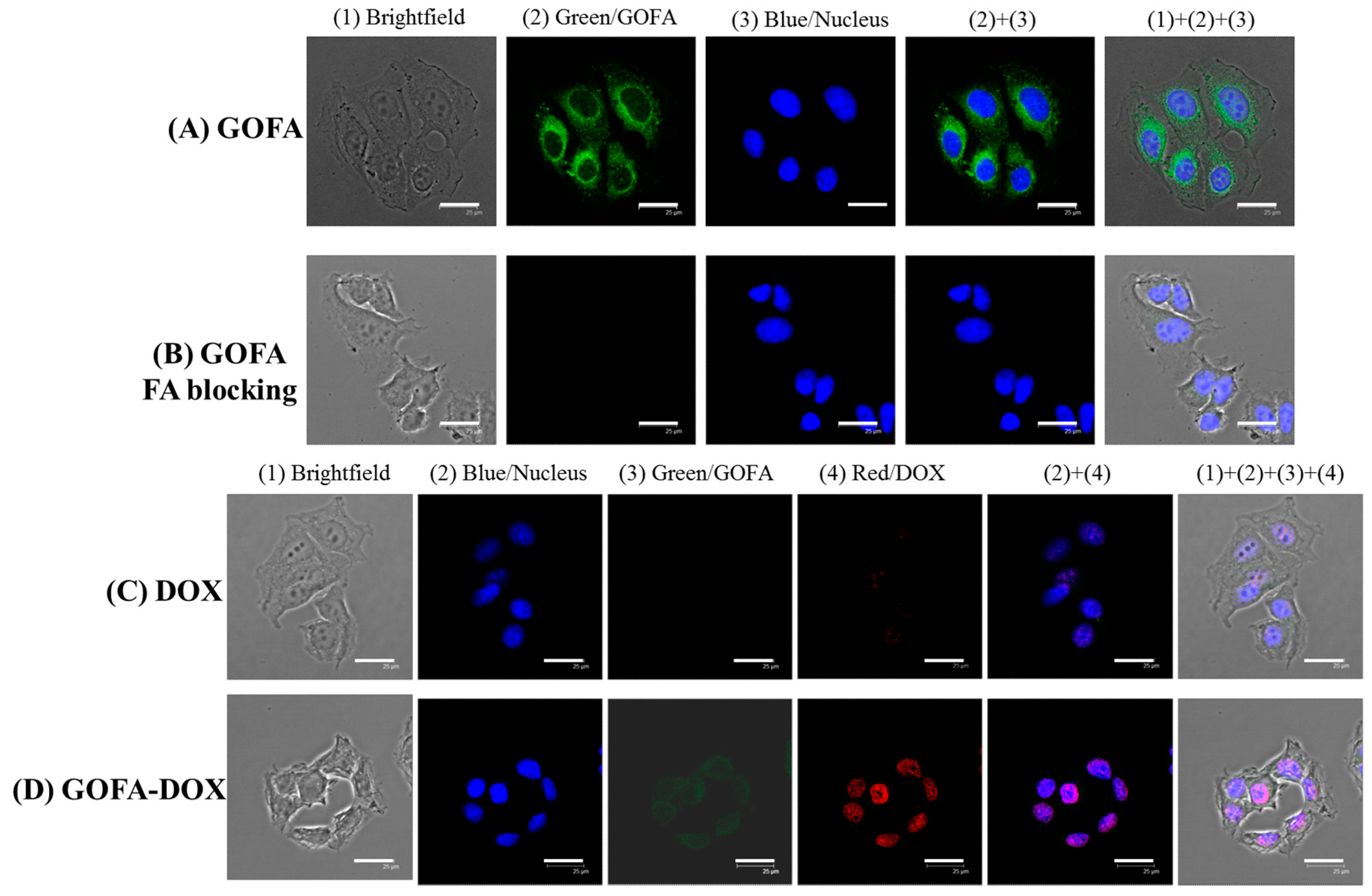
2.4. In Vitro Cell Culture
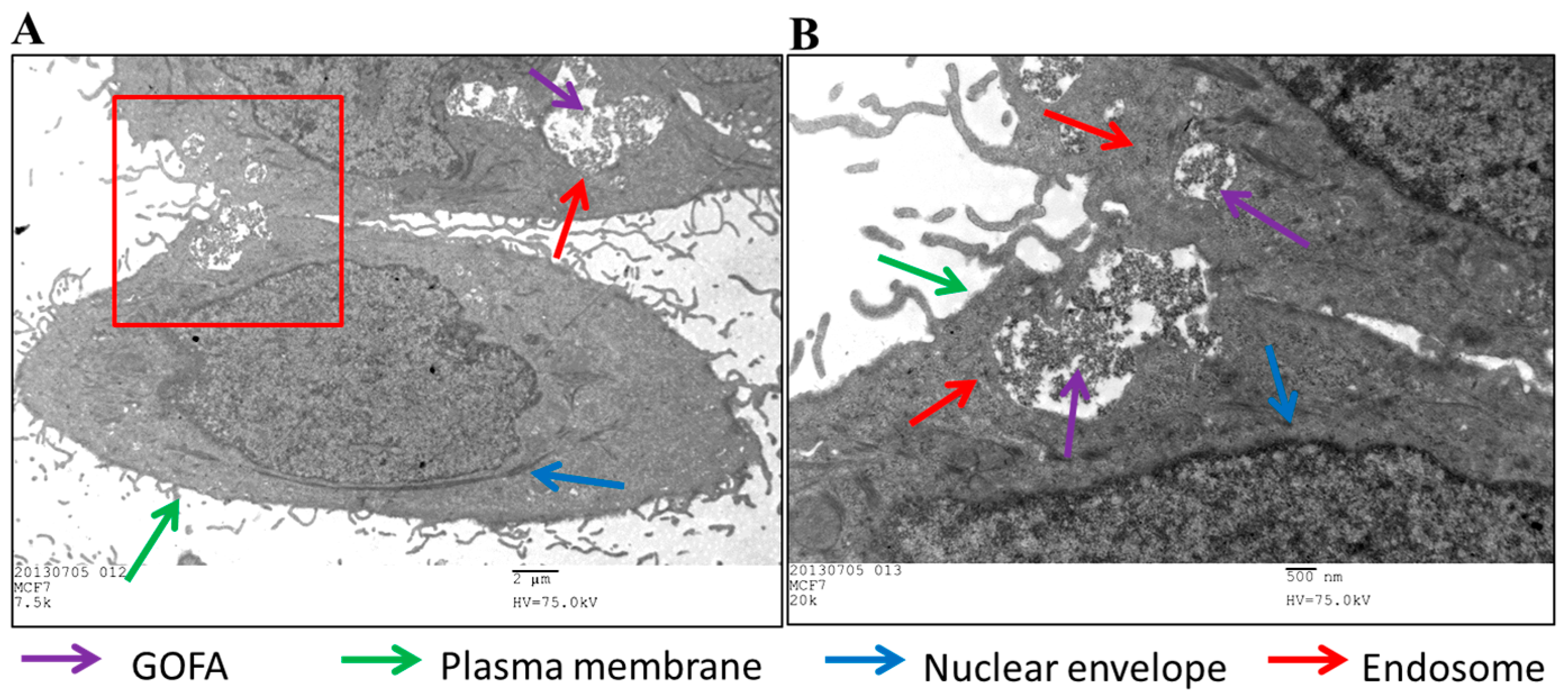
2.4.1. Cellular Uptake
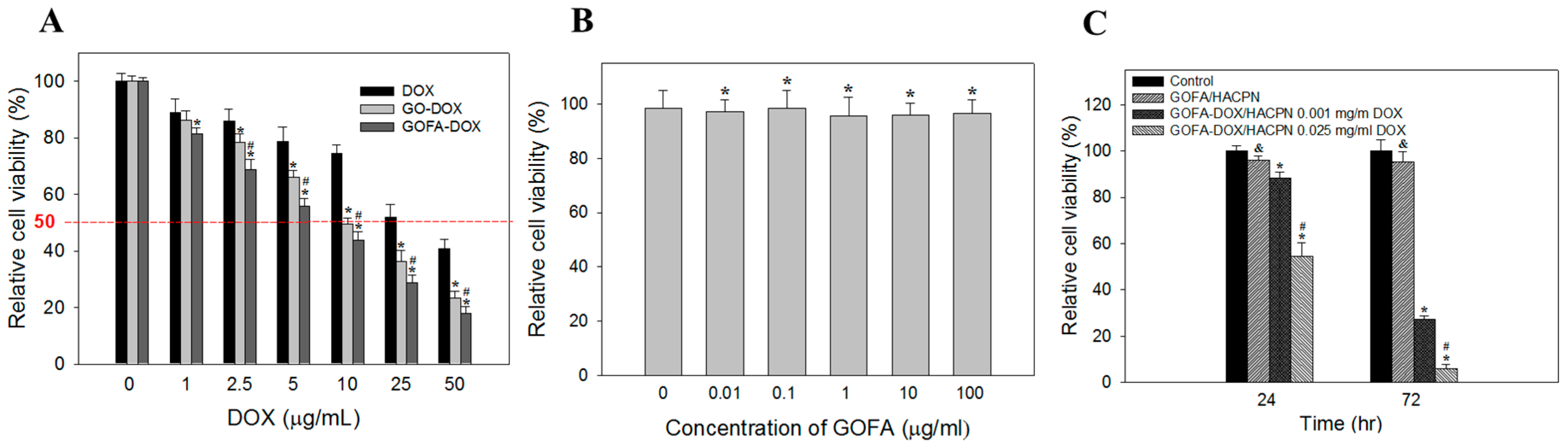
2.4.2. In Vitro Cytotoxicity and Biocompatibility Studies
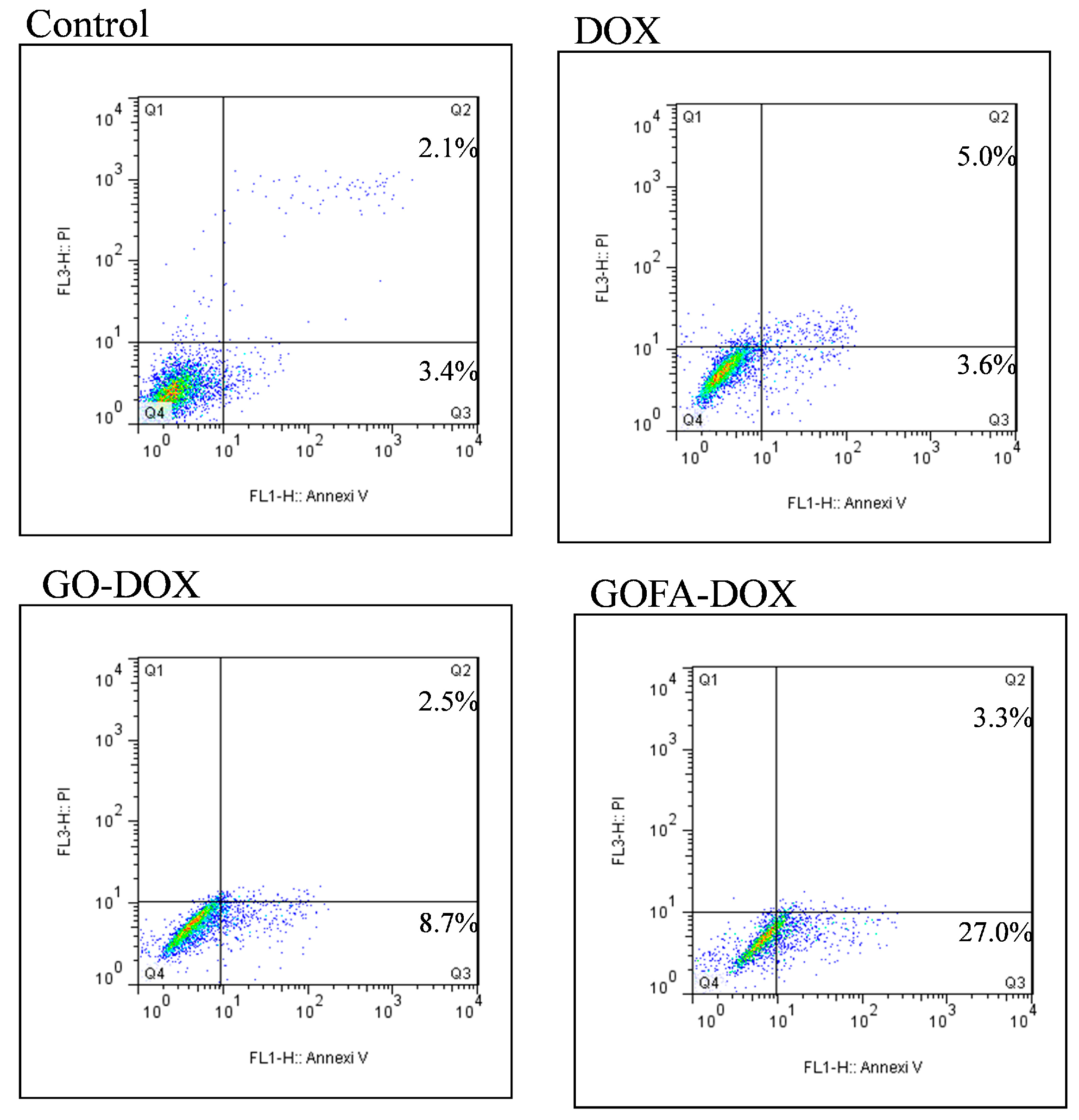
2.4.3. Cell Apoptosis Induced by DOX In Vitro
2.5. Animal Study
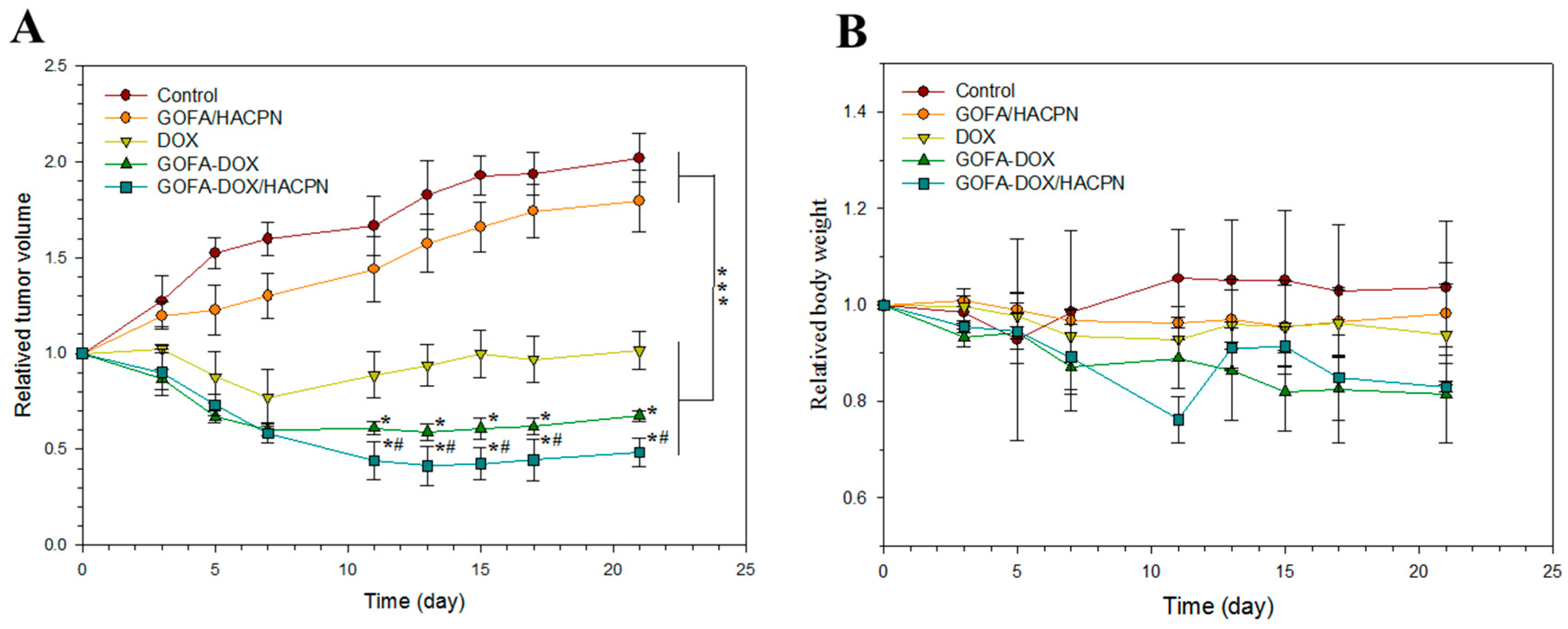
2.5.1. Antitumor Effect
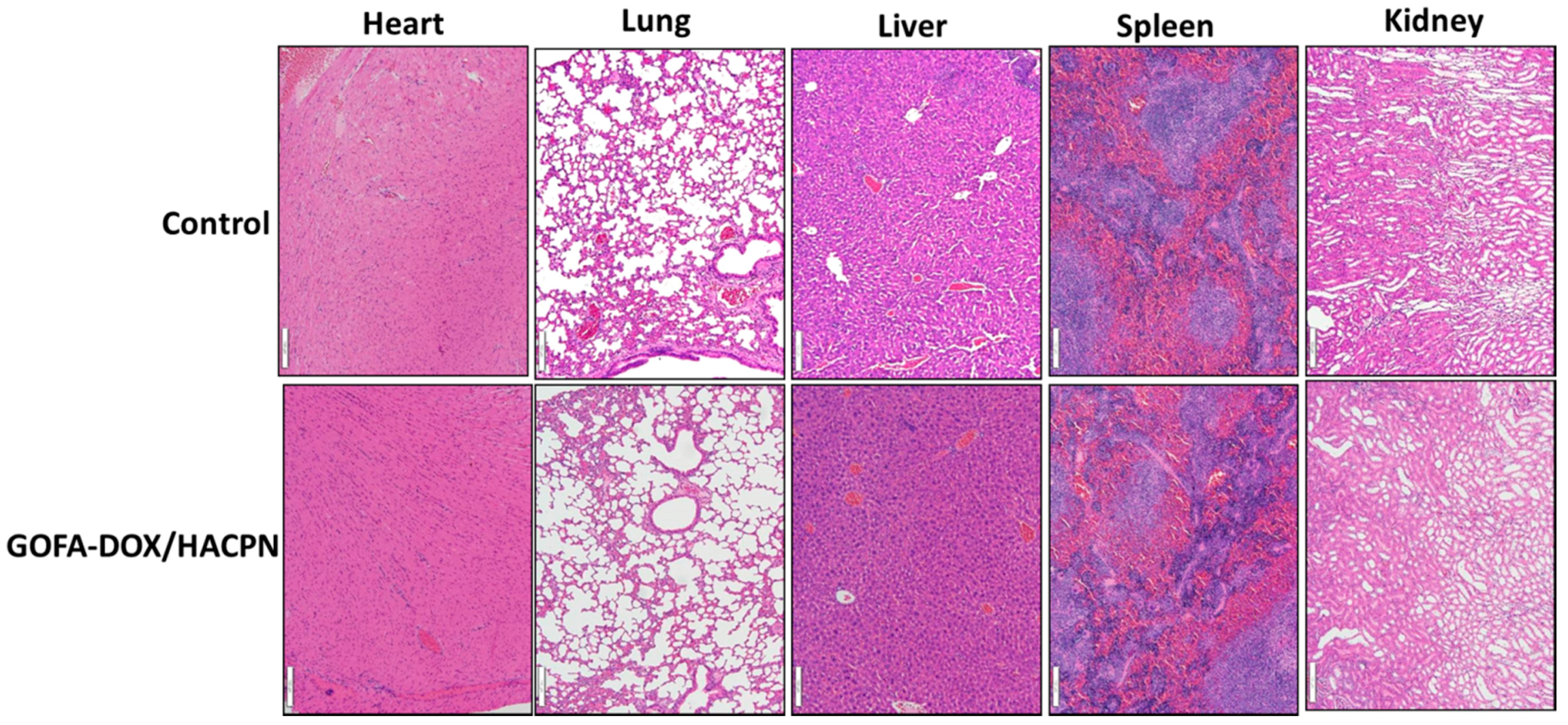
2.5.2. Histological and Systemic Toxicity Analysis
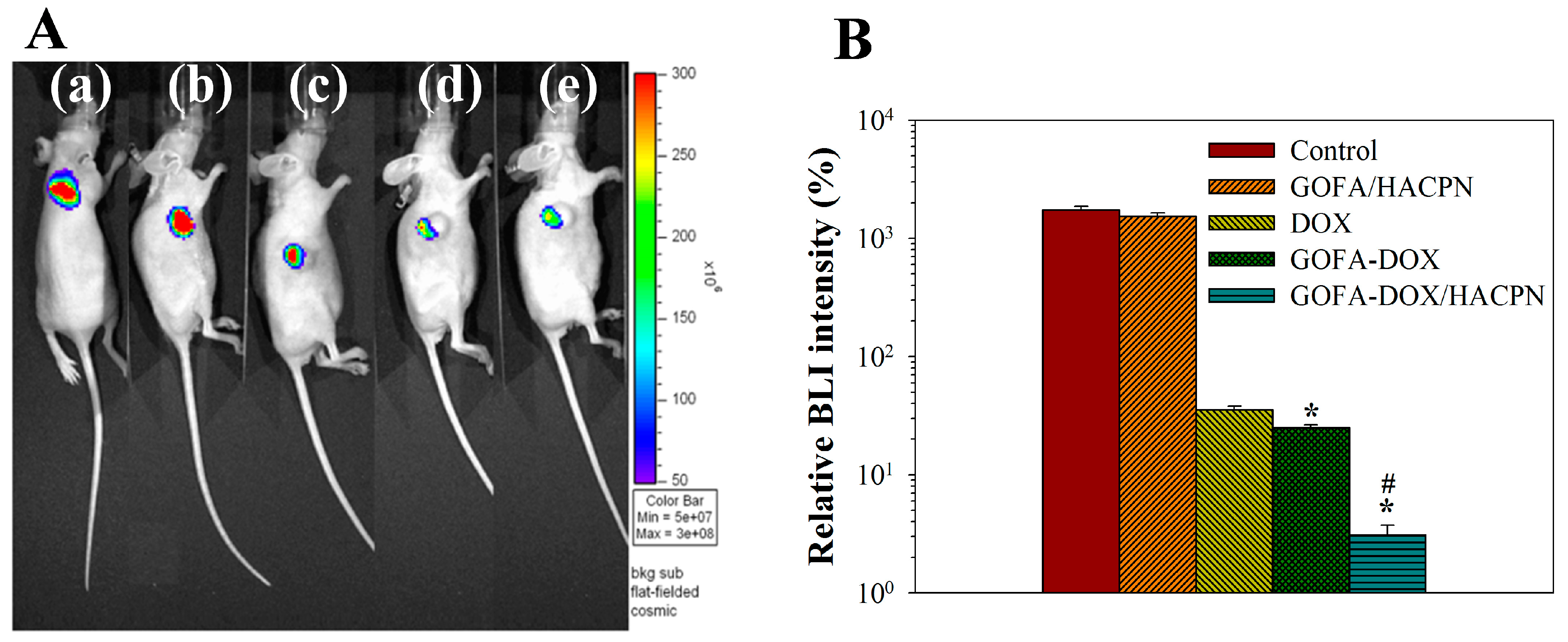
2.5.3. IVIS for Bioluminescence Imaging (BLI) Intensity
3. Materials and Methods
3.1. Materials
3.2. Preparation and Characterization of GO and GOFA
3.2.1. Preparation of GO, GOFA and Quantum Dot (QD)-Labeled GO and GOFA
3.2.2. Characterization of GO, GOFA and GOFA-DOX
3.3. Preparation and Characterization of HACPN Hydrogel
3.3.1. Synthesis of HACPN Hydrogel
3.3.2. Characterization of HACPN Hydrogel
3.4. DOX Loading and Release
3.4.1. Loading of DOX on GOFA
3.4.2. In Vitro DOX Release from GOFA-DOX and GOFA-DOX/HACPN
3.5. In Vitro Cell Culture
3.5.1. Cell Line and Cell Culture Condition
3.5.2. Intracellular Uptake
3.5.3. In Vitro Cytotoxicity Assessment
3.5.4. Analysis of Apoptosis Using Annexin V and Propidium Iodide Staining
3.6. Animal Studies
3.6.1. Xenograft Tumor Mouse Model
3.6.2. In Vivo Antitumor Efficacy
3.6.3. Histological, Immunohistochemical, Hematologic and Biochemical Analysis
3.6.4. Bioluminescence Imaging (BLI) for In Vivo Evaluation of Anti-Tumor Efficacy
3.7. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Govindan, B.; Swarna Latha, B.; Nagamony, P.; Ahmed, F.; Saifi, M.A.; Harrath, A.H.; Alwasel, S.; Mansour, L.; Alsharaeh, E.H. Designed synthesis of nanostructured magnetic hydroxyapatite based drug nanocarrier for anti-cancer drug delivery toward the treatment of human epidermoid carcinoma. Nanomaterials 2017, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carmona, M.; Colilla, M.; Vallet-Regí, M. Smart mesoporous nanomaterials for antitumor therapy. Nanomaterials 2015, 5, 1906–1937. [Google Scholar] [CrossRef] [PubMed]
