Vectors for Glioblastoma Gene Therapy: Viral & Non-Viral Delivery Strategies



Abstract
:1. Introduction
2. Gene Expression in GBM
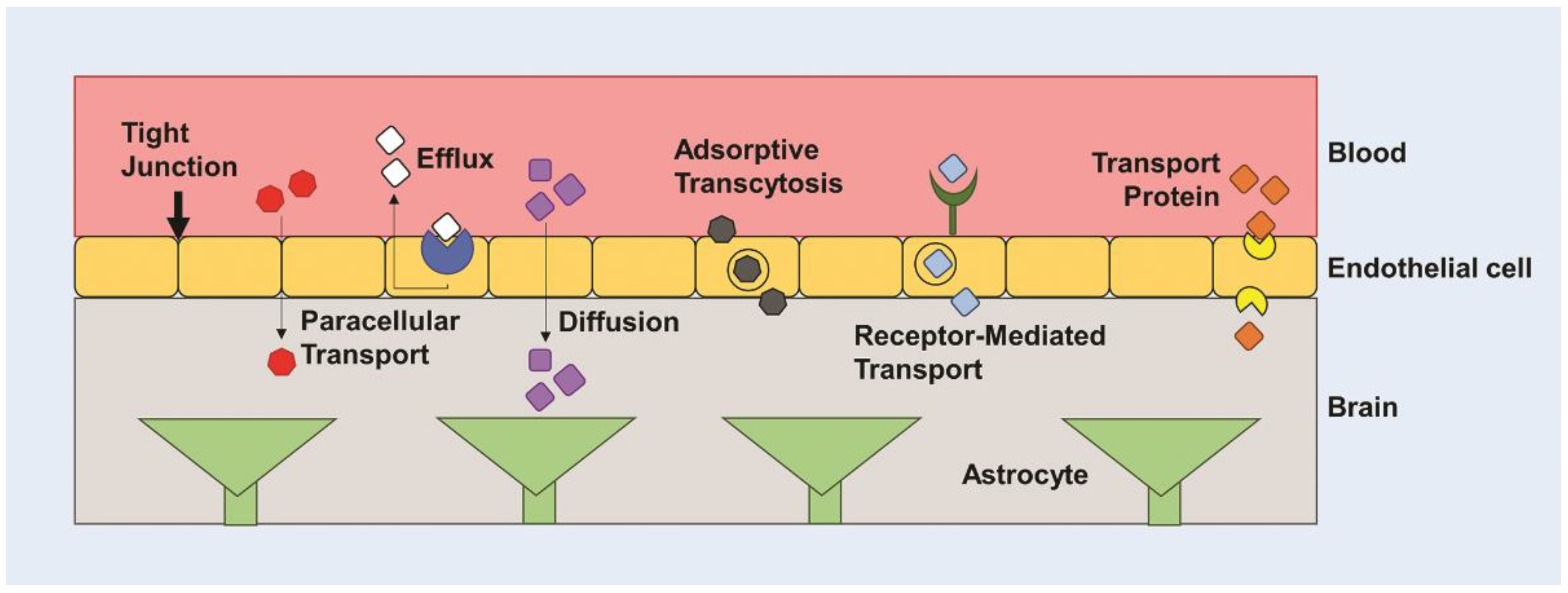
3. Barriers to Drug and Gene Delivery
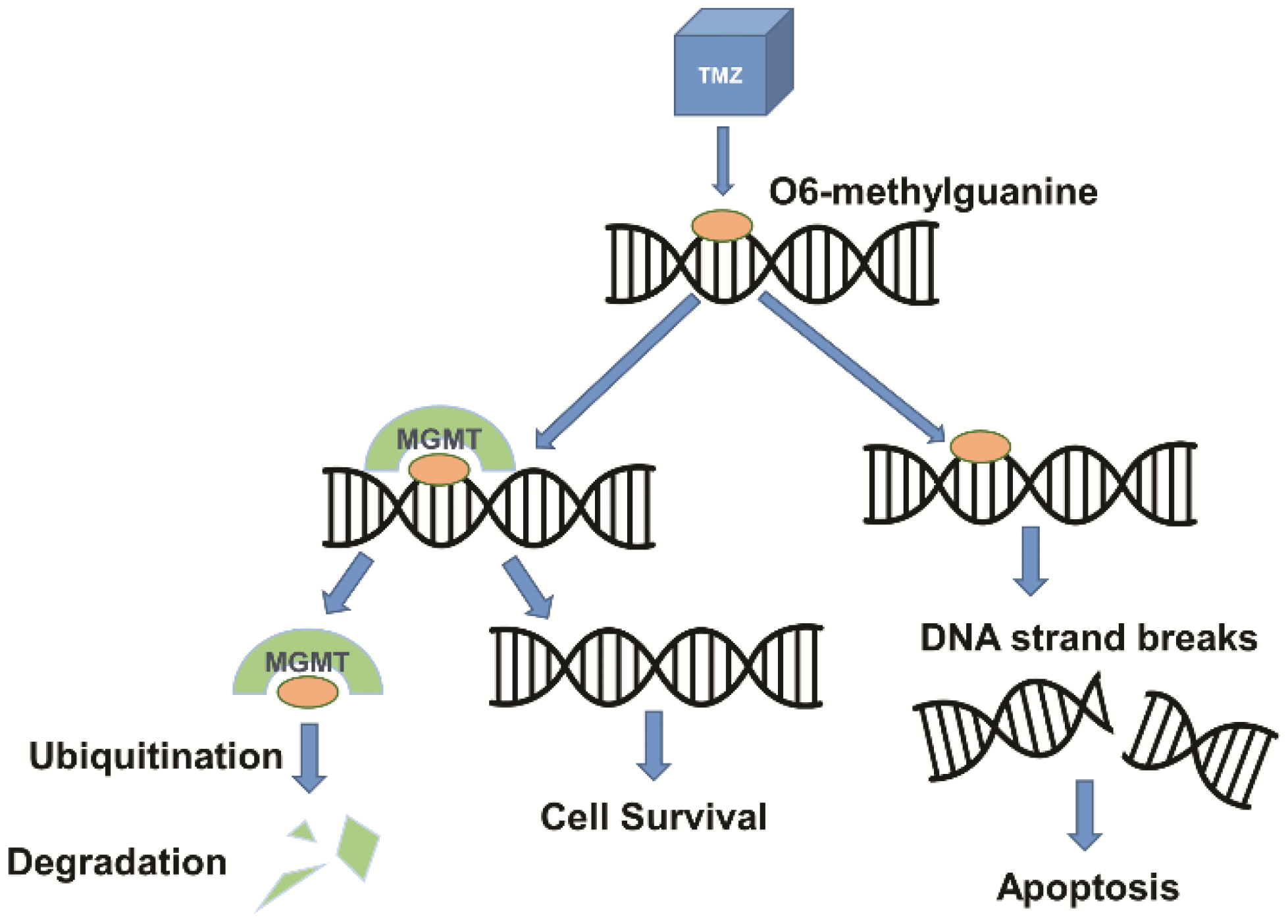
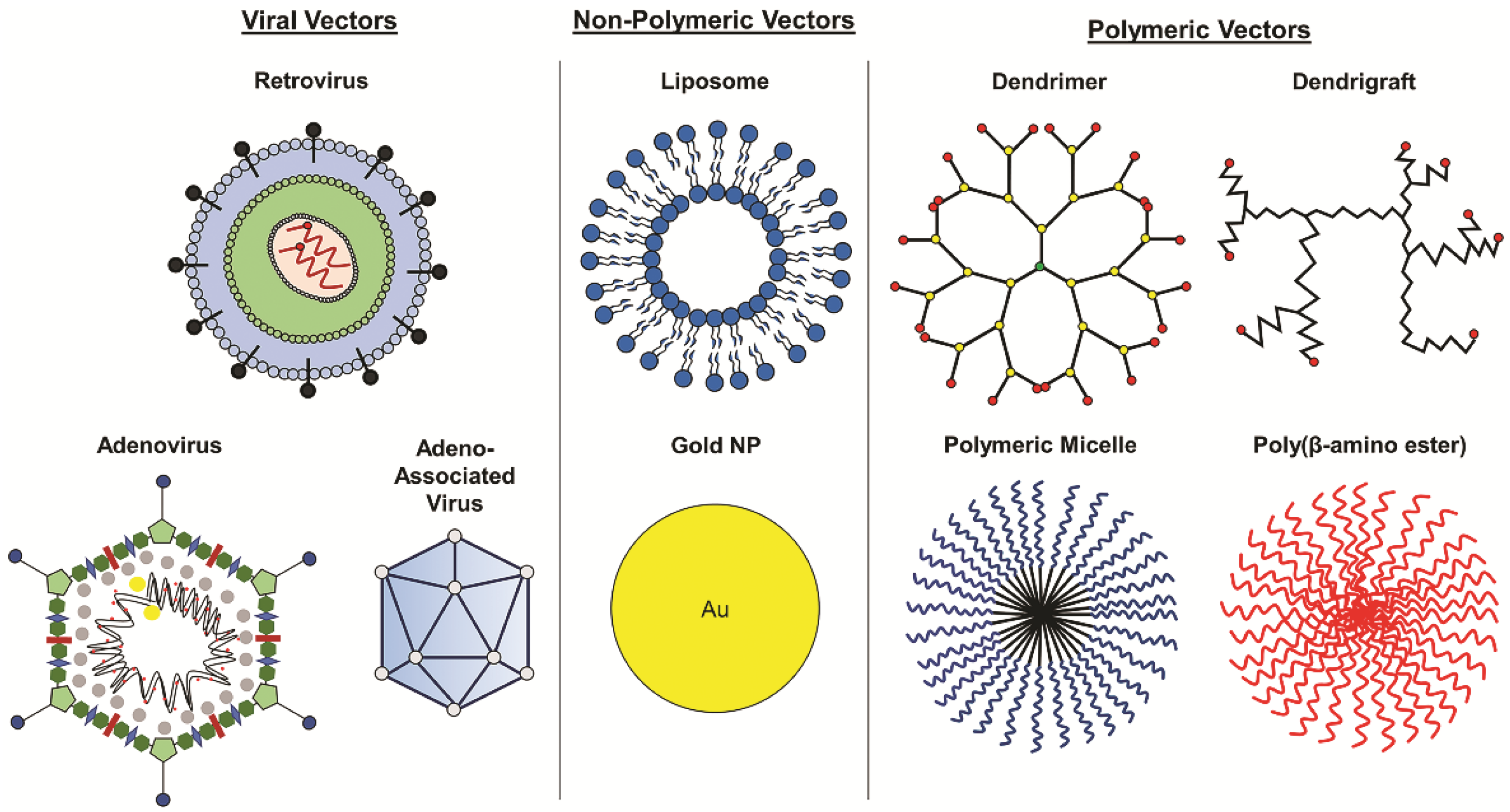
4. Vectors for Glioblastoma Gene Therapy
4.1. Viral Vectors
4.2. Non-Viral Vectors
4.3. Polymeric Delivery Systems
4.3.1. Dendrimers
4.3.2. Dendrigraft
4.3.3. Polymeric Micelles
4.3.4. Poly(β-amino ester)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C. Glioblastoma multiforme: the terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. J. Neurooncol. 2013, 15, 788–796. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hottinger, A.F.; Stupp, R.; Homicsko, K. Standards of care and novel approaches in the management of glioblastoma multiforme. Chin. J. Cancer 2014, 33, 32–39. [Google Scholar] [CrossRef]
- Bota, D.A.; Desjardins, A.; Quinn, J.A.; Affronti, M.L.; Friedman, H.S. Interstitial chemotherapy with biodegradable BCNU (Gliadel®) wafers in the treatment of malignant gliomas. Ther. Clin. Risk Manag. 2007, 3, 707–715. [Google Scholar] [PubMed]
- Chowdhary, S.A.; Ryken, T.; Newton, H.B. Survival outcomes and safety of carmustine wafers in the treatment of high-grade gliomas: a meta-analysis. J. Neurooncol. 2015, 122, 367–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Labagnara, M.; Friedman, M.; Kwasnicki, A.; Murali, R. Glioblastoma: Molecular pathways, stem cells and therapeutic targets. Cancers (Basel) 2015, 7, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Verhaak, R.; Hoadley, K.; Purdon, E.; Getz, G. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 19, 38–46. [Google Scholar] [CrossRef]
- Kleihues, P.; Ohgaki, H. Primary and secondary glioblastomas: from concept to clinical diagnosis. Neurol. Oncol. 1999, 1, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Albert, L.; McKenna, E.; Karsy, M.; Rajdev, P.; Braun, A.; Murali, R. Deciphering the signaling pathways of cancer stem cells of glioblastoma multiforme: Role of Akt/mTOR and MAPK pathways. Adv. Enzyme Regul. 2011, 51, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ling, Z.-Q. Role of isocitrate dehydrogenase 1/2 (IDH 1/2) gene mutations in human tumors. Histol. Histopathol. 2015, 30, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res.-Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Yoshino, A.; Ogino, A.; Yachi, K.; Ohta, T.; Fukushima, T.; Watanabe, T.; Kaatayama, Y. Effect of IFN-beta on human glioma cell lines with temozolomide resistance. Int. J. Oncol. 2009, 151, 414–420. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Tomar, T.; Eggens-Meijer, E.; Bhat, K.; Copray, S.; Walenkamp, A.M.E.; Boddeke, E.; Balasubramanyian, V.; Wagemakers, M.; et al. TGF-beta is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014, 5, e1443. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.-H.; Park, H.J.; George, J.; Yamamoto, K.; Gallup, A.D.; Graber, J.H.; Chen, Y.; Jiang, W.; Steindler, D.; Neilson, E.G.; et al. S100A4 is a biomarker and regulator of glioma stem cells that is critical for mesenchymal transition in glioblastoma. Cancer Res. 2017, 77, 5360–5373. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Zhou, J.; Patel, T.R.; Sirianni, R.W.; Strohbehn, G.; Zheng, M.-Q.; Duong, N.; Schafbauer, T.; Huttner, A.J.; Huang, Y.; Carson, R.E.; et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc. Natl. Acad. Sci. USA 2013, 110, 11751–11756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. The blood-brain barrier and neurotherapeutics. NeuroRx 2005, 2, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Kastin, A.J. Changing the chemokine gradient: CINC1 crosses the blood–brain barrier. J. Neuroimmunol. 2001, 115, 64–70. [Google Scholar] [CrossRef]
- Karanth, H.; Rayasa, M. Nanotechnology in Brain Targeting. Int. J. Pharm. Sci. Nanotechnol. 2008, 1, 924. [Google Scholar]
- Masserini, M. Nanoparticles for brain drug delivery. ISRN Biochem. 2013, 2013, 238428. [Google Scholar] [CrossRef]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Kim, S.S.; Harford, J.B.; Pirollo, K.F.; Chang, E.H. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: The promise of nanomedicine. Biochem. Biophys. Res. Commun. 2015, 468, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Séhédic, D.; Cikankowitz, A.; Hindré, F.; Davodeau, F.; Garcion, E. Nanomedicine to overcome radioresistance in glioblastoma stem-like cells and surviving clones. Trends Pharmacol. Sci. 2015, 36, 236–252. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. BBA-Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Horio, M.; Gottesman, M.M.; Pastan, I. ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3580–3584. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Wagenaar, E.; Mol, C.A.A.M.; Van Deemter, L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Investig. 1996, 97, 2517–2524. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Okura, H.; Smith, C.A.; Rutka, J.T. Gene therapy for malignant glioma. Mol. Cell. Ther. 2014, 2, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, R.; Takase-Yoden, S. Gene expression of neurotropic retrovirus in the CNS. Prog. Brain Res. 1995, 105, 255–262. [Google Scholar] [PubMed]
