Magnetic Graphene Oxide for Dual Targeted Delivery of Doxorubicin and Photothermal Therapy




Abstract
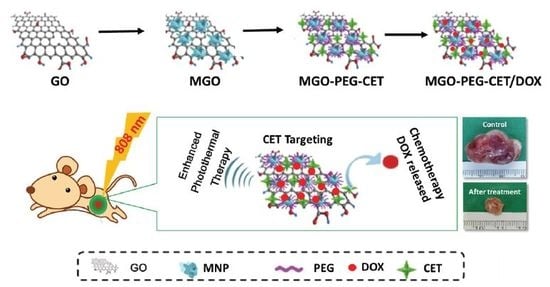
:
1. Introduction
2. Results and Discussion
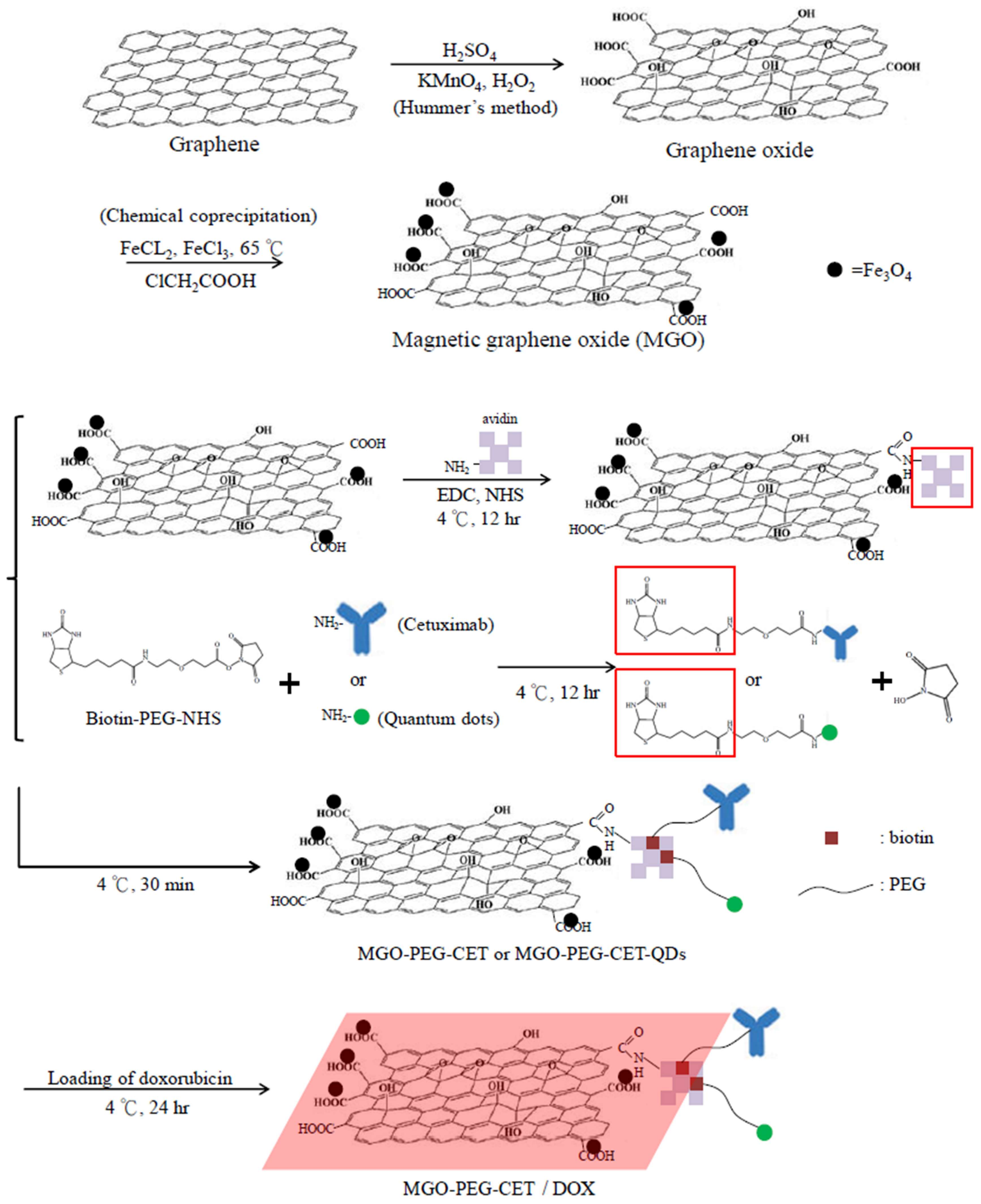
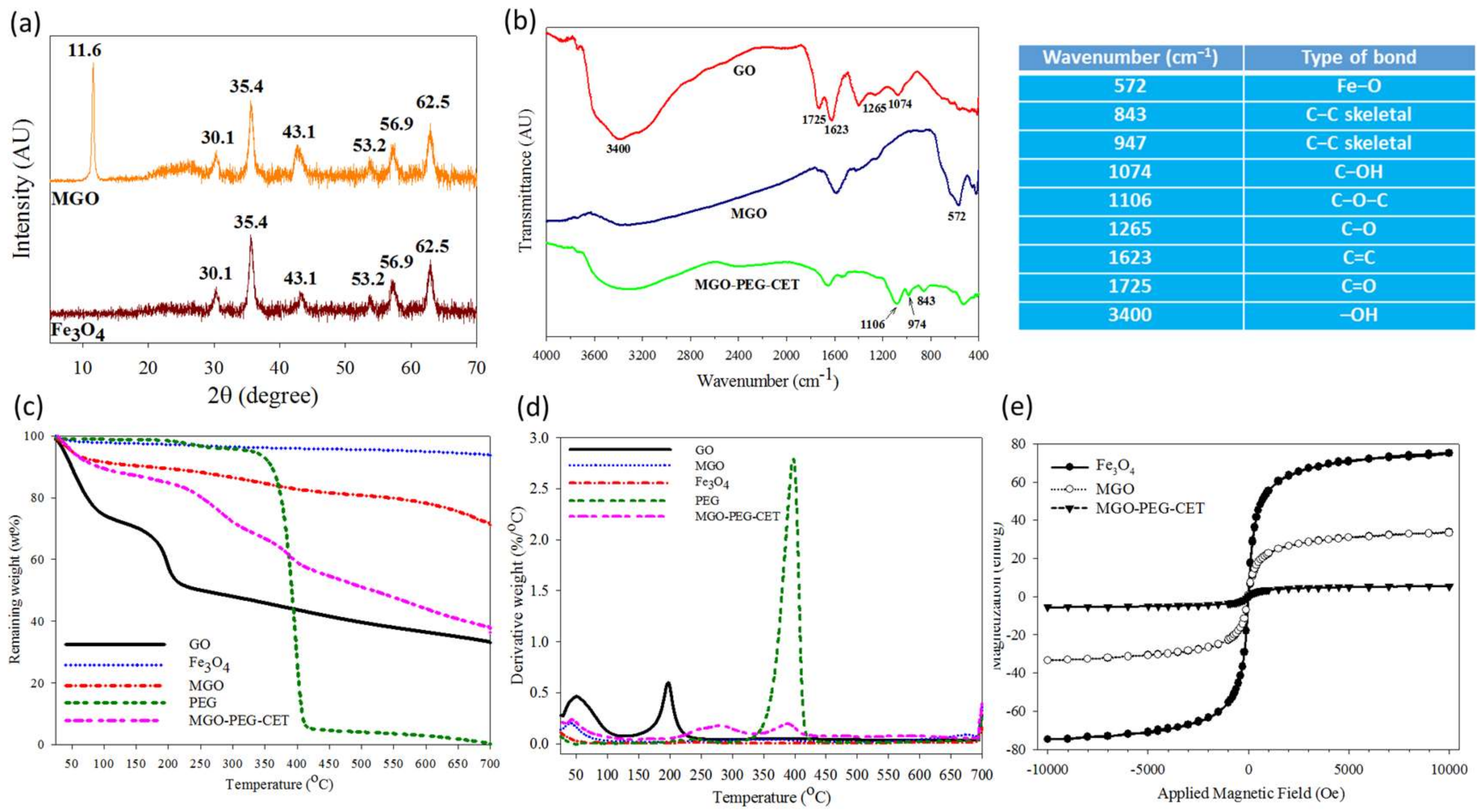
2.1. Preparation and Characteriazation of MGO, MGO-PEG-CET and MGO-PEG-CET/DOX
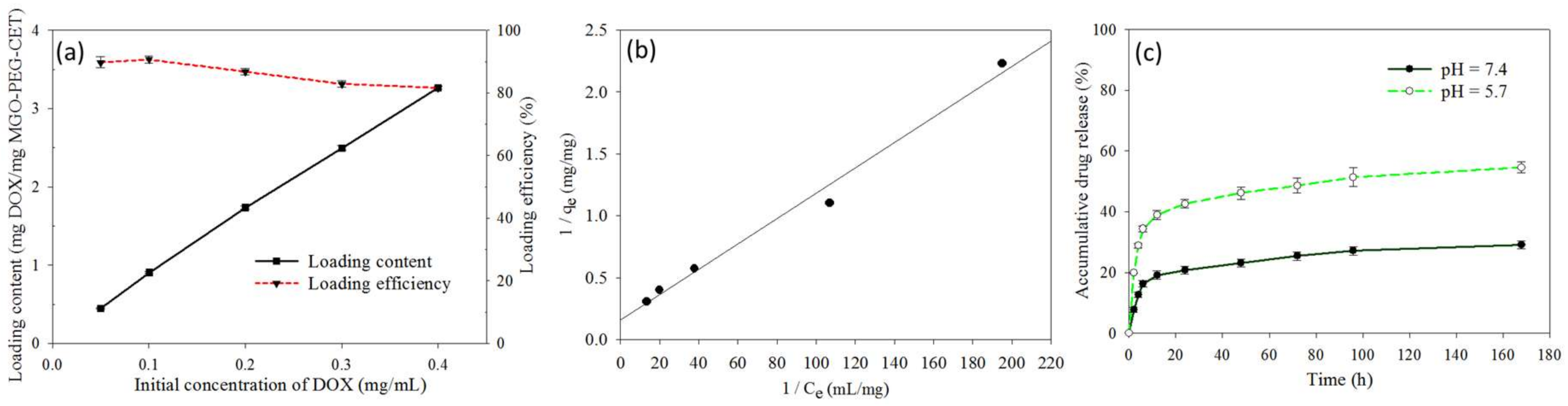
2.2. Drug Loading and Release
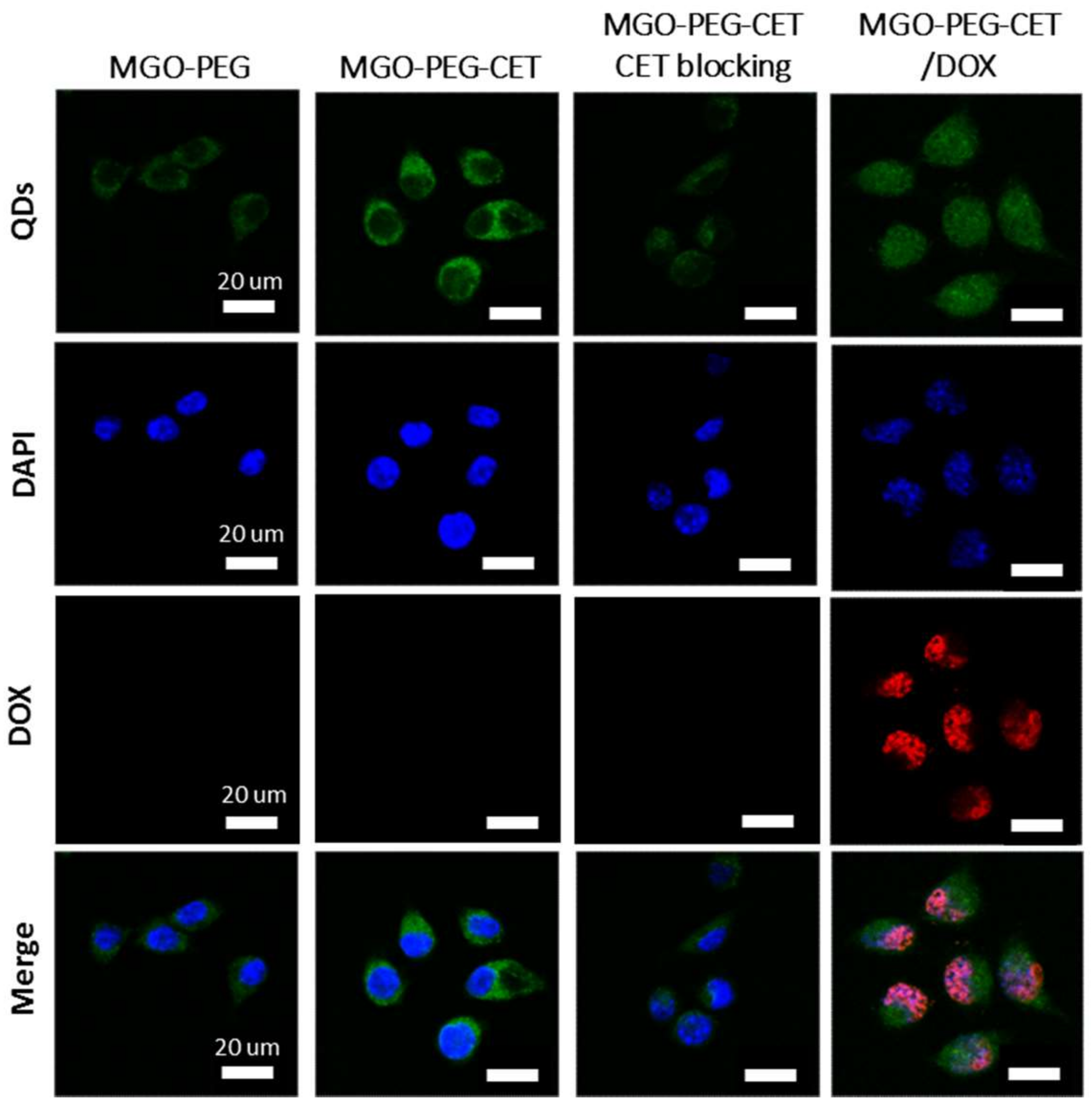
2.3. Intracelular Uptake
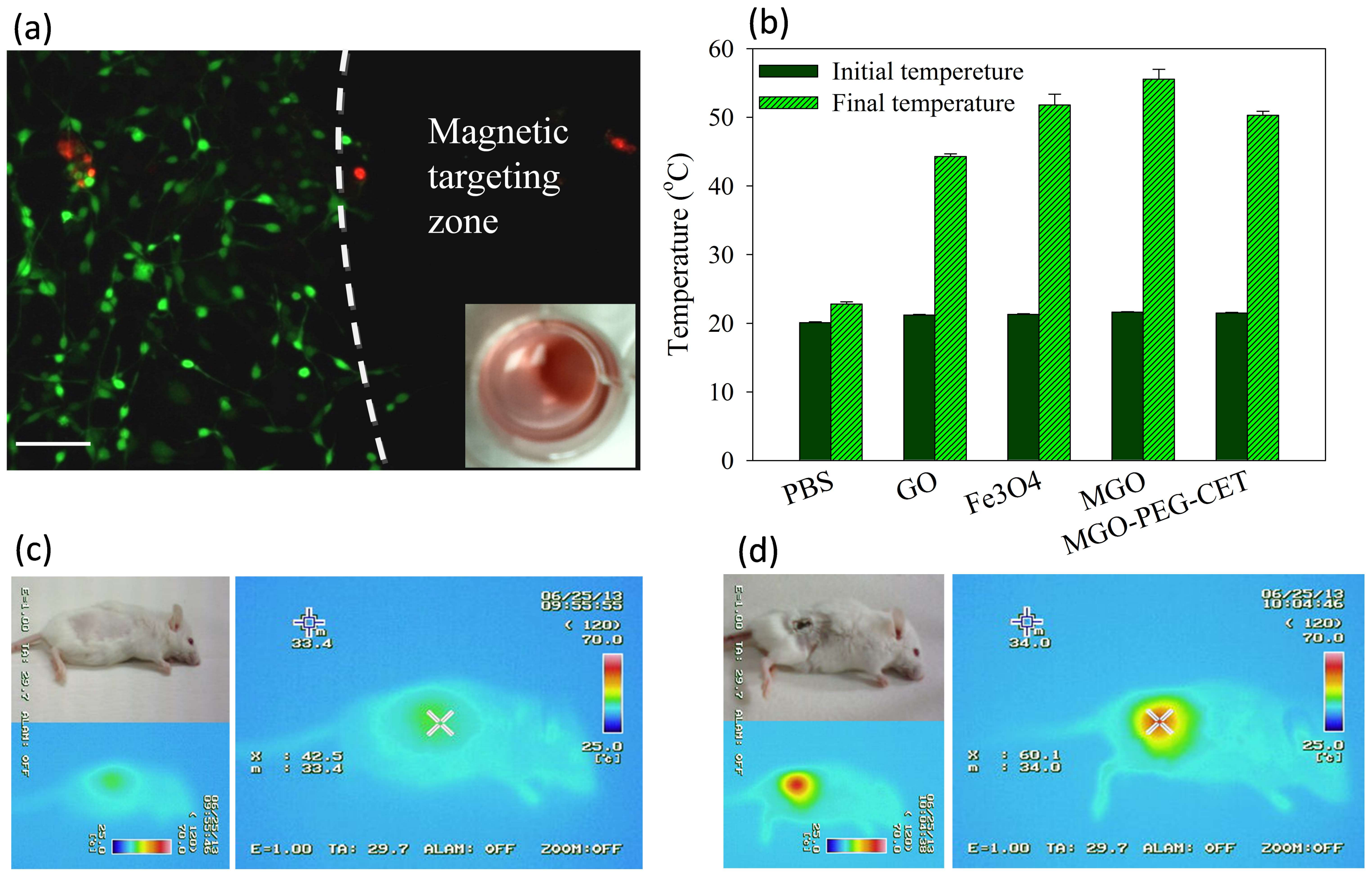
2.4. Magnetic Targeting and Laser-Induced Hyperthermia
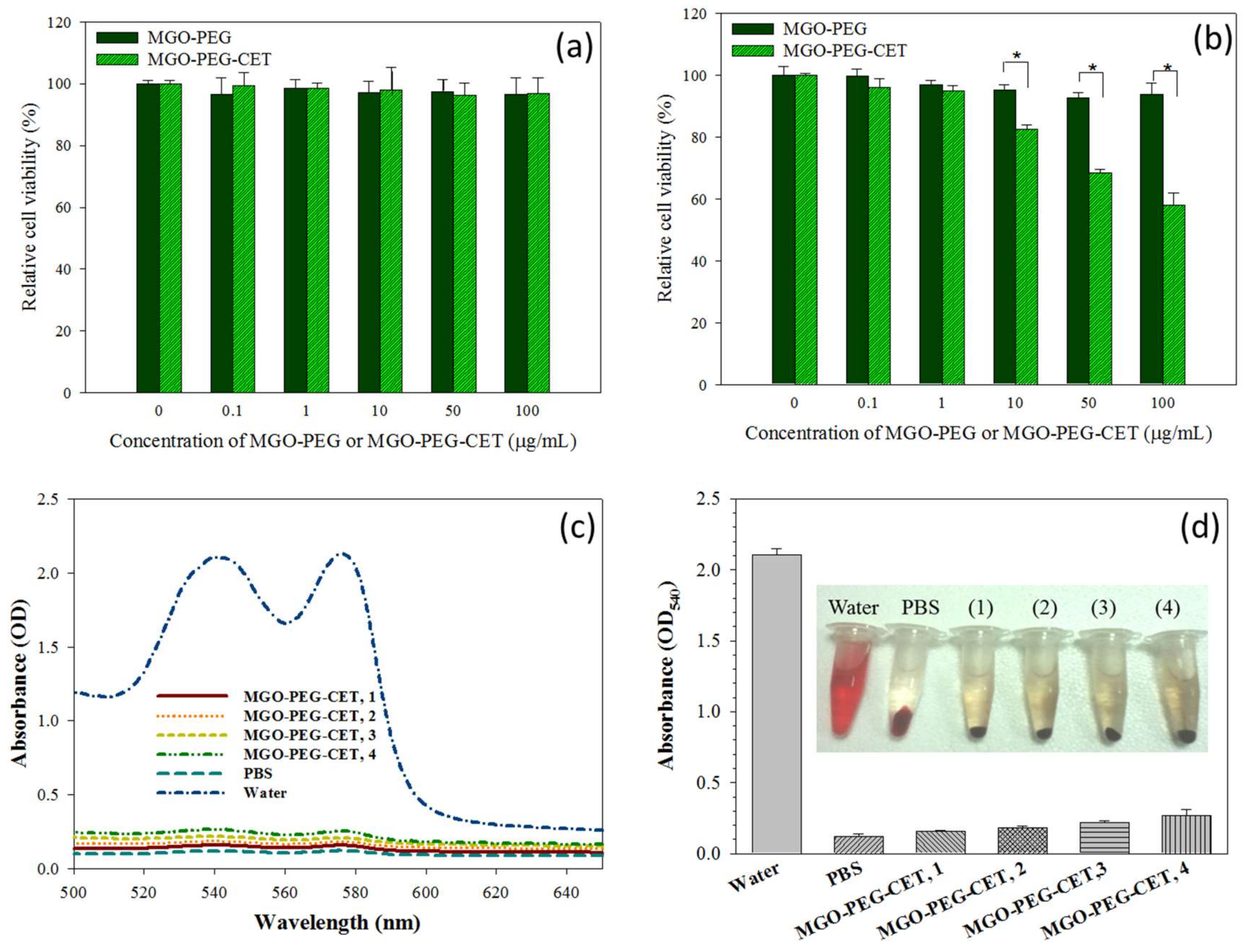
2.5. Biocompatibility of Nanocarriers
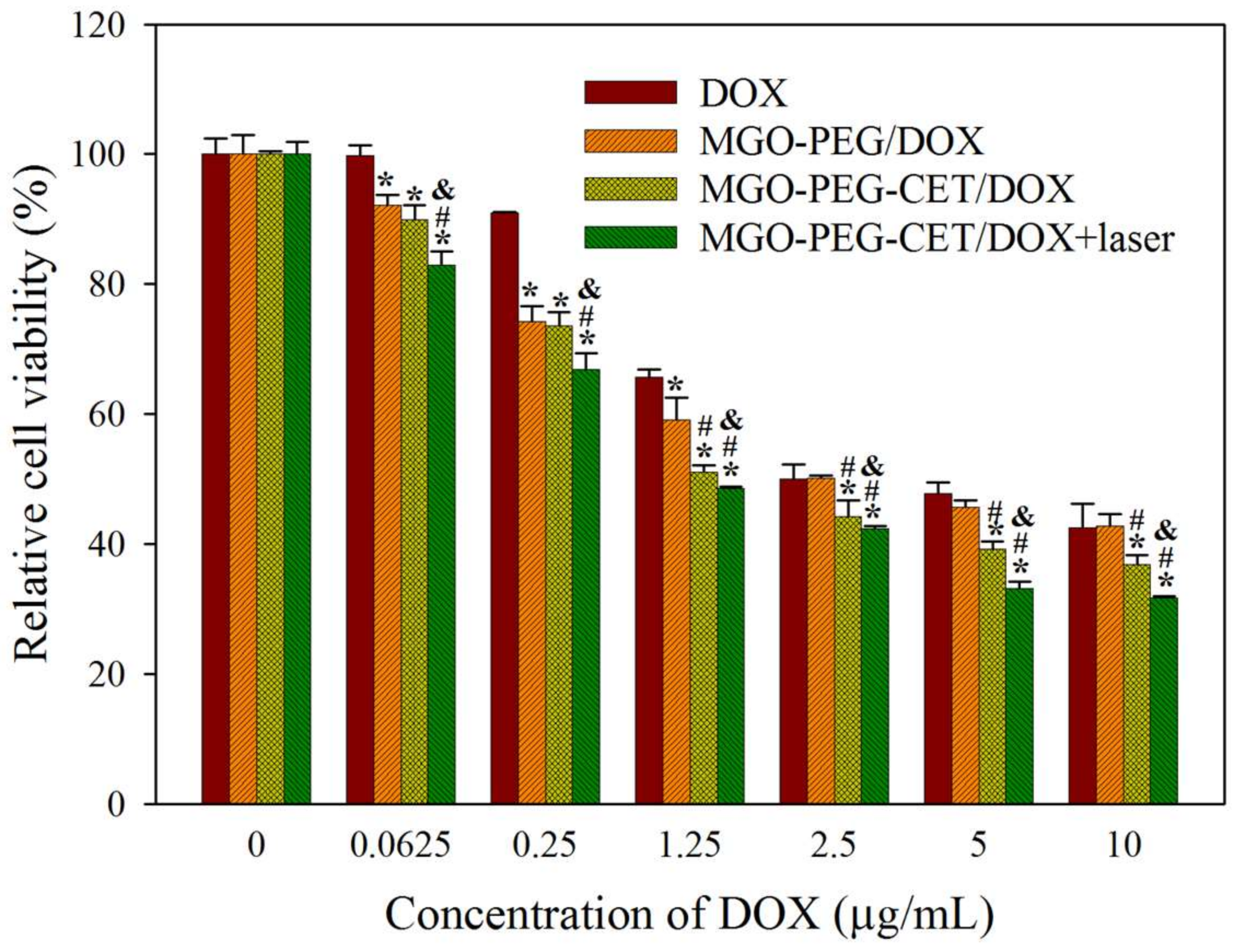
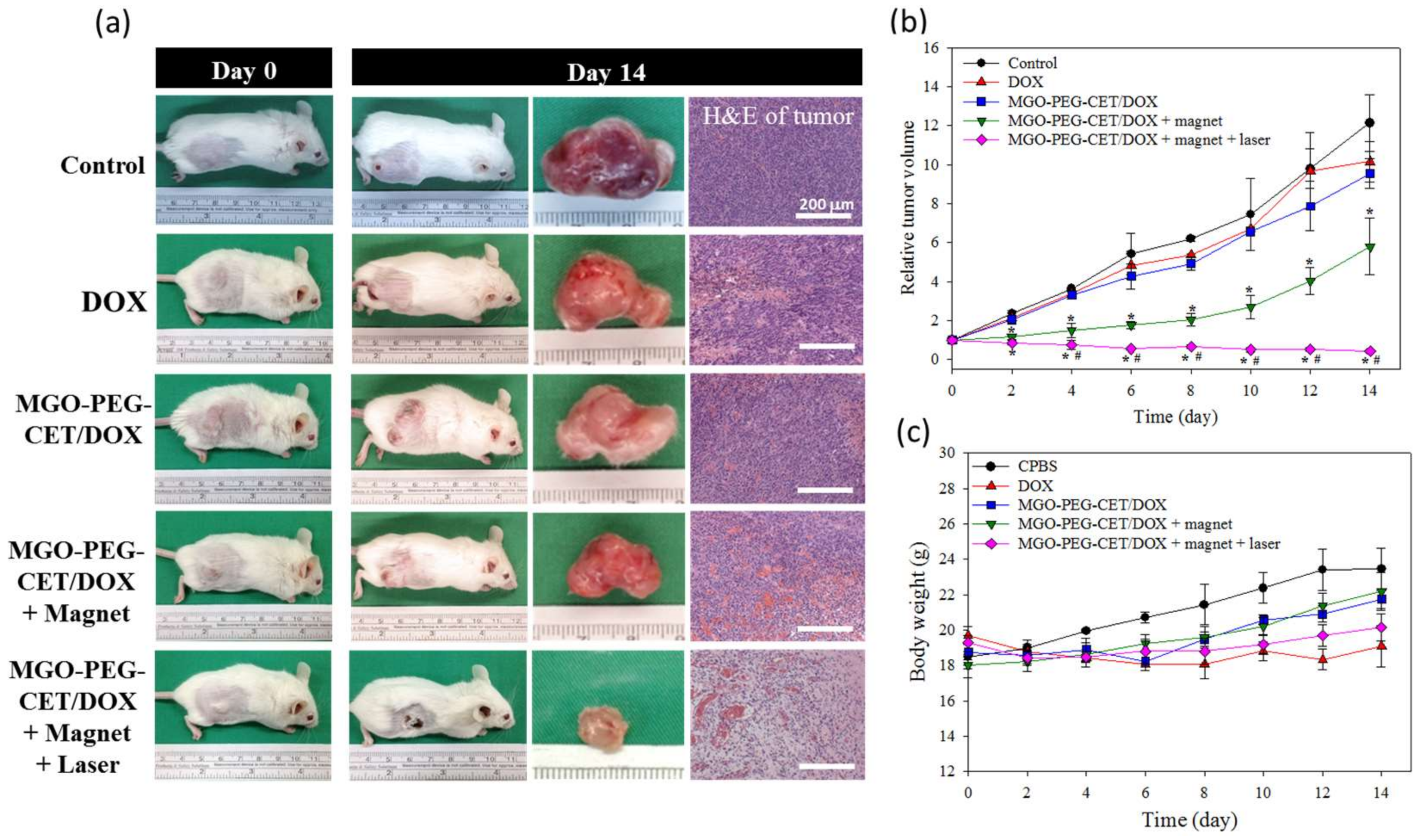
2.6. The Efficacy of Combined Therapy In Vitro and In Vivo
3. Materials and Methods
3.1. Materials
3.2. Preparation of Magnetic Graphene Oxide (MGO)
3.3. Preparation of MGO-PEG-CET and MGO-PEG-CET-QDs
3.4. Physico-Chemical Properties of Nanocarriers
3.5. Drug Loading and Release
3.6. Intracellular Uptake
3.7. Photothermla Effect
3.8. Magnetic Guidance In Vitro
3.9. Blood Compatibility Analysis
3.10. In Vitro Cytotoxicity
3.11. Mouse Subcutaneous Tumor Model
3.12. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Organization, W.H. Cancer. Available online: http://www.who.int/cancer/en/ (accessed on 19 October 2017).
- Feng, S.S.; Chien, S. Chemotherapeutic engineering: Application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem. Eng. Sci. 2003, 58, 4087–4114. [Google Scholar] [CrossRef]
- Skalickova, S.; Loffelmann, M.; Gargulak, M.; Kepinska, M.; Docekalova, M.; Uhlirova, D.; Stankova, M.; Fernandez, C.; Milnerowicz, H.; Ruttkay-Nedecky, B.; et al. Zinc-modified nanotransporter of doxorubicin for targeted prostate cancer delivery. Nanomaterials 2017, 7, 435. [Google Scholar] [CrossRef] [PubMed]
- Au, J.L.; Jang, S.H.; Zheng, J.; Chen, C.T.; Song, S.; Hu, L.; Wientjes, M.G. Determinants of drug delivery and transport to solid tumors. J. Controll. Release 2001, 74, 31–46. [Google Scholar] [CrossRef]
- Lammers, T.; Aime, S.; Hennink, W.E.; Storm, G.; Kiessling, F. Theranostic nanomedicine. Acc. Chem. Res. 2011, 44, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Quader, S.; Kataoka, K. Nanomaterial-enabled cancer therapy. Mol. Ther. 2017, 25, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.; Langer, R.; Jia, X. Nanostructured materials for applications in drug delivery and tissue engineering. J. Biomater. Sci. Polym. Ed. 2007, 18, 241–268. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Lu, Y.J.; Wei, K.C.; Ma, C.C.; Yang, S.Y.; Chen, J.P. Dual targeted delivery of doxorubicin to cancer cells using folate-conjugated magnetic multi-walled carbon nanotubes. Colloids Surf. B Biointerfaces 2012, 89, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McCallion, C.; Burthem, J.; Rees-Unwin, K.; Golovanov, A.; Pluen, A. Graphene in therapeutics delivery: Problems, solutions and future opportunities. Eur. J. Pharm. Biopharm. 2016, 104, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Leroueil, P.R.; Majoros, I.J.; Orr, B.G.; Baker, J.R.; Holl, M.M.B. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem. Biol. 2007, 14, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Montet, X.; Funovics, M.; Montet-Abou, K.; Weissleder, R.; Josephson, L. Multivalent effects of rgd peptides obtained by nanoparticle display. J. Med. Chem. 2006, 49, 6087–6093. [Google Scholar] [CrossRef] [PubMed]
- Mejias, R.; Perez-Yague, S.; Gutierrez, L.; Cabrera, L.I.; Spada, R.; Acedo, P.; Serna, C.J.; Lazaro, F.J.; Villanueva, A.; Morales, M.P.; et al. Dimercaptosuccinic acid-coated magnetite nanoparticles for magnetically guided in vivo delivery of interferon gamma for cancer immunotherapy. Biomaterials 2011, 32, 2938–2952. [Google Scholar] [CrossRef] [PubMed]
- Sanson, C.; Diou, O.; Thevenot, J.; Ibarboure, E.; Soum, A.; Brulet, A.; Miraux, S.; Thiaudiere, E.; Tan, S.; Brisson, A.; et al. Doxorubicin loaded magnetic polymersomes: Theranostic nanocarriers for mr imaging and magneto-chemotherapy. ACS Nano 2011, 5, 1122–1140. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Chen, J.P.; Wei, K.C.; Chen, J.Y.; Huang, C.W.; Liao, Z.X. Release of doxorubicin by a folate-grafted, chitosan-coated magnetic nanoparticle. Nanomaterials 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.G.; Papakonstantinou, P.; McMullan, M.; Chu, M.; Stamboulis, A.; Potenza, A.; Dhesi, S.S.; Marchetto, H. Catalyst-free efficient growth, orientation and biosensing properties of multilayer graphene nanoflake films with sharp edge planes. Adv. Funct. Mater. 2008, 18, 3506–3514. [Google Scholar] [CrossRef]
- Yang, X.Y.; Zhang, X.Y.; Ma, Y.F.; Huang, Y.; Wang, Y.S.; Chen, Y.S. Superparamagnetic graphene oxide-Fe3O4 nanoparticles hybrid for controlled targeted drug carriers. J. Mater. Chem. 2009, 19, 2710–2714. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano. Res. 2008, 1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Act. Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Jiang, T.; Lv, Y.; Wu, Y.; He, F.; Zhuo, R.X. Amphiphilic copolymers with pendent carboxyl groups for high-efficiency loading and controlled release of doxorubicin. Colloids Surf. B Biointerfaces 2015, 132, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Felber, A.E.; Dufresne, M.H.; Leroux, J.C. Ph-sensitive vesicles, polymeric micelles, and nanospheres prepared with polycarboxylates. Adv. Drug Deliv. Rev. 2012, 64, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Simoes, S.; Moreira, J.N.; Fonseca, C.; Duzgunes, N.; de Lima, M.C. On the formulation of ph-sensitive liposomes with long circulation times. Adv. Drug Deliv. Rev. 2004, 56, 947–965. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhang, H.; Huang, D.; Feng, S.; Fujita, M.; Gao, X.D. Chitosan-functionalized graphene oxide as a potential immunoadjuvant. Nanomaterials 2017, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Seabra, A.B.; Paula, A.J.; de Lima, R.; Alves, O.L.; Durán, N. Nanotoxicity of graphene and graphene oxide. Chem. Res. Toxicol. 2014, 27, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zong, C.; Shen, H.; Liu, M.; Chen, B.; Ren, B.; Zhang, Z. Mechanism of cellular uptake of graphene oxide studied by surface-enhanced raman spectroscopy. Small 2012, 8, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Wei, W.; Yue, Z.; Wang, B.; Luo, N.; Gao, Y.; Ma, D.; Ma, G.; Su, Z. The role of the lateral dimension of graphene oxide in the regulation of cellular responses. Biomaterials 2012, 33, 4013–4021. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wan, J.M.; Zhang, S.; Tian, B.; Zhang, Y.J.; Liu, Z. The influence of surface chemistry and size of nanoscale graphene oxide on photothermal therapy of cancer using ultra-low laser power. Biomaterials 2012, 33, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, Z.; Huang, D.; Liu, Z.; Guo, X.; Zhong, H. Synergistic effect of chemo-photothermal therapy using pegylated graphene oxide. Biomaterials 2011, 32, 8555–8561. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.X.; Tao, H.Q.; Yang, K.; Feng, L.Z.; Cheng, L.; Shi, X.Z.; Li, Y.G.; Guo, L.; Liu, Z. A functionalized graphene oxide-iron oxide nanocomposite for magnetically targeted drug delivery, photothermal therapy, and magnetic resonance imaging. Nano Res. 2012, 5, 199–212. [Google Scholar] [CrossRef]
- Huang, Y.S.; Lu, Y.J.; Chen, J.P. Magnetic graphene oxide as a carrier for targeted delivery of chemotherapy drugs in cancer therapy. J. Magn. Magn. Mater. 2017, 427, 34–40. [Google Scholar] [CrossRef]
- Park, J.W.; Mok, H.; Park, T.G. Epidermal growth factor (EGF) receptor targeted delivery of pegylated adenovirus. Biochem. Bioph. Res. Commun. 2008, 366, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle pegylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Höög, J.L.; Gluenz, E.; Vaughan, S.; Gull, K. Chapter 8—Ultrastructural investigation methods for trypanosoma brucei. In Methods in Cell Biology; Müller-Reichert, T., Ed.; Academic Press: Camgridge, MA, USA, 2010; Volume 96, pp. 175–196. [Google Scholar]
- Jokar, S.; Pourjavadi, A.; Adeli, M. Albumin-graphene oxide conjugates; carriers for anticancer drugs. RSC Adv. 2014, 4, 33001–33006. [Google Scholar] [CrossRef]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by epr effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.H.; Chou, M.Y.; Chu, I.M. Cetuximab-conjugated iron oxide nanoparticles for cancer imaging and therapy. Int. J. Nanomed. 2015, 10, 3663–3685. [Google Scholar]
- Mahmoud, W.E. Morphology and physical properties of poly(ethylene oxide) loaded graphene nanocomposites prepared by two different techniques. Eur. Polym. J. 2011, 47, 1534–1540. [Google Scholar] [CrossRef]
- Murugan, A.V.; Muraliganth, T.; Manthiram, A. Rapid, facile microwave-solvothermal synthesis of graphene nanosheets and their polyaniline nanocomposites for energy strorage. Chem. Mater. 2009, 21, 5004–5006. [Google Scholar] [CrossRef]
- Liang, R.P.; Liu, C.M.; Meng, X.Y.; Wang, J.W.; Qiu, J.D. A novel open-tubular capillary electrochromatography using beta-cyclodextrin functionalized graphene oxide-magnetic nanocomposites as tunable stationary phase. J. Chromatogr. A 2012, 1266, 95–102. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Gao, C. Supraparamagnetic, conductive, and processable multifunctional graphene nanosheets coated with high-density Fe3O4 nanoparticles. ACS Appl. Mater. Interfaces 2010, 2, 3201–3210. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Kolhe, P.; Kannan, R.M. Improvement in ductility of chitosan through blending and copolymerization with peg: Ftir investigation of molecular interactions. Biomacromolecules 2003, 4, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.F.; Shi, M.; Ma, H.W.; Yan, B.; Li, N.; Ye, M.X. Hydrothermal synthesis of magnetic reduced graphene oxide sheets. Mater. Res. Bull. 2011, 46, 2077–2083. [Google Scholar] [CrossRef]
- Xu, L.Q.; Wang, L.; Zhang, B.; Lim, C.H.; Chen, Y.; Neoh, K.G.; Kang, E.T.; Fu, G.D. Functionalization of reduced graphene oxide nanosheets via stacking interactions with the fluorescent and water-soluble perylene bisimide-containing polymers. Polymer 2011, 52, 2376–2383. [Google Scholar] [CrossRef]
- Ghosh, S.; Badruddoza, A.Z.M.; Hidajat, K.; Uddin, M.S. Adsorptive removal of emerging contaminants from water using superparamagnetic Fe3O4 nanoparticles bearing aminated beta-cyclodextrin. J. Environ. Chem. Eng. 2013, 1, 122–130. [Google Scholar] [CrossRef]
- Wang, C.; Feng, L.; Yang, H.; Xin, G.; Li, W.; Zheng, J.; Tian, W.; Li, X. Graphene oxide stabilized polyethylene glycol for heat storage. Phys. Chem. Chem. Phys. 2012, 14, 13233–13238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.P.; Yang, P.C.; Ma, Y.H.; Tu, S.J.; Lu, Y.J. Targeted delivery of tissue plasminogen activator by binding to silica-coated magnetic nanoparticle. Int. J. Nanomed. 2012, 7, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.L.; Chen, J.P. Preparation of thermosensitive magnetic liposome encapsulated recombinant tissue plasminogen activator for targeted thrombolysis. J. Magn. Magn. Mater. 2017, 427, 188–194. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, S.T.; Wang, Y.; Liu, Y.; Wang, H. Adsorption and desorption of doxorubicin on oxidized carbon nanotubes. Colloids Surf. B Biointerfaces 2012, 97, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Hayashi, H.; Iijima, M.; Nagasaki, Y. Endosomal release and intracellular delivery of anticancer drugs using ph-sensitive PEGylated nanogels. J. Mater. Chem. 2007, 17, 3720–3725. [Google Scholar] [CrossRef]
- Lee, M.; Jeong, J.; Kim, D. Intracellular uptake and ph-dependent release of doxorubicin from the self-assembled micelles based on amphiphilic polyaspartamide graft copolymers. Biomacromolecules 2015, 16, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.; Olin, H. Carbon nanomaterials as drug carriers: Real time drug release investigation. Mater. Sci. Eng. C Mater. Biol. Appl. 2012, 32, 1247–1252. [Google Scholar] [CrossRef]
- Cai, W.; Chen, K.; He, L.; Cao, Q.; Koong, A.; Chen, X. Quantitative PET of EGFR expression in xenograft-bearing mice using 64Cu-labeled cetuximab, a chimeric anti-egfr monoclonal antibody. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Su, G.; Li, L.; Gilbertson, B.O.; Yu, L.H.; Zhang, Q.; Sun, Y.P.; Yan, B. Size-dependent cell uptake of protein-coated graphene oxide nanosheets. ACS Appl. Mater. Interfaces 2012, 4, 2259–2266. [Google Scholar] [CrossRef] [PubMed]
- Chertok, B.; David, A.E.; Yang, V.C. Polyethyleneimine-modified iron oxide nanoparticles for brain tumor drug delivery using magnetic targeting and intra-carotid administration. Biomaterials 2010, 31, 6317–6324. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, S.; Zhang, G.; Sun, X.; Lee, S.-T.; Liu, Z. Graphene in mice: Ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano Lett. 2010, 10, 3318–3323. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Shao, Y.; Peng, J.; Dai, X.; Li, H.; Wu, Q.; Shi, D. Near-infrared laser light mediated cancer therapy by photothermal effect of Fe3O4 magnetic nanoparticles. Biomaterials 2013, 34, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Maiello, E.; Gebbia, V.; Manzione, L.; Giuliani, F.; Morelli, F.; Arcara, C.; Grimaldi, A.; Colucci, G. Clinical results of EGFR-targeted therapies in advancedcolorectal cancer. EJC Suppl. 2008, 6, 64–69. [Google Scholar] [CrossRef]
- Rivera, F.; Vega-Villegas, M.E.; Lopez-Brea, M.F. Cetuximab, its clinical use and future perspectives. Anticancer Drugs 2008, 19, 99–113. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, D.P.; Hirsch, L.R.; Halas, N.J.; Payne, J.D.; West, J.L. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004, 209, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kirui, D.K.; Khalidov, I.; Wang, Y.; Batt, C.A. Targeted near-IR hybrid magnetic nanoparticles for in vivo cancer therapy and imaging. Nanomedicine 2013, 9, 702–711. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Particle Size (nm) | Polydispersity Index | Zeta Potential (mV) |
|---|---|---|---|
| GO | 136.4 ± 5.7 | 0.18 ± 0.05 | −44.3 ± 2.8 |
| MGO | 205.5 ± 19.9 * | 0.28 ± 0.03 * | −35.1 ± 0.9 * |
| MGO-PEG-CET | 215.7 ± 18.4 * | 0.29 ± 0.03 * | −19.8 ± 0.4 *,# |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.-J.; Lin, P.-Y.; Huang, P.-H.; Kuo, C.-Y.; Shalumon, K.T.; Chen, M.-Y.; Chen, J.-P. Magnetic Graphene Oxide for Dual Targeted Delivery of Doxorubicin and Photothermal Therapy. Nanomaterials 2018, 8, 193. https://doi.org/10.3390/nano8040193
Lu Y-J, Lin P-Y, Huang P-H, Kuo C-Y, Shalumon KT, Chen M-Y, Chen J-P. Magnetic Graphene Oxide for Dual Targeted Delivery of Doxorubicin and Photothermal Therapy. Nanomaterials. 2018; 8(4):193. https://doi.org/10.3390/nano8040193
Chicago/Turabian StyleLu, Yu-Jen, Pin-Yi Lin, Pei-Han Huang, Chang-Yi Kuo, K.T. Shalumon, Mao-Yu Chen, and Jyh-Ping Chen. 2018. "Magnetic Graphene Oxide for Dual Targeted Delivery of Doxorubicin and Photothermal Therapy" Nanomaterials 8, no. 4: 193. https://doi.org/10.3390/nano8040193
APA StyleLu, Y. -J., Lin, P. -Y., Huang, P. -H., Kuo, C. -Y., Shalumon, K. T., Chen, M. -Y., & Chen, J. -P. (2018). Magnetic Graphene Oxide for Dual Targeted Delivery of Doxorubicin and Photothermal Therapy. Nanomaterials, 8(4), 193. https://doi.org/10.3390/nano8040193