- Debbage, P. Targeted drugs and nanomedicine: Present and future. Curr. Pharm. Des. 2009, 15, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-J.; Yang, H.-W.; Hung, S.-C.; Huang, C.-Y.; Li, S.-M.; Ma, C.; Chen, P.-Y.; Tsai, H.-C.; Wei, K.-C.; Chen, J.-P. Improving thermal stability and efficacy of BCNU in treating glioma cells using PAA-functionalized graphene oxide. Int. J. Nanomed. 2012, 7, 1737–1747. [Google Scholar]
- Wang, Y.; Li, Z.; Wang, J.; Li, J.; Lin, Y. Graphene and graphene oxide: Biofunctionalization and applications in biotechnology. Trends Biotechnol. 2011, 29, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Ma, Y.; Huang, Y.; Wang, Y.; Chen, Y. Superparamagnetic graphene oxide–Fe3O4 nanoparticles hybrid for controlled targeted drug carriers. J. Mater. Chem. 2009, 19, 2710–2714. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Robinson, J.T.; Sun, X.; Dai, H. Pegylated nanographene oxide for delivery of water-insoluble cancer drugs. J. Am. Chem. Soc. 2008, 130, 10876–10877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xia, J.; Zhao, Q.; Liu, L.; Zhang, Z. Functional graphene oxide as a nanocarrier for controlled loading and targeted delivery of mixed anticancer drugs. Small 2010, 6, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Liu, Z.; Ma, Y.; Huang, Y.; Chen, Y. High-efficiency loading and controlled release of doxorubicin hydrochloride on graphene oxide. J. Phys. Chem. C 2008, 112, 17554–17558. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, B.; Liu, J.; Zhang, P.; Li, Z.; Luan, Y. Green fabricated reduced graphene oxide: Evaluation of its application as nano-carrier for pH-sensitive drug delivery. Int. J. Pharm. 2015, 496, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; An, S.S.; Hulme, J. Current applications of graphene oxide in nanomedicine. Int. J. Nanomed. 2015, 10, 9–24. [Google Scholar]