- Peltékian, E.; Garcia, L.; Danos, O. Neurotropism and Retrograde Axonal Transport of a Canine Adenoviral Vector: A Tool for Targeting Key Structures Undergoing Neurodegenerative Processes. Mol. Ther. 2002, 5, 25–32. [Google Scholar] [CrossRef]
- Braun, E. Neurotropism of herpes simplex virus type 1 in brain organ cultures. J. Gen. Virol. 2006, 87, 2827–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crommentuijn, M.H.W.; Maguire, C.A.; Niers, J.M.; Vandertop, W.P.; Badr, C.E.; Wurdinger, T.; Tannous, B.A. Intracranial AAV-sTRAIL combined with lanatoside C prolongs survival in an orthotopic xenograft mouse model of invasive glioblastoma. Mol. Oncol. 2016, 10, 625–634. [Google Scholar] [CrossRef] [PubMed]
- GuhaSarkar, D.; Su, Q.; Gao, G.; Sena-Esteves, M. Systemic AAV9-IFNbeta gene delivery treats highly invasive glioblastoma. Neurol. Oncol. 2016, 18, 1508–1518. [Google Scholar] [CrossRef]
- Meijer, D.H.; Maguire, C.A.; LeRoy, S.G.; Sena-Esteves, M. Controlling brain tumor growth by intraventricular administration of an AAV vector encoding IFN-beta. Cancer Gene Ther. 2009, 16, 664–671. [Google Scholar] [CrossRef]
- Rainov, N. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef]
- Ram, Z.; Culver, K.W.; Oshiro, E.M.; Viola, J.J.; DeVroom, H.L.; Otto, E.; Long, Z.; Chiang, Y.; McGarrity, G.J.; Muul, L.M.; et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat. Med. 1997, 3, 1354–1361. [Google Scholar] [CrossRef]
- Huang, T.T.; Hlavaty, J.; Ostertag, D.; Espinoza, F.L.; Martin, B.; Petznek, H.; Rodriguez-Aguirre, M.; Ibanez, C.E.; Kasahara, N.; Gunzburg, W.; et al. Toca 511 gene transfer and 5-fluorocytosine in combination with temozolomide demonstrates synergistic therapeutic efficacy in a temozolomide-sensitive glioblastoma model. Cancer Gene Ther. 2013, 20, 544–551. [Google Scholar] [CrossRef]
- Takahashi, M.; Valdes, G.; Hiraoka, K.; Inagaki, A.; Kamijima, S.; Micewicz, E.; Gruber, H.E.; Robbins, J.M.; Jolly, D.J.; McBride, W.H.; et al. Radiosensitization of gliomas by intracellular generation of 5-fluorouracil potentiates prodrug activator gene therapy with a retroviral replicating vector. Cancer Gene Ther. 2014, 21, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Aghi, M.; Vogelbaum, M.A.; Kesari, S.; Chen, C.C.; Liau, L.M.; Piccioni, D.; Portnow, J.; Chang, S.; Robbins, J.M.; Boyce, T.; et al. AT-02 Intratumoral delivery of the retroviral replicating vector (RRV) TOCA 511 in subjects with recurrent high grade glioma: Interim report of phase I study (NCT 01156584). Neurol. Oncol. 2014, 16. [Google Scholar] [CrossRef]
- Lang, F.F.; Bruner, J.M.; Fuller, G.N.; Aldape, K.; Prados, M.D.; Chang, S.; Berger, M.S.; McDermott, M.W.; Kunwar, S.M.; Junck, L.R.; et al. Phase I Trial of Adenovirus-Mediated p53 Gene Therapy for Recurrent Glioma: Biological and Clinical Results. J. Clin. Oncol. 2003, 21, 2508–2518. [Google Scholar] [CrossRef]
- Sandmair, A.M.; Loimas, S.; Puranen, P.; Immonen, A.; Kossila, M.; Puranen, M.; Hurskainen, H.; Tyynela, K.; Turunen, M.; Vanninen, R.; et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum. Gene Ther. 2000, 11, 2197–2205. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; et al. Phase IB Study of Gene-Mediated Cytotoxic Immunotherapy Adjuvant to Up-Front Surgery and Intensive Timing Radiation for Malignant Glioma. J. Clin. Oncol. 2011, 29, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neurol. Oncol. 2016, 18, 1137–1145. [Google Scholar] [CrossRef]
- Kieran, M.W.; Goumnerova, L.; Manley, P.; Chi, S.N.; Marcus, K.; Manzanera, A.G.; Aguilar-Cordova, E.; DiPatri, A.J.; Tomita, T.; Lulla, R.; et al. EPT-14 Phase I study of gene mediated cytotoxic immunotherapy with AdV-tk as adjuvant to surgery and radiation therapy for pediatric malignant glioma and recurrent ependymoma. Neurol. Oncol. 2016, 18, iii26–iii27. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccin. Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Kim, S.-S.; Rait, A.; Kim, E.; Pirollo, K.F.; Nishida, M.; Farkas, N.; Dagata, J.A.; Chang, E.H. A Nanoparticle Carrying the p53 Gene Targets Tumors Including Cancer Stem Cells, Sensitizes Glioblastoma to Chemotherapy and Improves Survival. ACS Nano 2014, 8, 5494–5514. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-S.; Rait, A.; Kim, E.; Pirollo, K.F.; Chang, E.H. A Tumor-targeting p53 Nanodelivery System Limits Chemoresistance to Temozolomide Prolonging Survival in a Mouse Model of Glioblastoma Multiforme. Nanomedicine 2015, 11, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.A.; Day, E.S.; Ko, C.H.; Hurley, L.A.; Luciano, J.P.; Kouri, F.M.; Merkel, T.J.; Luthi, A.J.; Patel, P.C.; Cutler, J.I.; et al. Spherical Nucleic Acid Nanoparticle Conjugates as an RNAi-Based Therapy for Glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.J.; Yoo, J.Y.; Shu, D.; Li, H.; Zhang, J.; Yu, J.-G.; Jaime-Ramirez, A.C.; Acunzo, M.; Romano, G.; Cui, R.; et al. RNA Nanoparticle-Based Targeted Therapy for Glioblastoma through Inhibition of Oncogenic miR-21. Mol. Ther. 2017, 25, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Pi, F.; Sharma, A.; Rajabi, M.; Haque, F.; Shu, D.; Leggas, M.; Evers, B.M.; Guo, P. Stable RNA nanoparticles as potential new generation drugs for cancer therapy. Adv. Drug Deliv. Rev. 2014, 66, 74–89. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Yuan, X.; Li, F.; Pu, P.; Yu, S.; Shen, C.; Zhang, Z.; Zhang, Y. Evaluation of folate-PAMAM for the delivery of antisense oligonucleotides to rat C6 glioma cells in vitro and in vivo. J. Biomed. Mater. Res. A 2010, 93, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, G.; Su, Z.; Jiang, Z.; Chen, L.; Wang, J.; Yu, S.; Liu, Z. Poly(amido amine) is an ideal carrier of miR-7 for enhancing gene silencing effects on the EGFR pathway in U251 glioma cells. Oncol. Rep. 2013, 29, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Waite, C.L.; Roth, C.M. PAMAM-RGD Conjugates Enhance siRNA Delivery Through a Multicellular Spheroid Model of Malignant Glioma. Bioconjug. Chem. 2009, 20, 1908–1916. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.; Green, E.S.; Kim, G.Y.; Song, S.J.; Mun, J.Y.; Lee, S.; Park, J.I.; Park, J.S.; Ko, K.S.; Han, J.; et al. Dipeptide-functionalized Polyamidoamine dendrimer-mediated apoptin gene delivery facilitates apoptosis of human primary glioma cells. Int. J. Pharm. 2016, 515, 186–200. [Google Scholar] [CrossRef]
- Gao, S.; Li, J.; Jiang, C.; Hong, B.; Hao, B. Plasmid pORF-hTRAIL targeting to glioma using transferrin-modified polyamidoamine dendrimer. Drug Des. Dev. Ther. 2016, 10, 1–11. [Google Scholar] [CrossRef]
- Huang, R.; Ke, W.; Han, L.; Li, J.; Liu, S.; Jiang, C. Targeted delivery of chlorotoxin-modified DNA-loaded nanoparticles to glioma via intravenous administration. Biomaterials 2011, 32, 2399–2406. [Google Scholar] [CrossRef]
- Huang, S.; Li, J.; Han, L.; Liu, S.; Ma, H.; Huang, R.; Jiang, C. Dual targeting effect of Angiopep-2-modified, DNA-loaded nanoparticles for glioma. Biomaterials 2011, 32, 6832–6838. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Fan, J.; Wang, Z.; Zeng, X.; Sun, Y.; Song, P.; Ju, D. The role of autophagy in the neurotoxicity of cationic PAMAM dendrimers. Biomaterials 2014, 35, 7588–7597. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Shao, K.; Huang, R.; Ye, L.; Lou, J.; Jiang, C. A leptin derived 30-amino-acid peptide modified pegylated poly-L-lysine dendrigraft for brain targeted gene delivery. Biomaterials 2010, 31, 5246–5257. [Google Scholar] [CrossRef]
- Liu, S.; Guo, Y.; Huang, R.; Li, J.; Huang, S.; Kuang, Y.; Han, L.; Jiang, C. Gene and doxorubicin co-delivery system for targeting therapy of glioma. Biomaterials 2012, 33, 4907–4916. [Google Scholar] [CrossRef]
