
Computational Predictions for Single Chain Chalcogenide-Based One-Dimensional Materials

Abstract
:1. Introduction
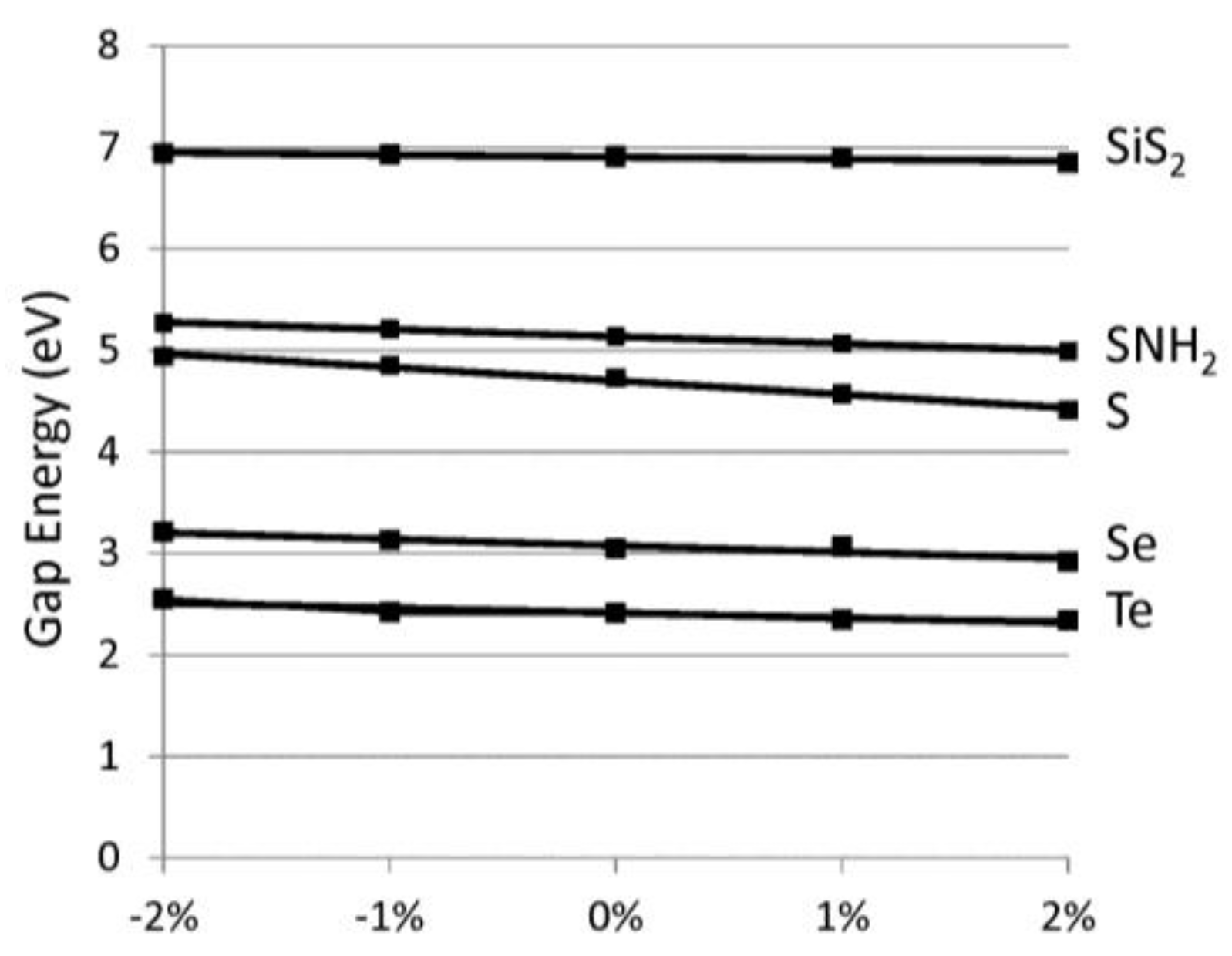
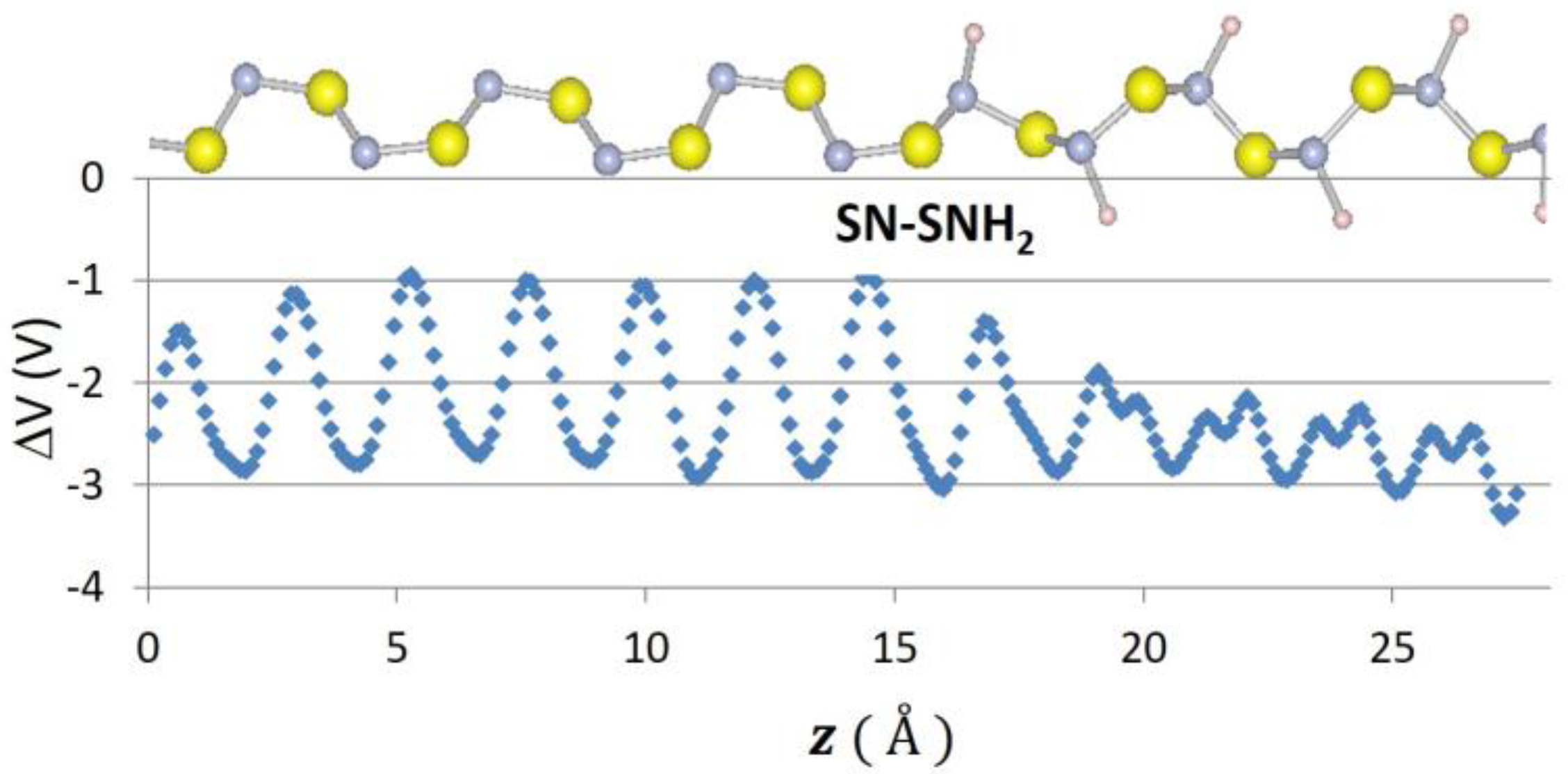
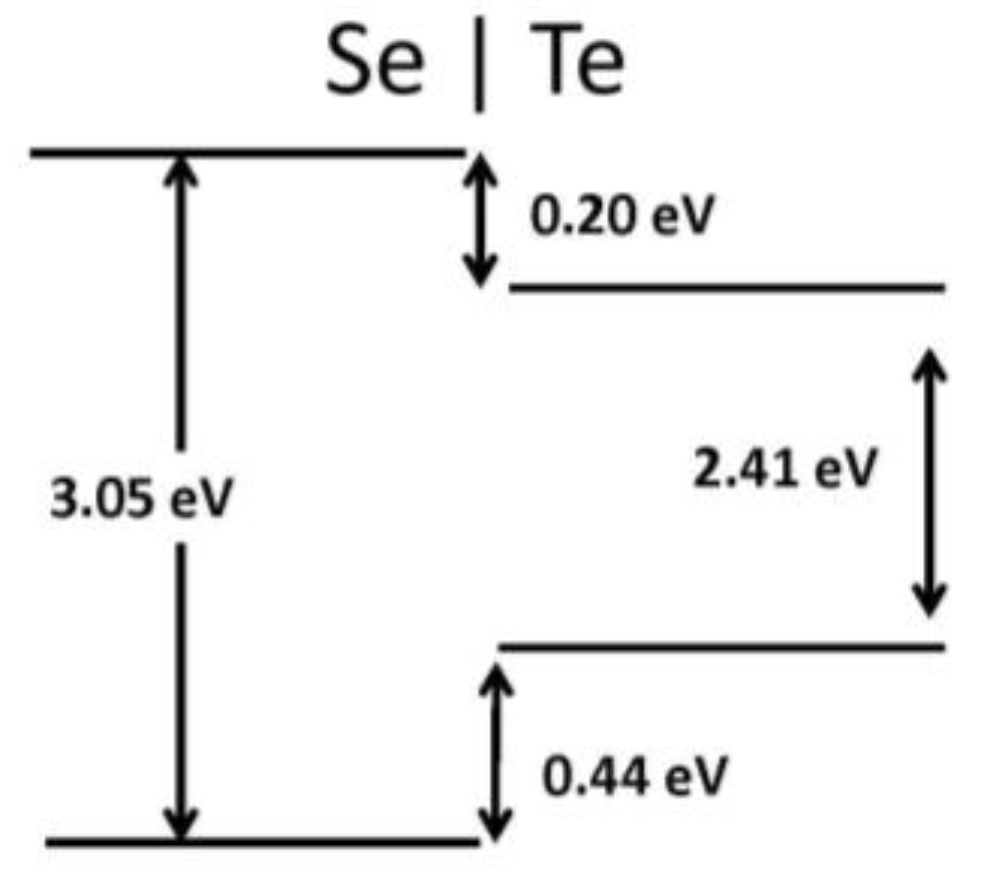
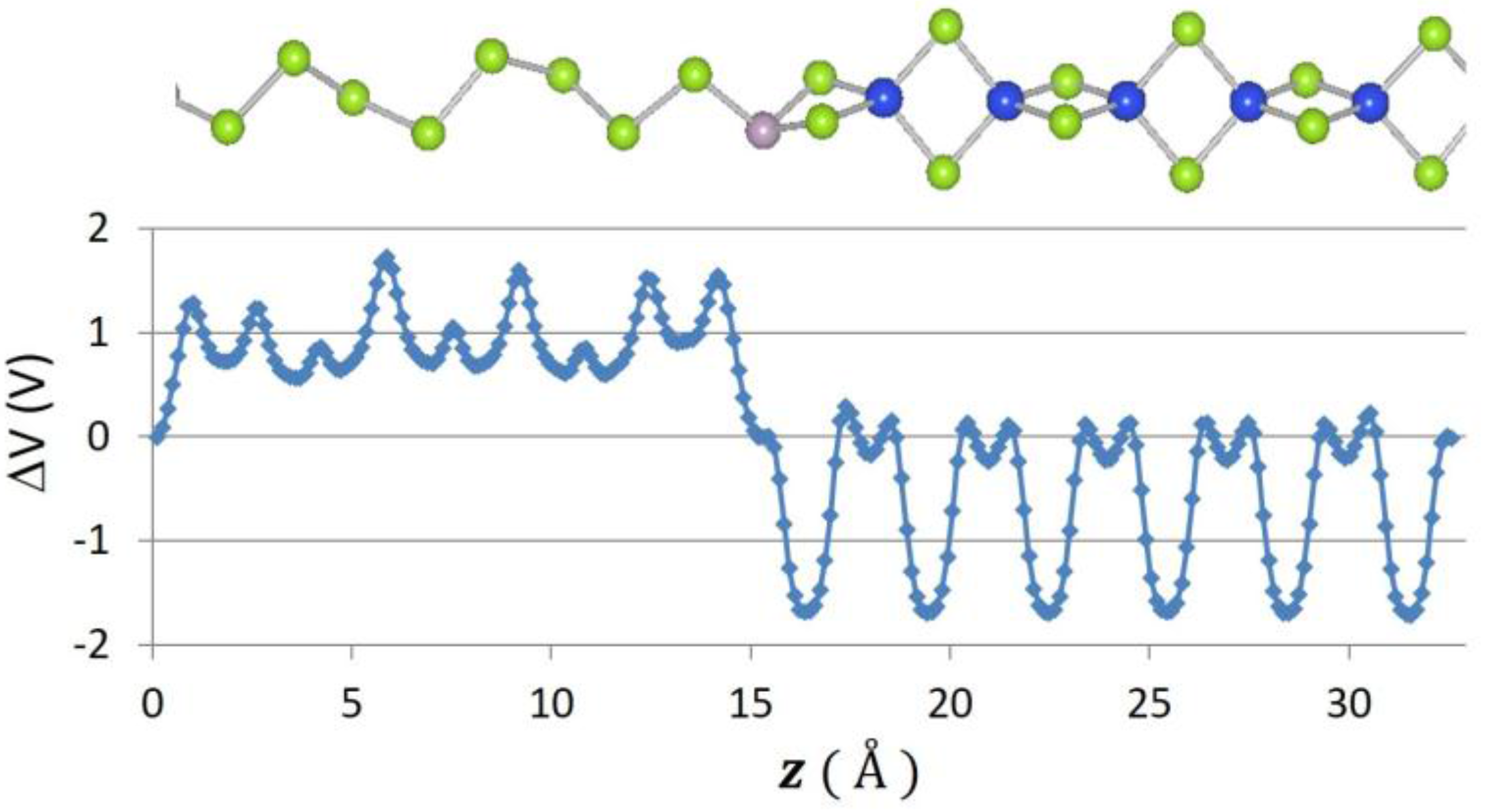
2. Results
3. Discussion
4. Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Novoselov, K.S.; Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Hennig, R.G. Computational prediction of two-dimensional group-IV mono-chalcogenides. Appl. Phys. Lett. 2014, 105, 042103. [Google Scholar] [CrossRef]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tománek, D.; Ye, P.D. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Soklaski, R.; Liang, Y.; Yang, L. Layer-controlled band gap and anisotropic excitons in few-layer black phosphorus. Phys. Rev. B 2014, 89, 235319. [Google Scholar] [CrossRef]
- Chernikov, A.; Berkelbach, T.C.; Hill, H.M.; Rigosi, A.; Li, Y.; Aslan, O.B.; Reichman, D.R.; Hybertsen, M.S.; Heinz, T.F. Exciton Binding Energy and Nonhydrogenic Rydberg Series in Monolayer. Phys. Rev. Lett. 2014, 113, 076802. [Google Scholar] [CrossRef] [PubMed]
- Conley, H.J.; Wang, B.; Ziegler, J.I.; Haglund, R.F.; Pantelides, S.T.; Bolotin, K.I. Bandgap Engineering of Strained Monolayer and Bilayer MoS2. Nano Lett. 2013, 13, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Mathew, K.; Zhuang, H.L.; Hennig, R.G. Computational Screening of 2D Materials for Photocatalysis. J. Phys. Chem. Lett. 2015, 6, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cretu, O.; Zhou, W.; Suenaga, K.; Prasai, D.; Bolotin, K.I.; Cuong, N.T.; Otani, M.; Okada, S.; Lupini, A.R.; et al. Flexible metallic nanowires with self-adaptive contacts to semiconducting transition-metal dichalcogenide monolayers. Nat. Nano Lett. 2014, 9, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Butun, S.; Tongay, S.; Aydin, K. Enhanced Light Emission from Large-Area Monolayer MoS2 Using Plasmonic Nanodisc Arrays. Nano Lett. 2015, 15, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.L.; Hennig, R.G. Theoretical perspective of photocatalytic properties of single-layer SnS2. Phys. Rev. B 2013, 88, 115314. [Google Scholar] [CrossRef]
- Leonard, F.; Talin, A.A. Electrical contacts to one- and two-dimensional nanomaterials. Nat. Nano 2011, 6, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Bom, N.M.; Soares, G.V.; de Oliveira Junior, M.H.; Lopes, J.M.J.; Riechert, H.; Radtke, C. Water Incorporation in Graphene Transferred onto SiO2/Si Investigated by Isotopic Labeling. J. Phys. Chem. C 2016, 120, 201–206. [Google Scholar] [CrossRef]
- Van’t Erve, O.M.J.; Friedman, A.L.; Li, C.H.; Robinson, J.T.; Connell, J.; Lauhon, L.J.; Jonker, B.T. Spin transport and Hanle effect in silicon nanowires using graphene tunnel barriers. Nat. Commun. 2015, 6, 7541. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, B.R.; Alhassan, S.M.; Pantelides, S.T. Large excitonic effects in group-IV sulfide monolayers. Phys. Rev. B 2015, 92, 235405. [Google Scholar] [CrossRef]
- Yin, J.; Li, J.; Hang, Y.; Yu, J.; Tai, G.; Li, X.; Zhang, Z.; Guo, W. Boron Nitride Nanostructures: Fabrication, Functionalization and Applications. Small 2016, 12, 2942–2968. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Pignedoli, C.A.; Talirz, L.; Ruffieux, P.; Söde, H.; Liang, L.; Meunier, V.; Berger, R.; Li, R.; Feng, X.; et al. Graphene nanoribbon heterojunctions. Nat. Nano Lett. 2014, 9, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Bäppler, S.A.; Plasser, F.; Wormit, M.; Dreuw, A. Exciton analysis of many-body wave functions: Bridging the gap between the quasiparticle and molecular orbital pictures. Phys. Rev. A 2014, 90, 052521. [Google Scholar] [CrossRef]
- Tellurium (Te) Effective Masses. Non-Tetrahedrally Bonded Elements and Binary Compounds I; Madelung, O., Rössler, U., Schulz, M., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 1–6. [Google Scholar]
- Kivelson, S.; Chapman, O.L. Polyacene and a new class of quasi-one-dimensional conductors. Phys. Rev. B 1983, 28, 7236–7243. [Google Scholar] [CrossRef]
- Mishima, A.; Kimura, M. Superconductivity of the quasi-one-dimensional semiconductor—Polyacene. Synth. Metals 1985, 11, 75–84. [Google Scholar] [CrossRef]
- Springborg, M.; Jones, R.O. Sulfur and selenium helices: Structure and electronic properties. J. Chem. Phys. 1988, 88, 2652–2658. [Google Scholar] [CrossRef]
- Lovinger, A.J.; Padden, F.J., Jr.; Davis, D.D. Structure of poly(p-phenylene sulphide). Polymer 1988, 29, 229–232. [Google Scholar] [CrossRef]
- Sharma, V.; Wang, C.; Lorenzini, R.G.; Ma, R.; Zhu, Q.; Sinkovits, D.W.; Pilania, G.; Oganov, A.R.; Kumar, S.; Sotzing, G.A.; et al. Rational design of all organic polymer dielectrics. Nat. Commun. 2014, 5, 4845. [Google Scholar] [CrossRef] [PubMed]
- Seel, M.; Collins, T.C.; Martino, F.; Rai, D.K.; Ladik, J. SNx with hydrogen impurities in the coherent-potential approximation. Phys. Rev. B 1978, 18, 6460–6464. [Google Scholar] [CrossRef]
- Amin, B.; Singh, N.; Schwingenschlögl, U. Heterostructures of transition metal dichalcogenides. Phys. Rev. B 2015, 92, 075439. [Google Scholar] [CrossRef]
- Ramasubramaniam, A. Large excitonic effects in monolayers of molybdenum and tungsten dichalcogenides. Phys. Rev. B 2012, 86, 115409. [Google Scholar] [CrossRef]
- Viljas, J.K.; Pauly, F.; Cuevas, J.C. Modeling elastic and photoassisted transport in organic molecular wires: Length dependence and current-voltage characteristics. Phys. Rev. B 2008, 77, 155119. [Google Scholar] [CrossRef]
- Chen, X.; Tian, F.; Persson, C.; Duan, W.; Chen, N.-X. Interlayer interactions in graphites. Sci. Rep. 2013, 3, 3046. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sutter, E.; Shi, N.N.; Zheng, J.; Yang, T.; Englund, D.; Gao, H.-J.; Sutter, P. Reliable Exfoliation of Large-Area High-Quality Flakes of Graphene and Other Two-Dimensional Materials. ACS Nano 2015, 9, 10612–10620. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lan, H.; Peng, L.; Suenaga, K.; Iijima, S. Deriving Carbon Atomic Chains from Graphene. Phys. Rev. Lett. 2009, 102, 205501. [Google Scholar] [CrossRef] [PubMed]
- Arbiol, J.; de la Mata, M.; Eickhoff, M.; Morral, A.F.I. Bandgap engineering in a nanowire: Self-assembled 0, 1 and 2D quantum structures. Mater. Today 2013, 16, 213–219. [Google Scholar] [CrossRef]
- Stuke, J. Review of optical and electrical properties of amorphous semiconductors. J. Non-Cryst. Solids 1970, 4, 1–26. [Google Scholar] [CrossRef]
- Tanioka, K.; Yamazaki, J.; Shidara, K.; Taketoshi, K.; Kawamura, T.; Ishioka, S.; Takasaki, Y. An avalanche-mode amorphous Selenium photoconductive layer for use as a camera tube target. IEEE Electron. Device Lett. 1987, 8, 392–394. [Google Scholar] [CrossRef]
- Frey, J.B.; Belev, G.; Tousignant, O.; Mani, H.; Laperriere, L.; Kasap, S.O. Dark current in multilayer stabilized amorphous selenium based photoconductive X-ray detectors. J. Appl. Phys. 2012, 112, 014502. [Google Scholar] [CrossRef]
- Wegscheider, W.; Pfeiffer, L.N.; Dignam, M.M.; Pinczuk, A.; West, K.W.; McCall, S.L.; Hull, R. Lasing from excitons in quantum wires. Phys. Rev. Lett. 1993, 71, 4071–4074. [Google Scholar] [CrossRef] [PubMed]
- Yoshita, M.; Hayamizu, Y.; Akiyama, H.; Pfeiffer, L.N.; West, K.W. Fabrication and microscopic characterization of a single quantum-wire laser with high uniformity. Phys. E Low-Dimens. Syst. Nanostruct. 2004, 21, 230–235. [Google Scholar] [CrossRef]
- Ostojic, G.N.; Zaric, S.; Kono, J.; Moore, V.C.; Hauge, R.H.; Smalley, R.E. Stability of High-Density One-Dimensional Excitons in Carbon Nanotubes under High Laser Excitation. Phys. Rev. Lett. 2005, 94, 097401. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical gga-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef]
- Shishkin, M.; Kresse, G. Implementation and performance of the frequency-dependent GW method within the PAW framework. Phys. Rev. B 2006, 74, 035101. [Google Scholar] [CrossRef]
- Fuchs, F.; Furthmüller, J.; Bechstedt, F.; Shishkin, M.; Kresse, G. Quasiparticle band structure based on a generalized Kohn-Sham scheme. Phys. Rev. B 2007, 76, 115109. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; Martin, R.M. Theoretical calculations of heterojunction discontinuities in the Si/Ge system. Phys. Rev. B 1986, 34, 5621–5634. [Google Scholar] [CrossRef]
- Tuttle, B.R. Theoretical investigation of the valence-band offset between Si(001) and SiO2. Phys. Rev. B 2004, 70, 125322. [Google Scholar] [CrossRef]
- Shaltaf, R.; Rignanese, G.M.; Gonze, X.; Giustino, F.; Pasquarello, A. Band Offsets at the SiSiO2 Interface from Many-Body Perturbation Theory. Phys. Rev. Lett. 2008, 100, 186401. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | ao (Å) | Esm (eV/Å) | Eexf. (eV/at.) | Egap (eV) | Dgap (eV/%) |
|---|---|---|---|---|---|
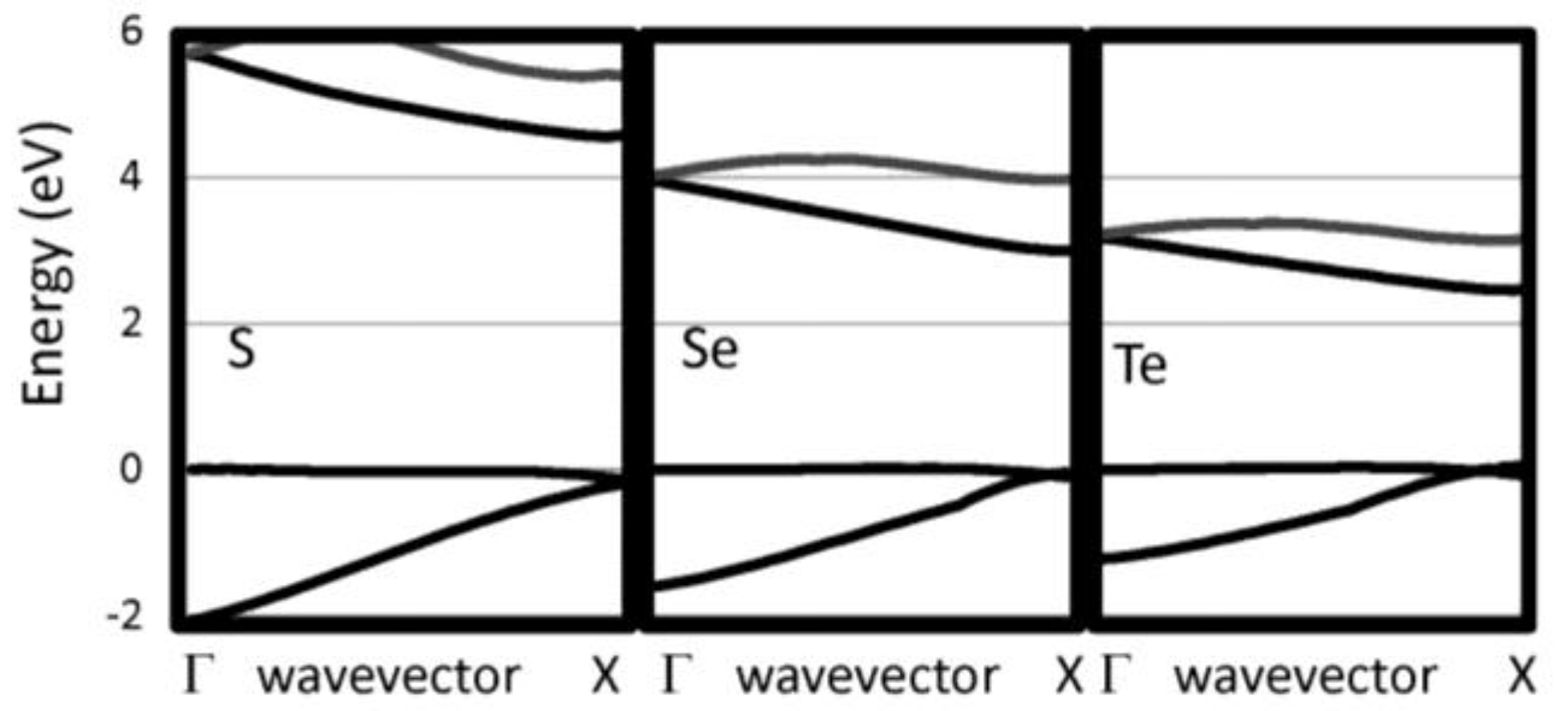
| S | 4.404 | 6.78 | 0.08 | 4.73 (4.60) | −0.13 |
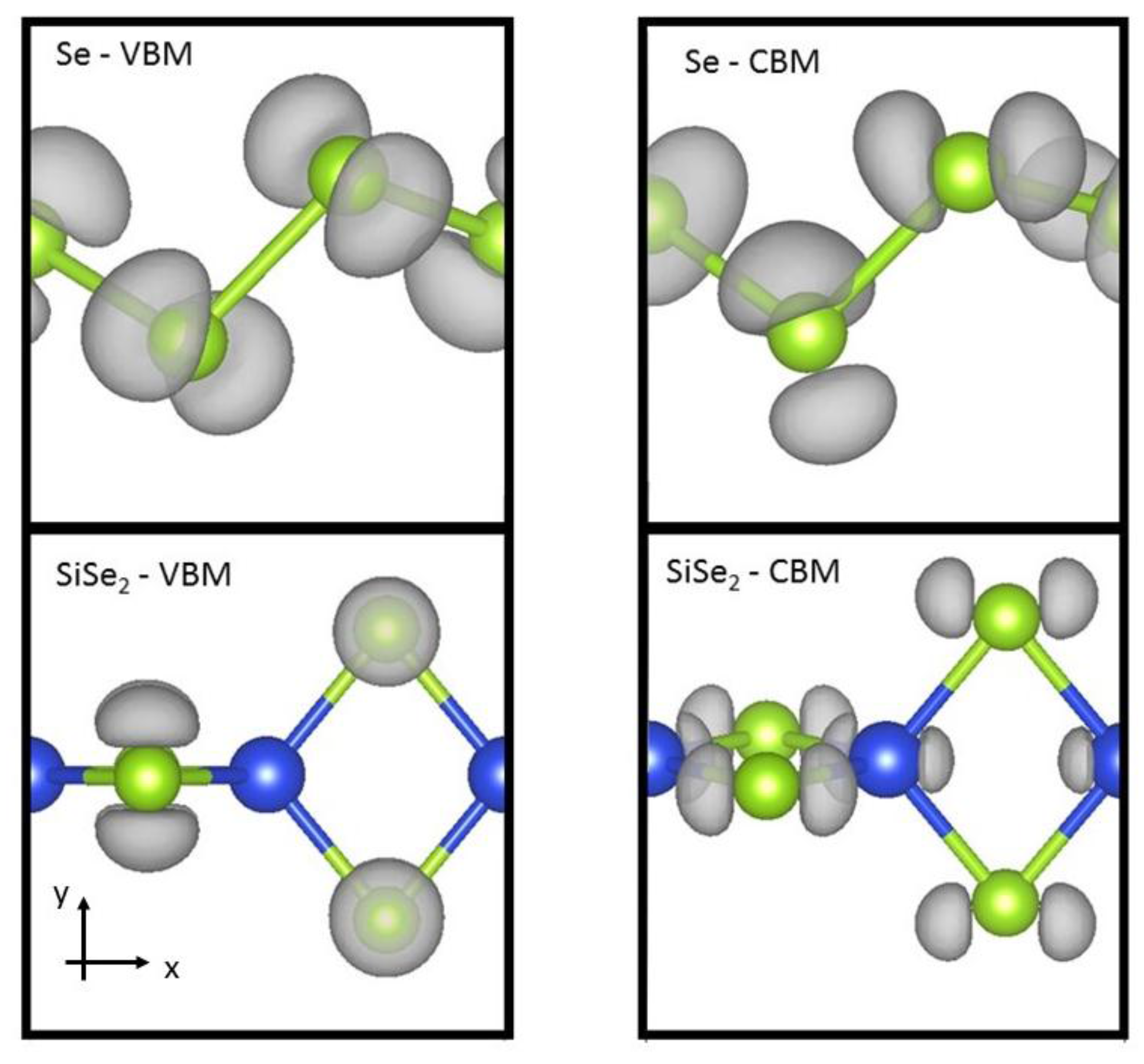
| Se | 4.966 | 4.32 | 0.20 | 3.05 (3.00) | −0.06 |
| Te | 5.682 | 2.78 | 0.27 | 2.41 | −0.05 |
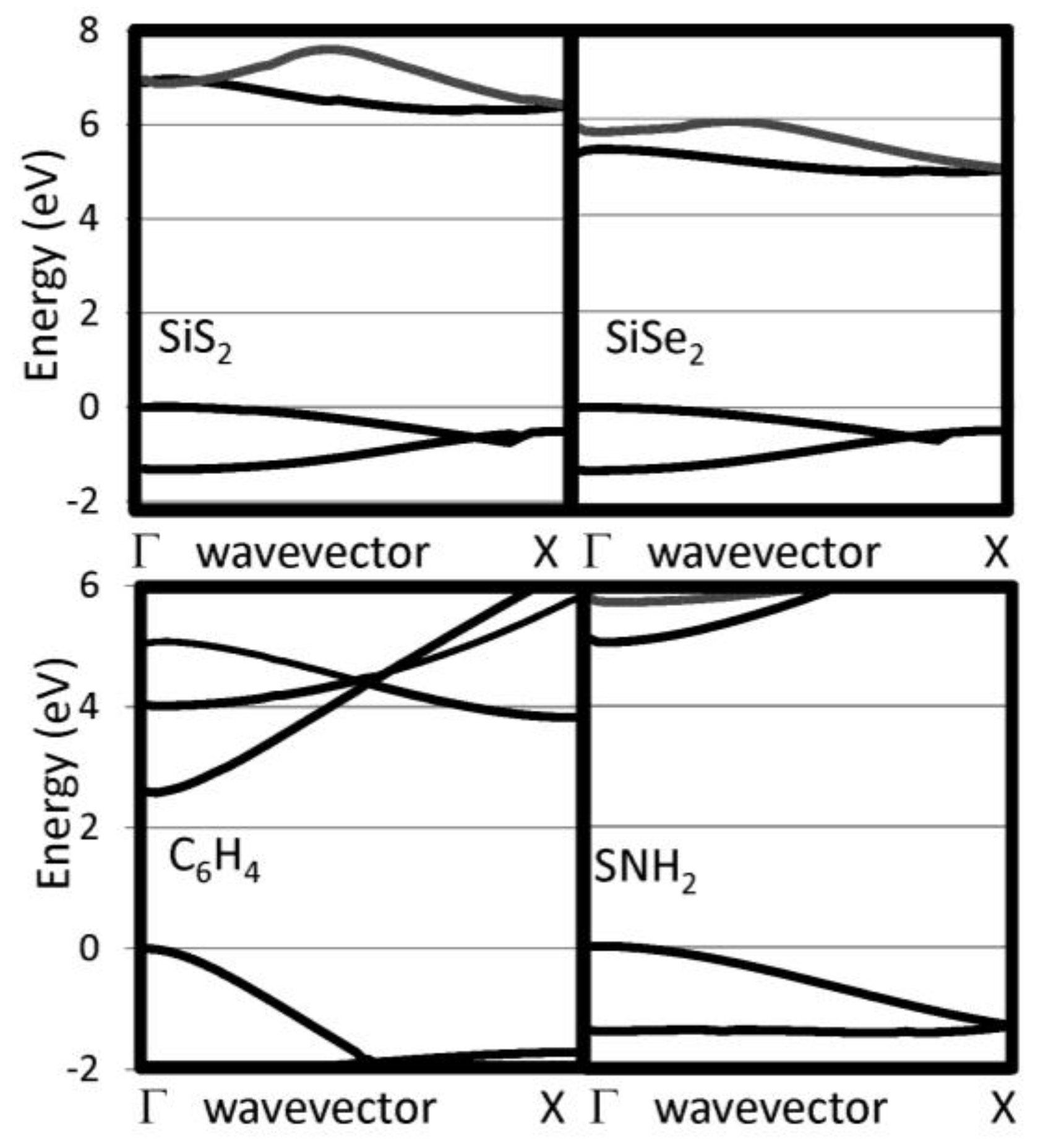
| SiS2 | 5.588 | 10.56 | 0.07 | 6.91 (6.34) | −0.02 |
| SiSe2 | 5.939 | 8.19 | 0.09 | 5.25 (4.92) | −0.11 |
| C6H4 | 4.364 | 31.07 | 0.12 | 2.60 | +0.11 |
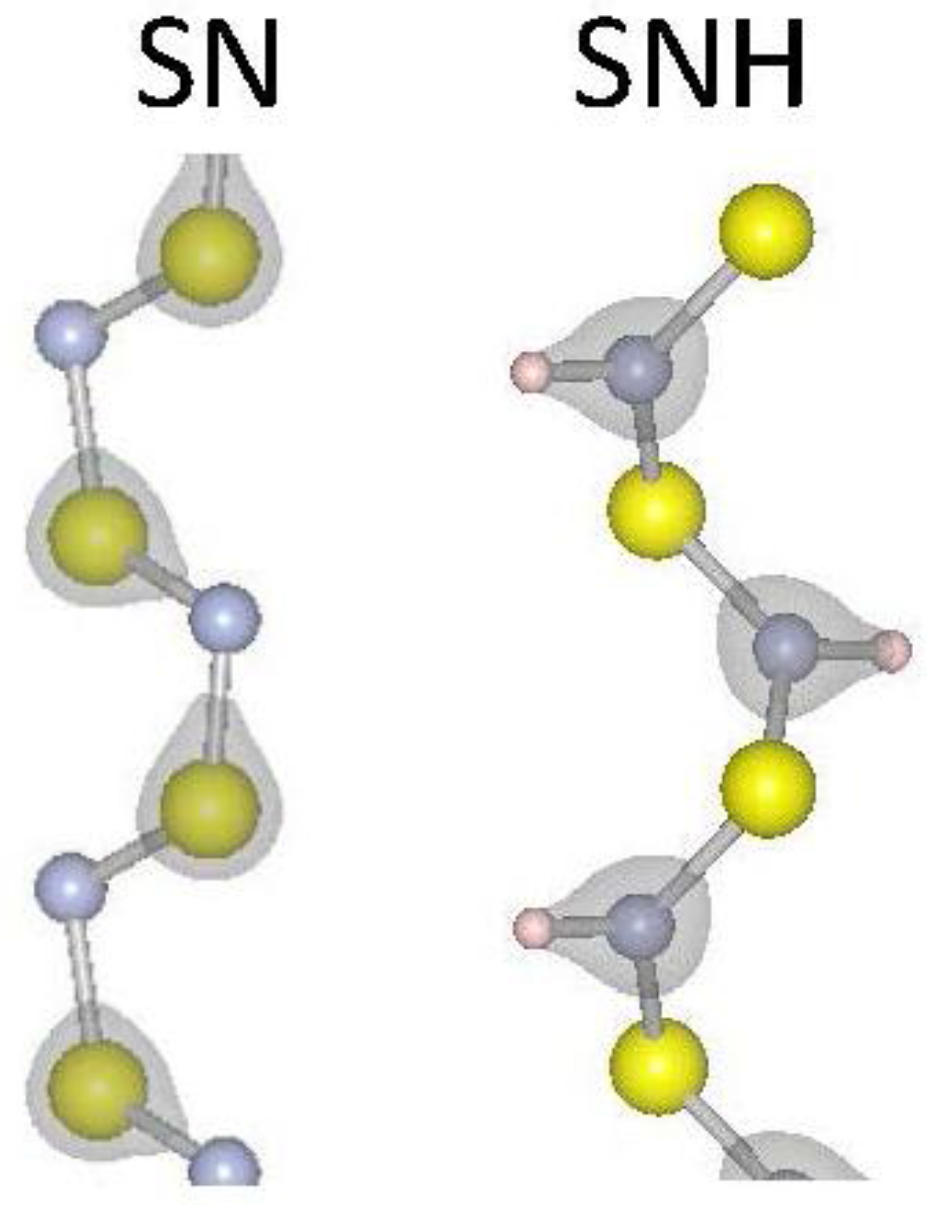
| SNH | 4.700 | 3.10 | 0.14 | 5.14 | −0.07 |
| SN | 4.556 | 5.06 | 0.05 | N/A | N/A |
| C4H2 | 2.468 | 50.9 | 0.18 | N/A | N/A |
| Material | VBO (eV) | CBO (eV) |
|---|---|---|
| Te–Se | 0.44 | 0.20 |
| SiSe2–SiS2 | 1.00 | 0.42 |
| SN–SNH2 | 1.93 | 3.21 |
| Se–P–SiSe2 | 0.13 | 1.72 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuttle, B.; Alhassan, S.; Pantelides, S. Computational Predictions for Single Chain Chalcogenide-Based One-Dimensional Materials. Nanomaterials 2017, 7, 115. https://doi.org/10.3390/nano7050115
Tuttle B, Alhassan S, Pantelides S. Computational Predictions for Single Chain Chalcogenide-Based One-Dimensional Materials. Nanomaterials. 2017; 7(5):115. https://doi.org/10.3390/nano7050115
Chicago/Turabian StyleTuttle, Blair, Saeed Alhassan, and Sokrates Pantelides. 2017. "Computational Predictions for Single Chain Chalcogenide-Based One-Dimensional Materials" Nanomaterials 7, no. 5: 115. https://doi.org/10.3390/nano7050115
APA StyleTuttle, B., Alhassan, S., & Pantelides, S. (2017). Computational Predictions for Single Chain Chalcogenide-Based One-Dimensional Materials. Nanomaterials, 7(5), 115. https://doi.org/10.3390/nano7050115