- Bae, K.H.; Chung, H.J.; Park, T.G. Nanomaterials for cancer therapy and imaging. Mol. Cells 2011, 31, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Wang, X.; Nie, S.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhao, N. A Targeted nanoprobe based on carbon nanotubes-natural biopolymer chitosan composites. Nanomaterials 2016, 6, 216. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.K.; Mallick, S.K.; Santra, S.; Maiti, T.K.; Ghosh, S.K.; Pramanik, P. In vitro evaluation of folic acid modified carboxymethyl chitosan nanoparticles loaded with doxorubicin for targeted delivery. J. Mater. Sci. Mater. Med. 2010, 21, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Chen, J.P.; Wei, K.C.; Chen, J.Y.; Huang, C.W.; Liao, Z.X. Release of doxorubicin by a folate-grafted, chitosan-coated magnetic nanoparticle. Nanomaterials 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Wei, K.C.; Ma, C.C.; Yang, S.Y.; Chen, J.P. Dual targeted delivery of doxorubicin to cancer cells using folate-conjugated magnetic multi-walled carbon nanotubes. Colloids Surf. B Biointerfaces 2012, 89, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liow, S.S.; Dou, Q.; Kai, D.; Karim, A.A.; Zhang, K.; Xu, F.; Loh, X.J. Thermogels: In situ gelling biomaterial. ACS Biomater. Sci. Eng. 2016, 2, 295–316. [Google Scholar] [CrossRef]
- Rzaev, Z.M.; Dincer, S.; Pişkin, E. Functional copolymers of N-isopropylacrylamide for bioengineering applications. Prog. Polym. Sci. 2007, 32, 534–595. [Google Scholar] [CrossRef]
- Okano, T.; Yamada, N.; Sakai, H.; Sakurai, Y. A novel recovery system for cultured cells using plasma-treated polystyrene dishes grafted with poly(N-isopropylacrylamide). J. Biomed. Mater. Res. 1993, 27, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Gil, E.S.; Hudson, S.M. Stimuli-responsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Fakhari, A.; Subramony, J.A. Engineered in-situ depot-forming hydrogels for intratumoral drug delivery. J. Control. Release 2015, 220, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.B.; Colson, Y.L.; Grinstaff, M.W. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods and wafers. J. Control. Release 2012, 159, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Kempe, S.; Mäder, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J.; Li, J. Biodegradable thermosensitive copolymer hydrogels for drug delivery. Expert Opin. Ther. Pat. 2007, 17, 965–977. [Google Scholar] [CrossRef]
- Wu, W.; Chen, H.; Shan, F.; Zhou, J.; Sun, X.; Zhang, L.; Gong, T. A novel doxorubicin-loaded in situ forming gel based high concentration of phospholipid for intratumoral drug delivery. Mol. Pharm. 2014, 11, 3378–3385. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-P.; Cheng, T.-H. Preparation and evaluation of thermo-reversible copolymer hydrogels containing chitosan and hyaluronic acid as injectable cell carriers. Polymer 2009, 50, 107–116. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Lu, Y.-J.; Chen, J.-P. Magnetic graphene oxide as a carrier for targeted delivery of chemotherapy drugs in cancer therapy. J. Magn. Magn. Mater. 2017, 427, 34–40. [Google Scholar] [CrossRef]
- Zhi, F.; Dong, H.; Jia, X.; Guo, W.; Lu, H.; Yang, Y.; Ju, H.; Zhang, X.; Hu, Y. Functionalized graphene oxide mediated adriamycin delivery and miR-21 gene silencing to overcome tumor multidrug resistance in vitro. PLoS ONE 2013, 8, e60034. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Zhu, C.-L.; Li, J.; Liu, J.-J.; Chen, X.; Yang, H.-H. Using graphene to protect DNA from cleavage during cellular delivery. Chem. Commun. 2010, 46, 3116–3118. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.P.; Cheng, T.H. Thermo-responsive chitosan-graft-poly(N-isopropylacrylamide) injectable hydrogel for cultivation of chondrocytes and meniscus cells. Macromol. Biosci. 2006, 6, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Chen, J.P.; Leu, Y.L.; Hu, J.W. Temperature-sensitive hydrogels composed of chitosan and hyaluronic acid as injectable carriers for drug delivery. Eur. J. Pharm. Biopharm. 2008, 68, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Feil, H.; Bae, Y.H.; Feijen, J.; Kim, S.W. Effect of comonomer hydrophilicity and ionization on the lower critical solution temperature of N-isopropylacrylamide copolymers. Macromolecules 1993, 26, 2496–2500. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, J.; Ding, Y.; Ye, X. Effect of urea on phase transition of poly(N-isopropylacrylamide) investigated by differential scanning calorimetry. J. Phys. Chem. B 2014, 118, 9460–9466. [Google Scholar] [CrossRef] [PubMed]
- Kamath, K.R.; Park, K. Biodegradable hydrogels in drug delivery. Adv. Drug Deliv. Rev. 1993, 11, 59–84. [Google Scholar] [CrossRef]
- Cho, J.K.; Hong, K.Y.; Park, J.W.; Yang, H.K.; Song, S.C. Injectable delivery system of 2-methoxyestradiol for breast cancer therapy using biodegradable thermosensitive poly(organophosphazene) hydrogel. J. Drug Target. 2011, 19, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wu, R.A.; Zhao, L.; Hu, Z.; Guo, S.; Pan, X.; Zou, H. Folate and iron difunctionalized multiwall carbon nanotubes as dual-targeted drug nanocarrier to cancer cells. Carbon 2011, 49, 1797–1805. [Google Scholar] [CrossRef]
- Zhou, T.; Zhou, X.; Xing, D. Controlled release of doxorubicin from graphene oxide based charge-reversal nanocarrier. Biomaterials 2014, 35, 4185–4194. [Google Scholar] [CrossRef] [PubMed]
- Depan, D.; Shah, J.; Misra, R. Controlled release of drug from folate-decorated and graphene mediated drug delivery system: Synthesis, loading efficiency and drug release response. Mater. Sci. Eng. C 2011, 31, 1305–1312. [Google Scholar] [CrossRef]
- Saul, J.M.; Annapragada, A.; Natarajan, J.V.; Bellamkonda, R.V. Controlled targeting of liposomal doxorubicin via the folate receptor in vitro. J. Control. Release 2003, 92, 49–67. [Google Scholar] [CrossRef]
- Wong, H.L.; Rauth, A.M.; Bendayan, R.; Manias, J.L.; Ramaswamy, M.; Liu, Z.; Erhan, S.Z.; Wu, X.Y. A new polymer–lipid hybrid nanoparticle system increases cytotoxicity of doxorubicin against multidrug-resistant human breast cancer cells. Pharm. Res. 2006, 23, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Manaspon, C.; Viravaidya-Pasuwat, K.; Pimpha, N. Preparation of folate-conjugated Pluronic F127/chitosan core-shell nanoparticles encapsulating doxorubicin for breast cancer treatment. J. Nanomater. 2012, 2012, 22. [Google Scholar] [CrossRef]
- Lee, D.-G.; Ponvel, K.M.; Kim, M.; Hwang, S.; Ahn, I.-S.; Lee, C.-H. Immobilization of lipase on hydrophobic nano-sized magnetite particles. J. Mol. Catal. B Enzym. 2009, 57, 62–66. [Google Scholar] [CrossRef]
- Zunino, F.; Di Marco, A.; Zaccara, A.; Luoni, G. The inhibition of RNA polymerase by daunomycin. Chem.-Biol. Interact. 1974, 9, 25–36. [Google Scholar] [CrossRef]
- Frederick, C.A.; Williams, L.D.; Ughetto, G.; Van der Marel, G.A.; Van Boom, J.H.; Rich, A.; Wang, A.-J. Structural comparison of anticancer drug-DNA complexes: Adriamycin and daunomycin. Biochemistry 1990, 29, 2538–2549. [Google Scholar] [CrossRef] [PubMed]
- Meyn, M.S. Ataxia-telangiectasia and cellular responses to DNA damage. Cancer Res. 1995, 55, 5991–6001. [Google Scholar] [PubMed]
- Kim, J.I.; Lee, B.S.; Chun, C.; Cho, J.-K.; Kim, S.-Y.; Song, S.-C. Long-term theranostic hydrogel system for solid tumors. Biomaterials 2012, 33, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Frishman, W.H.; Yee, H.C.M.; Keefe, D.; Sung, H.M.; Liu, L.L.; Einzig, A.I.; Dutcher, J. Cardiovascular toxicity with cancer chemotherapy. Curr. Probl. Cancer 1997, 21, 301–360. [Google Scholar] [CrossRef]
- Luo, J.W.; Zhang, T.; Zhang, Q.; Cao, X.; Zeng, X.; Fu, Y.; Zhang, Z.R.; Gong, T. A novel injectable phospholipid gel co-loaded with doxorubicin and bromotetrandrine for resistant breast cancer treatment by intratumoral injection. Colloids Surf. B Biointerfaces 2016, 140, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Seabra, A.B.; Paula, A.J.; de Lima, R.; Alves, O.L.; Durán, N. Nanotoxicity of graphene and graphene oxide. Chem. Res. Toxicol. 2014, 27, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Kean, T.; Thanou, M. Biodegradation, biodistribution and toxicity of chitosan. Adv. Drug Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Fakhari, A.; Berkland, C. Applications and emerging trends of hyaluronic acid in tissue engineering, as a dermal filler and in osteoarthritis treatment. Acta Biomater. 2013, 9, 7081–7092. [Google Scholar] [CrossRef] [PubMed]
- Kohori, F.; Sakai, K.; Aoyagi, T.; Yokoyama, M.; Sakurai, Y.; Okano, T. Preparation and characterization of thermally responsive block copolymer micelles comprising poly(N-isopropylacrylamide-b-dl-lactide). J. Control. Release 1998, 55, 87–98. [Google Scholar] [CrossRef]
- Patenaude, M.; Hoare, T. Injectable, degradable thermoresponsive poly(N-isopropylacrylamide) hydrogels. ACS Macro Lett. 2012, 1, 409–413. [Google Scholar] [CrossRef]
- Tiffen, J.C.; Bailey, C.G.; Ng, C.; Rasko, J.E.; Holst, J. Luciferase expression and bioluminescence does not affect tumor cell growth in vitro or in vivo. Mol. Cancer 2010, 9, 299. [Google Scholar] [CrossRef] [PubMed]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Huang, P.; Xu, C.; Lin, J.; Wang, C.; Wang, X.; Zhang, C.; Zhou, X.; Guo, S.; Cui, D. Folic acid-conjugated graphene oxide loaded with photosensitizers for targeting photodynamic therapy. Theranostics 2011, 1, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-T.; Chen, C.-T.; Chen, J.-P. Osteogenic differentiation and ectopic bone formation of canine bone marrow-derived mesenchymal stem cells in injectable thermo-responsive polymer hydrogel. Tissue Eng. Part C Methods 2011, 17, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Grenha, A.; Seijo, B.; Remunán-López, C. Microencapsulated chitosan nanoparticles for lung protein delivery. Eur. J. Pharm. Sci. 2005, 25, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hartner, W.C.; Gillespie, J.W.; Praveen, K.P.; Yang, S.; Mei, L.A.; Petrenko, V.A.; Torchilin, V.P. Enhanced tumor delivery and antitumor activity in vivo of liposomal doxorubicin modified with MCF-7-specific phage fusion protein. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Long, Q.; Wu, Q.; Shi, S.; Dai, M.; Liu, Y.; Liu, L.; Gong, C.; Qian, Z.; Wei, Y. Improving therapeutic effect in ovarian peritoneal carcinomatosis with honokiol nanoparticles in a thermosensitive hydrogel composite. RSC Adv. 2012, 2, 7759–7771. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Unit | Control | GOFA-DOX/HACPN |
|---|---|---|---|
| Hematology | |||
| WBC | 103 cells/μL | 5.78 ± 2.34 | 2.9 ± 0.6 * |
| RBC | 106 cells/μL | 8.57 ± 0.51 | 8.5 ± 0.2 * |
| HGB | g/dL | 13.30 ± 0.87 | 13.2 ± 0.5 * |
| HCT | % | 41.77 ± 2.23 | 39.1 ± 2.5 * |
| PLT | 103 cells/μL | 334.30 ± 29.8 | 279.5 ± 37.5 * |
| Clinical Chemistry | |||
| AST | U/L | 254.5 ± 19.1 | 294 ± 39.4 * |
| ALT | U/L | 102.50 ± 0.71 | 113.8 ± 51.3 * |
| BUN | mg/dL | 28.40 ± 3.54 | 32.1 ± 3.1 * |
| CREA | mg/dL | 0.12 ± 0.01 | 0.13 ± 0.01 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fong, Y.T.; Chen, C.-H.; Chen, J.-P. Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy. Nanomaterials 2017, 7, 388. https://doi.org/10.3390/nano7110388
Fong YT, Chen C-H, Chen J-P. Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy. Nanomaterials. 2017; 7(11):388. https://doi.org/10.3390/nano7110388
Chicago/Turabian StyleFong, Yi Teng, Chih-Hao Chen, and Jyh-Ping Chen. 2017. "Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy" Nanomaterials 7, no. 11: 388. https://doi.org/10.3390/nano7110388
APA StyleFong, Y. T., Chen, C. -H., & Chen, J. -P. (2017). Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy. Nanomaterials, 7(11), 388. https://doi.org/10.3390/nano7110388