- Liu, Y.; He, X.; Kuang, Y.; An, S.; Wang, C.; Guo, Y.; Ma, H.; Lou, J.; Jiang, C. A bacteria deriving peptide modified dendrigraft poly-l-lysines (DGL) self-assembling nanoplatform for targeted gene delivery. Mol. Pharm. 2014, 11, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; An, S.; Guo, Y.; Huang, S.; Shao, K.; Liu, Y.; Li, J.; Ma, H.; Jiang, C. T7 peptide-functionalized nanoparticles utilizing RNA interference for glioma dual targeting. Int. J. Pharm. 2013, 454, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wang, K.; Wang, Y.; Wang, S.; Li, J.; Lou, J.; Ye, L.; Yan, X.; Lu, W.; Huang, R. Enhanced blood-brain barrier penetration and glioma therapy mediated by a new peptide modified gene delivery system. Biomaterials 2015, 37, 345–352. [Google Scholar] [CrossRef]
- Li, J.; Guo, Y.; Kuang, Y.; An, S.; Ma, H.; Jiang, C. Choline transporter-targeting and co-delivery system for glioma therapy. Biomaterials 2013, 34, 9142–9148. [Google Scholar] [CrossRef]
- Kodama, Y.; Nakamura, T.; Kurosaki, T.; Egashira, K.; Mine, T.; Nakagawa, H.; Muro, T.; Kitahara, T.; Higuchi, N.; Sasaki, H. Biodegradable nanoparticles composed of dendrigraft poly-L-lysine for gene delivery. Eur. J. Pharm. Biopharm. 2014, 87, 472–479. [Google Scholar] [CrossRef]
- Kodama, Y.; Kuramoto, H.; Mieda, Y.; Muro, T.; Nakagawa, H.; Kurosaki, T.; Sakaguchi, M.; Nakamura, T.; Kitahara, T.; Sasaki, H. Application of biodegradable dendrigraft poly-l-lysine to a small interfering RNA delivery system. J. Drug Target. 2017, 25, 49–57. [Google Scholar] [CrossRef]
- Tang, M.; Dong, H.; Li, Y.; Ren, T. Harnessing the PEG-cleavable strategy to balance cytotoxicity, intracellular release and the therapeutic effect of dendrigraft poly-l-lysine for cancer gene therapy. J. Mater. Chem. B 2016, 4, 1284–1295. [Google Scholar] [CrossRef]
- Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Nijsen, J.F.W.; Hennink, W.E. Polymeric Micelles in Anticancer Therapy: Targeting, Imaging and Triggered Release. Pharm. Res. 2010, 27, 2569–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Cao, N.; Chen, J.; Yu, X.; Shuai, X. Multifunctional nanocarrier mediated co-delivery of doxorubicin and siRNA for synergistic enhancement of glioma apoptosis in rat. Biomaterials 2012, 33, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; He, M.L.; Chan, C.Y.; Chen, Y.C.; Li, X.P.; Li, Y.; Zheng, D.; Lin, M.C.; Kung, H.F.; Shuai, X.T.; et al. The use of folate-PEG-grafted-hybranched-PEI nonviral vector for the inhibition of glioma growth in the rat. Biomaterials 2009, 30, 4014–4020. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Wei, X.; Qian, J.; Feng, L.; Zhu, J.; Lu, W. Co-delivery of TRAIL gene enhances the anti-glioblastoma effect of paclitaxel in vitro and in vivo. J. Control. Release 2012, 160, 630–636. [Google Scholar] [CrossRef]
- Wang, J.; Lei, Y.; Xie, C.; Lu, W.; Wagner, E.; Xie, Z.; Gao, J.; Zhang, X.; Yan, Z.; Liu, M. Retro-inverso CendR peptide-mediated polyethyleneimine for intracranial glioblastoma-targeting gene therapy. Bioconjug. Chem. 2014, 25, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, J.; Xie, C.; Wagner, E.; Lu, W.; Li, Y.; Wei, X.; Dong, J.; Liu, M. Glutathione-sensitive RGD-poly(ethylene glycol)-SS-polyethylenimine for intracranial glioblastoma targeted gene delivery. J. Gene Med. 2013, 15, 291–305. [Google Scholar] [CrossRef]
- Green, J.J.; Zugates, G.T.; Langer, R.; Anderson, D.G. Poly (β-amino esters): Procedures for Synthesis and Gene Delivery. Methods Mol. Biol. 2009, 480, 53–63. [Google Scholar] [CrossRef]
- Anderson, D.G.; Lynn, D.M.; Langer, R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angew Chem. Int. Ed. Engl. 2003, 42, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.Y.; Green, J.J. Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for siRNA and DNA delivery to human brain cancer. Adv Heal. Mater 2013, 2, 468–480. [Google Scholar] [CrossRef]
- Guerrero-Cazares, H.; Tzeng, S.Y.; Young, N.P.; Abutaleb, A.O.; Quinones-Hinojosa, A.; Green, J.J. Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo. ACS Nano 2014, 8, 5141–5153. [Google Scholar] [CrossRef] [PubMed]
- Mangraviti, A.; Tzeng, S.Y.; Kozielski, K.L.; Wang, Y.; Jin, Y.; Gullotti, D.; Pedone, M.; Buaron, N.; Liu, A.; Wilson, D.R.; et al. Polymeric Nanoparticles for Nonviral Gene Therapy Extend Brain Tumor Survival in Vivo. ACS Nano 2015, 9, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, P.; Song, E.; Zhang, C.; Berry, S.; Park, H.W.; Kim, Y.E.; Park, J.S.; Lee, S.; Suk, J.S.; Hanes, J. Biodegradable DNA Nanoparticles that Provide Widespread Gene Delivery in the Brain. Small 2016, 12, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Peng, D.Y.; Green, J.J. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol. Pharm. 2012, 9, 3375–3383. [Google Scholar] [CrossRef]
- Kozielski, K.L.; Tzeng, S.Y.; Green, J.J. A bioreducible linear poly(β-amino ester) for siRNA delivery. Chem. Commun. (Camb) 2013, 49, 5319–5321. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene Target | Effect | GBM Clinical Subtype | References |
|---|---|---|---|
| EGFR (epidermal growth factor receptor) | Reduction in apoptosis and increased uncontrolled cell proliferation | Classical | [13,14] |
| PTEN (phosphate and tensin homologue) | Activation of the P13K/Akt/mTOR pathway, leading to cell proliferation, migration and growth | Classical | [1,13,15,16] |
| PDGFRA (platelet derived growth factor receptor—alpha) | Increased tumor cell proliferation | Proneural | [11,13,14] |
| IDH-1 (isocitrate dehydrogenase 1) | Alters DNA and histone methylation | Proneural | [17,18] |
| Tumor suppressor p53 | Uncontrolled cell growth | Proneural, mesenchymal | [13,14,20] |
| NF-1 (neurofibromin 1) | Uncontrolled cell growth | Mesenchymal | [13] |
| Vector | Gene Therapy Agent | Mechanism | Combination Therapy | Clinical Trial Phase | Clinical Trial Number |
|---|---|---|---|---|---|
| Retrovirus | HSV-tk | Suicide gene therapy, HSV-tk converts ganciclovir to antiviral drug ganciclovir triphosphate | Ganciclovir | Phase I | NCT00001328 |
| Retrovirus | Toca 511 | Suicide gene therapy, CD converts prodrug 5-FC to anti-neoplastic 5-FU | Oral 5-FC | Phase II/III | NCT02414165 |
| Adenovirus | SCH-58500 | Tumor suppressor gene therapy, transfects p53 gene | N/A | Phase I | NCT00004080 |
| Adenovirus | Ad-p53 | Tumor suppressor gene therapy, transfects p53 gene | N/A | Phase I | NCT00004041 |
| Retro or adenovirus | HSV-tk | Suicide gene therapy, HSV-tk converts ganciclovir to antiviral drug ganciclovir triphosphate | Ganciclovir | Phase I | Sandmair et. al. |
| Adenovirus | AdV-tk | Gene-mediated cytotoxic immunotherapy, HSV-tk converts valacyclovir to antiviral drug acyclovir | Valacyclovir | Phase I | NCT00751270 |
| Adenovirus | AdV-tk | Gene-mediated cytotoxic immunotherapy, HSV-tk converts valacyclovir to antiviral drug acyclovir | Valacyclovir and radiation therapy | Phase IIa | NCT00589875 |
| Liposome | SGT-53 | Tumor suppressor gene therapy, transfects p53 gene | TMZ | Phase II | NCT02340156 |
| Spherical Nucleic Acid Gold NP | NU-0129 | RNAi gene therapy, transfects siRNAs targeting oncogene Bcl2L12 | N/A | Early Phase I | NCT03020017 |
| Vector | Advantages | Disadvantages |
|---|---|---|
| Viral | ||
| Adenovirus |
|
|
| Retrovirus |
|
|
| Adeno-associated virus |
|
|
| Non-Viral | ||
| Liposome |
|
|
| Gold nanoparticles |
|
|
| Dendrimer & Dendrigraft |
|
|
| Polymeric micelles |
|
|
| Poly(β-amino ester) |
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caffery, B.; Lee, J.S.; Alexander-Bryant, A.A. Vectors for Glioblastoma Gene Therapy: Viral & Non-Viral Delivery Strategies. Nanomaterials 2019, 9, 105. https://doi.org/10.3390/nano9010105
Caffery B, Lee JS, Alexander-Bryant AA. Vectors for Glioblastoma Gene Therapy: Viral & Non-Viral Delivery Strategies. Nanomaterials. 2019; 9(1):105. https://doi.org/10.3390/nano9010105
Chicago/Turabian StyleCaffery, