Coating Dependent In Vitro Biocompatibility of New Fe-Si Nanoparticles

 ,
, 


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Set-Up for Laser Pyrolysis
2.2. Synthesis and Coating of Hybrid Fe-Si Nanoparticles
2.3. Characterization of Hybrid Fe-Si Nanoparticles and Resulted Suspensions
2.4. Cell Line Culture and Treatment
2.5. Cell Viability and IC50 Evaluation
2.6. Cell Morphology and F-Actin Cytoskeleton Imaging
2.7. Lactate Dehydrogenase (LDH) Assay
2.8. Measurement of ROS Production
2.9. Protein Extraction
2.10. Quantification of GSH Content
2.11. Immunoblotting of Nuclear Factor E2-Related Factor 2 (Nrf-2)
2.12. Statistical Analysis
3. Results and Discussion
3.1. Hybrid Fe-Si Nanoparticles Preparation and Characterization
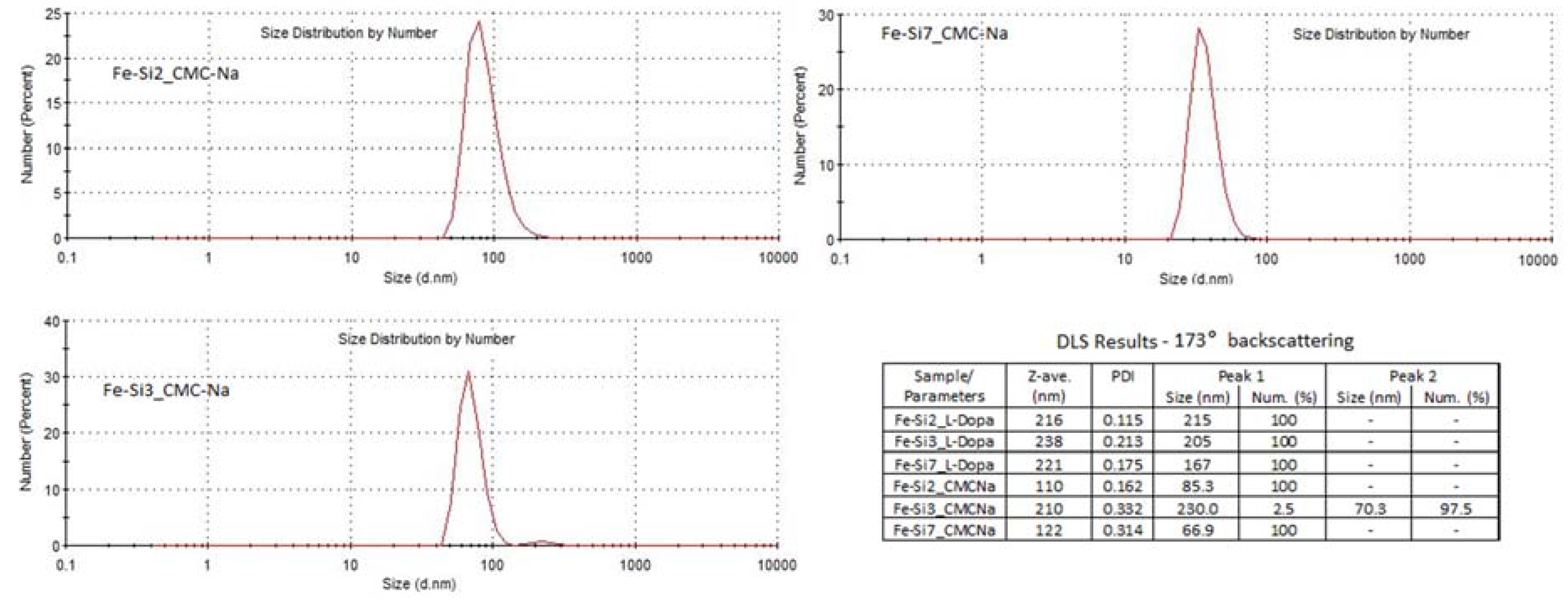
3.2. Coating of Hybrid Fe-Si NPs with l-DOPA or CMC-Na
3.3. Biocompatibility Assessment
3.3.1. Cytotoxicity
3.3.2. Cell Morphology and Dispersion of NPs in Culture
3.3.3. Oxidative Stress
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hosseini, F.; Panahifar, A.; Adeli, M.; Amiri, H.; Lascialfari, A.; Orsini, F.; Doschak, M.R.; Mahmoudi, M. Synthesis of pseudopolyrotaxanes-coated Superparamagnetic Iron Oxide Nanoparticles as new MRI contrast agent. Coll. Surf. B Biointerfaces 2013, 103, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, L.; Wang, C.; Ma, X.; Li, Y.; Liu, Z. Polymer encapsulated upconversion nanoparticle/iron oxide nanocomposites for multimodal imaging and magnetic targeted drugdelivery. Biomaterials 2011, 32, 9364–9373. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, L.; Gomaa, H.G.; Ragab, D.; Zhu, J. Magnetic nanoparticles for environmental and biomedical applications: A review. Particuology 2017, 30, 1–14. [Google Scholar] [CrossRef]
- Franke, K.; Kettering, M.; Lange, K.; Kaiser, W.; Hilger, I. The exposure of cancer cells to hyperthermia, iron oxide nanoparticles, and mitomycin C influences membrane multidrug resistance protein expression levels. Int. J. Nanomed. 2013, 8, 351–363. [Google Scholar]
- Zhuonan, L.; Huihui, Y.; Hao, Z.; Chuanjun, H.; Laifeng, L. Oil-field wastewater purification by magnetic separation technique using a novel magnetic nanoparticle. Cryogenics 2012, 52, 699–703. [Google Scholar]
- Taylor, R.; Coulombe, S.; Otanicar, T.; Phelan, P.; Gunawan, A.; Lv, W.; Rosengarten, G.; Prasher, R.; Tyagi, H. Small particles, big impacts: A review of the diverse applications of nanofluids. J. Appl. Phys. 2013, 113, 011301. [Google Scholar] [CrossRef]
- Peyghambarzadeh, S.M.; Hashemabadi, S.H.; Hoseini, S.M.; Seifi, J. Experimental study of heat transfer enhancement using water/ethylene glycol based nanofluids as a new coolant for car radiators. Int. Commun. Heat Mass Transf. 2011, 38, 1283–1290. [Google Scholar] [CrossRef]
- Liu, T.; Chen, X.; Di, Z.; Zhang, J.; Li, X.; Chen, J. Tunable magneto-optical wavelength filter of long-period fiber grating with magnetic fluids. Appl. Phys. Lett. 2007, 91, 121116. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Samiei, M.; Davaran, S. Magnetic nanoparticles: Preparation, physical properties, and applications in biomedicine. Nanoscale Res. Lett. 2012, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- El-Dakdouki, M.H.; El-Boubbou, K.; Xia, J.; Kavunja, H.; Huang, X. Methods for Magnetic Nanoparticle Synthesis and Functionalization. In Chemistry of Bioconjugates; Narain, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 281–314. ISBN 9781118775882. [Google Scholar]
- Majidi, S.; Sehrig, F.Z.; Farkhani, S.M.; Goloujeh, M.S.; Akbarzadeh, A. Current methods for synthesis of magnetic nanoparticles. Artif. Cell Nanomed. Biotechnol. 2014, 44, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Kudr, J.; Haddad, Y.; Richtera, L.; Heger, Z.; Cernak, M.; Adam, V.; Zitka, O. Magnetic Nanoparticles: From Design and Synthesis to Real World Applications. Nanomaterials 2017, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Mosayebi, J.; Kiyasatfar, M.; Laurent, S. Synthesis, Functionalization, and Design of Magnetic Nanoparticles for Theranostic Applications. Adv. Healthc. Mater. 2017, 6, 1700306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Layek, S.; Pandey, B.; Verma, H.C. Magnetic structure of Fe-Fe oxide nanoparticles made by electrodeposition. Int. J. Eng. Sci. Technol. 2010, 2, 66–72. [Google Scholar]
- Hadjipanayis, C.G.; Bonder, M.J.; Balakrishnan, S.; Wang, X.; Mao, H.; Hadjipanayis, G.C. Metallic Iron Nanoparticles for MRI Contrast Enhancement and Local Hyperthermia. Small 2008, 4, 1925–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvell, J.; Ayieta, E.; Gavrin, A.; Cheng, R.; Shah, V.R.; Sokol, P. Magnetic properties of iron nanoparticle. J. Appl. Phys. 2010, 107, 103913. [Google Scholar] [CrossRef]
- Bychkova, A.V.; Sorokina, O.N.; Rosenfeld, M.A.; Kovarski, A.L. Multifunctional biocompatible coatings on magnetic nanoparticles. Russ. Chem. Rev. 2012, 81, 1026–1050. [Google Scholar] [CrossRef]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R.N. Magnetic Iron Oxide Nanoparticles: Synthesis, Stabilization, Vectorization, Physicochemical Characterizations, and Biological Applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Demirer, G.S.; Okur, A.C.; Kizilel, S. Synthesis and design of biologically inspired biocompatible iron oxide nanoparticles for biomedical applications. J. Mater. Chem. B 2015, 3, 7831–7849. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Foy, S.P.; Jain, T.K.; Labhasetwar, V. PEG-Functionalized Magnetic Nanoparticles for Drug Delivery and Magnetic Resonance Imaging Applications. Pharm. Res. 2010, 27, 2283–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illés, E.; Tombácz, E.; Szekeres, M.; Tóth, I.Y.; Szabó, Á.; Iván, B. Novel carboxylated PEG-coating on magnetite nanoparticles designed for biomedical applications. J. Magn. Magn. Mater. 2015, 380, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.L.; Qi, X.R.; Maitani, Y.; Nagai, T. Preparation and characterization of superparamagnetic iron oxide nanoparticles stabilized by alginate. Int. J. Pharm. 2007, 333, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Uthaman, S.; Lee, S.J.; Cherukula, K.; Cho, C.S.; Park, I.K. Polysaccharide-Coated Magnetic Nanoparticles for Imaging and Gene Therapy. Biomed. Res. Int. 2015, 2015, 959175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ma, J.; Jia, N.; Zhao, Y.; Shen, H. Chitosan-coated magnetic nanoparticles as carriers of 5-fluorouracil: Preparation, characterization and cytotoxicity studies. Colloid Surf. B 2009, 68, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.J.; Cheng, T.H.; Chen, C.Y.; Hsu, S.C.; Cheng, T.L.; Liu, G.C.; Wang, Y.M. Targeted Herceptin-dextran iron oxide nanoparticles for noninvasive imaging of HER2/neu receptors using MRI. J. Biol. Inorg. Chem. 2009, 14, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jung, M.J.; Hwang, Y.H.; Lee, Y.J.; Lee, S.; Lee, D.Y.; Shin, H. Heparin-coated superparamagnetic iron oxide for in vivo MR imaging of human MSCs. Biomaterials 2012, 33, 4861–4871. [Google Scholar] [CrossRef] [PubMed]
- Dumitrache, F.; Morjan, I.; Fleaca, C.; Badoi, A.; Manda, G.; Pop, S.; Marta, D.S.; Huminic, G.; Huminic, A.; Vekas, L.; et al. Highly magnetic Fe2O3 nanoparticles synthesized by laser pyrolysis used for biological and heat transfer applications. Appl. Surf. Sci. 2015, 336, 297–303. [Google Scholar] [CrossRef]
- Comănescu, M.V.; Mocanu, M.A.; Anghelache, L.; Marinescu, B.; Dumitrache, F.; Bădoi, A.D.; Manda, G. Toxicity of l-DOPA coated iron oxide nanoparticles in intraperitoneal delivery setting-preliminary preclinical study. Rom. J. Morphol. Embryol. 2015, 56, 691–696. [Google Scholar] [PubMed]
- Costo, R.; Morales, M.P.; Veintemillas-Verdaguer, S. Improving magnetic properties of ultrasmall magnetic nanoparticles by biocompatible coatings. J. Appl. Phys. 2015, 117, 064311. [Google Scholar] [CrossRef] [Green Version]
- Huminic, A.; Huminic, G.; Fleaca, C.; Dumitrache, F.; Morjan, I. Thermal conductivity, viscosity and surface tension of nanofluids based on FeC nanoparticles. Powder Technol. 2015, 284, 78–84. [Google Scholar] [CrossRef]
- Doll, T.A.P.F.; Raman, S.; Dey, R.; Burkhard, P. Nanoscale assemblies and their biomedical applications. J. R. Soc. Interface 2013, 10, 20120740. [Google Scholar] [CrossRef] [PubMed]
- Pinto Reis, C.; Neufeld, R.J.; Ribeiro, A.J.; Veiga, F.; Nanoencapsulation, I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2006, 2, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar]
- Chavanpatil, M.D.; Khdair, A.; Patil, Y.; Handa, H.; Mao, G.; Panyam, J. Polymer- surfactant nanoparticles for sustained release of water-soluble drugs. J. Pharm. Sci. 2007, 96, 3379–3389. [Google Scholar] [CrossRef] [PubMed]
- Dauda, K.; Busari, Z.; Morenikeji, O.; Afolayan, F.; Oyeyemi, O.; Meena, J.; Sahu, D.; Panda, A. Poly(d,l-lactic-co-glycolic acid)-based artesunate nanoparticles: Formulation, antimalarial and toxicity assessments. J. Zhejiang Univ.-Sci. B 2017, 18, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Charyulu, R.N. Prolonged drug delivery system of an antifungal drug by association with polyamidoamine dendrimers. Int. J. Pharm. Investig. 2016, 6, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Naha, P.C.; Mukherjee, S.P.; Byrne, H.J. Toxicology of engineered nanoparticles: Focus on poly(amidoamine) dendrimers. Int. J. Environ. Res. Public Health 2018, 15, 338. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cheng, K.; Hu, X.; Ma, X.; Zhang, R.; Yang, M.; Lu, X.; Xing, L.; Huang, W.; Gambhir, S.S.; et al. Transferring biomarker into molecular probe: Melanin nanoparticle as a naturally active platform for multimodality imaging. J. Am. Chem. Soc. 2014, 136, 15185–15194. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, D.; Zhao, T.; Wei, X.; McMahon, S.; O’Keeffe Ahern, J.; Wang, W.; Greiser, U.; Rodriguez, B.J.; Wang, W. Intramolecular cyclization dominating homopolymerization of multivinyl monomers toward single-chain cyclized/knotted polymeric nanoparticles. Macromolecules 2015, 48, 6882–6889. [Google Scholar] [CrossRef]
- Cutlar, L.; Gao, Y.; Aied, A.; Greiser, U.; Murauer, E.M.; Zhou, D.; Wang, W. A knot polymer mediated non-viral gene transfection for skin cells. Biomater. Sci. 2016, 4, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-Y.; Gao, Y.; Cutlar, L.; O’Keeffe-Ahern, J.; Zhao, T.; Lin, F.-H.; Zhou, D.; McMahon, S.; Greiser, U.; Wang, W.; et al. Tailoring highly branched poly(β-amino ester)s: A synthetic platform for epidermal gene therapy. Chem. Commun. 2015, 5, 8473–8476. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Huang, J.-Y.; O’Keeffe Ahern, J.; Cutlar, L.; Zhou, D.G.; Lin, F.-H.; Wang, W. Highly branched poly(β-amino esters) for non-viral gene delivery: High transfection efficiency and low toxicity achieved by increasing molecular weight. Biomacromolecules 2016, 17, 3640–3647. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Gao, Y.; Aied, A.; Cutlar, L.; Igoucheva, O.; Newland, B.; Alexeeve, V.; Greiser, U.; Uitto, J.; Wang, W. Highly branched poly(β-amino ester)s for skin gene therapy. J. Control. Release 2016, 244, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Ertel, S.I.; Ratner, B.D.; Kaul, A.; Schway, M.B.; Horbett, T.A. In vitro study of the intrinsic toxicity of synthetic surfaces to cells. J. Biomed. Mater. Res. 1994, 28, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Morais, J.M.; Papadimitrakopoulos, F.; Burgess, D.J. Biomaterials/tissue interactions: Possible solutions to overcome foreign body response. AAPS J. 2010, 12, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Santerre, J.P.; Woodhouse, K.; Laroche, G.; Labow, R.S. Understanding the biodegradation of polyurethanes: From classical implants to tissue engineering materials. Biomaterials 2005, 26, 7457–7470. [Google Scholar] [CrossRef] [PubMed]
- Gatoo, M.A.; Naseem, S.; Arfat, M.Y.; Dar, A.M.; Qasim, K.; Zubair, S. Physicochemical Properties of Nanomaterials: Implication in Associated Toxic Manifestations. BioMed Res. Int. 2014, 2014, 498420. [Google Scholar] [CrossRef] [PubMed]
- Septiadi, D.; Crippa, F.; Moore, T.L.; Rothen-Rutishauser, B.; Petri-Fink, A. Nanoparticle-Cell Interaction: A Cell Mechanics Perspective. Adv. Mater. 2018, 30, 1704463. [Google Scholar] [CrossRef] [PubMed]
- Foglia, S.; Ledda, M.; Fioretti, D.; Iucci, G.; Papi, M.; Capellini, G.; Lisi, A. In vitro biocompatibility study of sub-5 nm silica-coated magnetic iron oxide fluorescent nanoparticles for potential biomedical application. Sci. Rep. 2017, 7, 46513. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Luccioni, H.L.; Latorre-Esteves, M.; Méndez-Vega, J.; Soto, O.; Rodríguez, A.R.; Rinaldi, C.; Torres-Lugo, M. Enhanced reduction in cell viability by hyperthermia induced by magnetic nanoparticles. Int. J. Nanomed. 2011, 6, 373–380. [Google Scholar]
- Lee, J.S.; Rodríguez-Luccioni, H.L.; Méndez, J.; Sood, A.K.; Lpez-Berestein, G.; Rinaldi, C.; Torres-Lugo, M. Hyperthermia induced by magnetic nanoparticles improves the effectiveness of the anticancer drug cis-diamminedichloroplatinum. J. Nanosci. Nanotechnol. 2011, 11, 4153–4157. [Google Scholar] [CrossRef] [PubMed]
- Moersdorf, D.; Hugounenq, P.; Phuoc, L.; Mamlouk-Chaouachi, H.; Felder-Flesch, D.; Begin-Colin, S.; Pourroy, G.; Bernhardt, I. Influence of magnetic iron oxide nanoparticles on red blood cells and Caco-2 cells. Adv. Biosci. Biotechnol. 2010, 1, 439–443. [Google Scholar] [CrossRef]
- Bannunah, A.M.; Vllasaliu, D.; Lord, J.; Stolnik, S. Mechanisms of Nanoparticle Internalization and Transport Across an Intestinal Epithelial Cell Model: Effect of Size and Surface Charge. Mol. Pharm. 2014, 11, 4363–4373. [Google Scholar] [CrossRef] [PubMed]
- Popovici, E.; Dumitrache, F.; Morjan, I.; Alexandrescu, R.; Ciupina, V.; Prodan, G.; Vekas, L.; Bica, D.; Marinica, O.; Vasile, E. Iron/iron oxides core-shell nanoparticles by laser pyrolysis: Structural characterization and enhanced particle dispersion. Appl. Surf. Sci. 2007, 254, 1048–1052. [Google Scholar] [CrossRef]
- Lacour, F.; Guillois, O.; Portier, X.; Perez, H.; HerlinBoime, N.; Reynaud, C. Laser Pyrolysis synthesis and characterization of luminescent silicon nanocrystals. Physica E 2007, 38, 11–15. [Google Scholar] [CrossRef]
- Wegner, K.; Pratsinis, S.E. Scale-up of nanoparticle synthesis in difusion fame reactors. Chem. Eng. Sci. 2003, 58, 4581–4589. [Google Scholar] [CrossRef]
- Veintemillas-Verdaguer, S.; Morales, M.P.; Serna, C.J. Effect of the oxidation conditions on the maghemites produced by laser pyrolysis. Appl. Organomet. Chem. 2001, 15, 365–372. [Google Scholar] [CrossRef]
- Kang, T.; Guan, R.; Chen, X.; Song, Y.; Jiang, H.; Zhao, J. In vitro toxicity of different-sized ZnO nanoparticles in Caco-2 cells. Nanoscale Res. Lett. 2013, 8, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Gavrila-Florescu, L.; Dumitrache, F.; Balas, M.; Fleaca, C.T.; Scarisoreanu, M.; Morjan, I.P.; Dutu, E.; Ilie, A.; Banici, A.M.; Locovei, C.; et al. Synthesis of Fe based core@ZnO shell nanopowder by laser pyrolysis for biomedical applications. Appl. Phys. A 2017, 123, 802. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of proteindye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Panariti, A.; Lettiero, B.; Alexandrescu, R.; Collini, M.; Sironi, L.; Chanana, M.; Morjan, I.; Wang, D.; Chirico, G.; Mieserrochi, G.; et al. Dynamic invesitgations of interaction of biocompatible iron oxide nanoparticles with epithelial cells for biomedical applications. J. Biomed. Nanotechnol. 2013, 9, 1–13. [Google Scholar] [CrossRef]
- Hong, P.; Xiao, Z.; Kaixun, H.; Huibi, X. Modification of Fe3O4 magnetic nanoparticles by l-DOPA or Dopamine as an enzyme support. J. Wuhan Univ. Technol. 2008, 23, 480–485. [Google Scholar]
- Azari, S.; Zou, L.; Cornelissen, E. Assessing the effect of surface modification of polyamide RO membrane by l-DOPA on the short range physiochemical interactions with biopolymer fouling on the membrane. Colloid Surf. B 2014, 120, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Yang, W.; Ju, Y.; Xin, C.; Wang, Y.; Deng, Y.; Mahmood, N.; Hou, Y. Biocompatibility of iron carbide and detection of metals ions signaling proteomic analysis via HPLC/ESI-Orbitrap. Nano Res. 2017, 10, 1912–1923. [Google Scholar] [CrossRef]
- Park, J.H.; Gu, L.; von Maltzahn, G.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Biodegradable Luminescent Porous Silicon Nanoparticles for In Vivo Applications. Nat. Mater. 2009, 8, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Ruizendaal, L.; Bhattacharjee, S.; Pournazari, K.; Rosso-Vasic, M.; de Haan, L.H.J.; Alink, G.M.; Marcelis, A.T.M.; Zuilhof, H. Synthesis and Cytotoxicity of Silicon Nanoparticles with Covalently Attached Organic Monolayers. Nanotoxicology 2009, 3, 339–347. [Google Scholar] [CrossRef]
- Luna-Martı´nez, J.F.; Reyes-Melo, E.; Gonzalez-Gonzalez, V.; Guerrero-Salazar, C.; Torres-Castro, A.; Sepulveda-Guzman, S. Synthesis and Characterization of a Magnetic Hybrid Material. Consisting of Iron Oxide in a Carboxymethyl Cellulose Matrix. J. Appl. Polym. Sci. 2012. [Google Scholar] [CrossRef]
- Sobik, M.; Pondman, K.M.; Erné, B.; Kuipers, B.; ten Haken, B.; Rogalla, H. Magnetic Nanoparticles for Diagnosis and Medical Therapy. In Carbon Nanotubes for Biomedical Applications. Carbon Nanostructures; Klingeler, R., Sim, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 85–95. ISBN 978-3-642-14802-6. [Google Scholar]
- Fleaca, C.T.; Dumitrache, F.; Morjan, I.; Niculescu, A.M.; Sandu, I.; Iliea, A.; Stamatin, I.; Iordache, A.; Vasile, E.; Prodan, G. Synthesis and characterization of polyaniline_Fe@C magnetic nanocomposite powder. Appl. Surf. Sci. 2015, 374, 213–221. [Google Scholar] [CrossRef]
- Suo, B.; Li, H.; Wang, Y.; Li, Z.; Pan, Z.; Ai, Z. Effects of ZnO nanoparticle-coated packaging film on pork meat quality during cold storage. J. Sci. Food Agric. 2017, 97, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.; Naik, T.; Paria, S. Organization of SiO2 and TiO2 Nanoparticles into Fractal Patterns on Glass Surface for the Generation of Superhydrophilicity. J. Phys. Chem. C 2017, 121, 2428–2436. [Google Scholar] [CrossRef]
- El-Hag Ali, A.; Abd El-Rehim, A.; Kamal, H.; Hegazy, D.; El-Sayed, A. Synthesis of carboxymethyl cellulose based drug carrier hydrogel using ionizing radiation for possible use as site specific delivery system. J. Macromol. Sci. Part A Pure Appl. Chem. 2008, 45, 628–634. [Google Scholar] [CrossRef]
- Basma, A.N.; Morris, E.J.; Nicklas, W.J.; Geller, H.M. l-DOPA cytotoxicity to PC12 cells in culture is via its autoxidation. J. Neurochem. 1995, 64, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Melamed, E.; Offen, D.; Shirvan, A.; Djaldetti, R.; Barzilai, A.; Ziv, I. Levodopa toxicity and apoptosis. Ann. Neurol. 1998, 44, S149–S154. [Google Scholar] [CrossRef] [PubMed]
- Lipski, J.; Nistico, R.; Berretta, N.; Guatteo, E.; Bernardi, G.; Mercuri, N.B. l-DOPA: A scapegoat for accelerated neurodegeneration in Parkinson’s disease? Prog. Neurobiol. 2011, 94, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.Y.; Li, Y.B.; Zhang, L.; Wang, X.J. Preparation and characterization of a novel composite containing carboxymethyl cellulose used for bone repair. Mater. Sci. Eng. C 2009, 29, 193–198. [Google Scholar] [CrossRef]
- Lee, S.Y.; Bang, S.; Kim, S.; Jo, S.Y.; Kim, B.C.; Hwang, Y.; Noh, I. Synthesis and in vitro characterizations of porous carboxymethyl cellulose-poly(ethylene oxide) hydrogel film. Biomater. Res. 2015, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Zanganeh, S.; Spitler, R.; Erfanzadeh, M.; Alkilany, A.M.; Mahmoudi, M. Protein corona: Opportunities and challenges. Int. J. Biochem. Cell Biol. 2016, 75, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonagh, B.H.; Singh, G.; Hak, S.; Bandyopadhyay, S.; Augestad, I.L.; Peddis, D.; Sandvig, I.; Sandvig, A.; Glomm, W.R. l-DOPA-Coated Manganese Oxide Nanoparticles as Dual MRI Contrast Agents and Drug-Delivery Vehicles. Small 2016, 12, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakulkhu, U.; Mahmoudi, M.; Maurizi, L.; Salaklang, J.; Hofmann, H. Protein Corona Composition of Superparamagnetic Iron Oxide Nanoparticles with Various Physico-Chemical Properties and Coatings. Sci. Rep. 2014, 4, 5020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Le Thanh, T.; Gong, J.; Kim, J.-H.; Kim, E.-J.; Chang, Y.-S. Carboxymethyl cellulose coating decreases toxicity and oxidizing capacity of nanoscale zerovalent iron. Chemosphere 2014, 104, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive Oxygen Species in Normal and Tumor Stem Cells. Adv. Cancer. Res. 2014, 122, 1–67. [Google Scholar] [PubMed] [Green Version]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in Cancer Cell Death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saddick, S.; Afifi, M.; Abu Zinada, O.A. Effect of Zinc nanoparticles on oxidative stress-related genes and antioxidant enzymes activity in the brain of Oreochromis niloticus and Tilapia zillii. Saudi J. Biol. Sci. 2015, 24, 1672–1678. [Google Scholar] [CrossRef]
- Hao, L.H.; Wang, Z.Y.; Xing, B.S. Effect of sub-acute exposure to TiO2 nanoparticles on oxidative stress and histopathological changes in Juvenile Carp (Cyprinus carpio). J. Environ. Sci. 2009, 21, 1459–1466. [Google Scholar] [CrossRef]
- Manke, A.; Wang, L.; Rojanasakul, Y. Mechanisms of Nanoparticle-Induced Oxidative Stress and Toxicity. BioMed Res. Int. 2013, 2013, 942916. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Patil, U.S.; Adireddy, S.; Jaiswal, A.; Mandava, S.; Lee, B.R.; Chrisey, D.B. In Vitro/In Vivo Toxicity Evaluation and Quantification of Iron Oxide Nanoparticles. Int. J. Mol. Sci. 2015, 16, 24417–24450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdolenova, Z.; Drlickova, M.; Henjum, K.; Runden-Pran, E.; Tulinska, J.; Bilanicova, D.; Pojana, G.; Kazimirova, A.; Barancokova, M.; Kuricova, M.; et al. Coating-dependent induction of cytotoxicity and genotoxicity of iron oxide nanoparticles. Nanotoxicology 2015, 9, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Malvindi, M.A.; de Matteis, V.; Galeone, A.; Brunetti, V.; Anyfantis, G.C.; Athanassiou, A.; Cingolani, R.; Pompa, P.P. Toxicity assessment of silica coated iron oxide nanoparticles and biocompatibility improvement by surface engineering. PLoS ONE 2014, 9, e85835. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Lagarde, F.; Maraloiu, V.A.; Blanchin, M.G.; Gendron, F.; Wilhelm, C.; Gazeau, F. Degradability of superparamagnetic nanoparticles in a model of intracellular environment: Follow-up of magnetic, structural and chemical properties. Nanotechnology 2010, 21, 395103. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Erogbogbo, F.; Yong, K.-T.; Ye, L.; Liu, J.; Hu, R.; Chen, H.; Hu, Y.; Yang, Y.; Yang, J.; et al. Assessing Clinical Prospects of Silicon Quantum Dots: Studies in Mice and Monkeys. ACS Nano 2013, 7, 7303–7310. [Google Scholar] [CrossRef] [PubMed]
- Erogbogo, F.; Yong, K.-T.; Roy, I.; Hu, R.; Law, W.-C.; Zhao, W.; Ding, H.; Wu, F.; Kumar, R.; Swihardt, M.; et al. In Vivo Targeted Cancer Imaging Sentinel Limph Node Mapping and Multi-Channel Imaging with Biocompatible Silicon Nanocrystals. ACS Nano 2011, 1, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Erogbogo, F.; Yong, K.-T.; Hu, R.; Law, W.-C.; Ding, H.; Chang, C.-W.; Prasad, P.N.; Swihardt, M. Biocompatibel Magnetofluorescent Probes: Silicon Quantum Dots Coupled with Superparamagnetic Iron (III) Oxide. ACS Nano 2010, 9, 5131–5138. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample/Parameters | Exhaust Geometry | D1 Flow | D2 Flow | Laser Power | Flame Temp. (°C) | |
|---|---|---|---|---|---|---|
| DC2H4/Fe(CO)5 (sccm) | DSiH4 (sccm) | DAr (sccm) | PL/Ar/PL/abs. (W) | |||
| Fe-Si2 | normal | 60 | 20 | 50 | 115/105 | 660 |
| Fe-Si3 | normal | 60 | 10 | 40 | 115/108 | 620 |
| Fe-Si7 | narrow | 60 | 5 | 55 | 115/110 | 540 |
| Element/Sample | C (atom %) | O (atom %) | Si (atom %) | Fe (atom %) |
|---|---|---|---|---|
| Fe-Si2 | 11.1 | 23.8 | 34.3 | 30.8 |
| Fe-Si3 | 13.4 | 33.1 | 26.7 | 26.8 |
| Fe-Si7 | 13.3 | 26.1 | 19.9 | 40.7 |
| Sample | Ms (emu/g) | Mr (emu/g) | Hc (kA/m) | µmax (emu/Oe) |
|---|---|---|---|---|
| Fe-Si2 | 21 | 2.85 | 5.37 | 5.22 × 10−4 |
| Fe-Si3 | 23 | 3.81 | 8.84 | 7.38 × 10−4 |
| Fe-Si7 | 47 | 10.53 | 12.88 | 7.87 × 10−4 |
| NP Sample | IC50 (µg/mL) |
|---|---|
| Fe-Si2 | 382.18 ± 10.06 |
| Fe-Si3 | 383.08 ± 11.86 |
| Fe-Si7 | 904 ± 15.02 |
| l-DOPA | 81.2 ± 4.81 |
| CMC-Na | 1339 ± 14.27 |
| Fe-Si2_l-DOPA | 355.86 ± 17.23 |
| Fe-Si3_l-DOPA | 247.73 ± 14.56 |
| Fe-Si7_l-DOPA | 682.37 ± 14.07 |
| Fe-Si2_CMC-Na | 219.10 ± 11.83 |
| Fe-Si3_CMC-Na | 153.48 ± 8.14 |
| Fe-Si7_CMC-Na | 124.10 ± 7.10 |
| Sample | Dose (µg/mL) | ROS Production (RFU) | |||
|---|---|---|---|---|---|
| 1 h | 2 h | 3 h | 4 h | ||
| Control | 0 | 14.26 ± 0.98 | 18.46 ± 3.45 | 20.67 ± 1.23 | 22.15 ± 1.34 |
| Fe-Si2 | 25 | 35.45 ± 3.2 ** | 63.19 ± 5.36 ** | 78.32 ± 6.49 ** | 82.37 ± 6.96 ** |
| 50 | 37.63 ± 2.06 ** | 67.93 ± 4.92 ** | 84.08 ± 5.5 ** | 88.79 ± 5.7 ** | |
| Fe-Si3 | 25 | 39.59 ± 3.42 ** | 70.43 ± 6.14 ** | 88.83 ± 6.38 ** | 92.41 ± 7.63 ** |
| 50 | 45.87 ± 10.6 * | 81.56 ± 17.35 * | 101.3 ± 20.49 * | 106.46 ± 22.26 * | |
| Fe-Si7 | 25 | 47.58 ± 5.76 ** | 80.44 ± 9.76 ** | 93.68 ± 11.33 ** | 101.24 ± 12.00 ** |
| 50 | 56.02 ± 8.8 * | 91.74 ± 12.63 * | 104.05 ± 12.82 ** | 114.15 ± 14.82 ** | |
| L-DOPA | 15 | 11.83 ± 0.48 | 16.93 ± 0.78 | 18.73 ± 1.13 | 20.32 ± 1.04 |
| 30 | 11.65 ± 0.62 | 16.63 ± 1.62 | 18.3 ± 1.99 | 19.91 ± 2.02 | |
| CMC-Na | 15 | 13.04 ± 0.59 | 19.36 ± 1.06 | 23.02 ± 1.74 | 24.43 ± 1.78 |
| 30 | 11.28 ± 0.62 | 16.85 ± 0.77 | 19.73 ± 0.86 | 21.1 ± 1.00 | |
| Fe-Si2_L-DOPA | 25 | 18.39 ± 1.85 * | 25.76 ± 2.35 * | 29.84 ± 2.99 * | 33.03 ± 3.26 * |
| 50 | 13.28 ± 1.08 | 18.23 ± 1.73 | 21.15 ± 1.89 | 23.79 ± 1.98 | |
| Fe-Si3_L-DOPA | 25 | 19.22 ± 0.42 | 27.64 ± 0.99 * | 32.91 ± 1.67 ** | 36.36 ± 2.20 ** |
| 50 | 14.81 ± 2.62 | 20.19 ± 3.92 | 22.86 ± 4.26 | 25.28 ± 4.85 | |
| Fe-Si7_L-DOPA | 25 | 19.23 ± 2.47 | 26.52 ± 3.26 | 31.17 ± 4.02 | 35.02 ± 4.70 |
| 50 | 15.11 ± 1.24 | 21.23 ± 1.94 | 24.42 ± 2.23 | 27.07 ± 2.56 | |
| Fe-Si2_CMC-Na | 25 | 16.56 ± 1.50 | 23.78 ± 2.11* | 28.69 ± 2.39* | 32.34 ± 2.45 ** |
| 50 | 15.53 ± 1.69 | 22.71 ± 2.57 | 28.36 ± 3.24* | 32.68 ± 3.74 * | |
| Fe-Si3_CMC-Na | 25 | 17.51 ± 1.02 * | 25.5 ± 1.48 * | 31.4 ± 1.82 ** | 35.9 ± 2.04 ** |
| 50 | 19.71 ± 0.60 * | 29.52 ± 0.95 * | 37.7 ± 1.27 ** | 43.68 ± 1.40 ** | |
| Fe-Si7_CMC-Na | 25 | 23.59 ± 3.00 * | 39.43 ± 4.53 ** | 50.29 ± 5.77 ** | 58 ± 6.61 ** |
| 50 | 25.23 ± 1.75 * | 42.66 ± 3.44 ** | 56.44 ± 4.65 ** | 67.36 ± 6.22 ** | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balas, M.; Dumitrache, F.; Badea, M.A.; Fleaca, C.; Badoi, A.; Tanasa, E.; Dinischiotu, A. Coating Dependent In Vitro Biocompatibility of New Fe-Si Nanoparticles. Nanomaterials 2018, 8, 495. https://doi.org/10.3390/nano8070495
Balas M, Dumitrache F, Badea MA, Fleaca C, Badoi A, Tanasa E, Dinischiotu A. Coating Dependent In Vitro Biocompatibility of New Fe-Si Nanoparticles. Nanomaterials. 2018; 8(7):495. https://doi.org/10.3390/nano8070495
Chicago/Turabian StyleBalas, Mihaela, Florian Dumitrache, Madalina Andreea Badea, Claudiu Fleaca, Anca Badoi, Eugenia Tanasa, and Anca Dinischiotu. 2018. "Coating Dependent In Vitro Biocompatibility of New Fe-Si Nanoparticles" Nanomaterials 8, no. 7: 495. https://doi.org/10.3390/nano8070495
APA StyleBalas, M., Dumitrache, F., Badea, M. A., Fleaca, C., Badoi, A., Tanasa, E., & Dinischiotu, A. (2018). Coating Dependent In Vitro Biocompatibility of New Fe-Si Nanoparticles. Nanomaterials, 8(7), 495. https://doi.org/10.3390/nano8070495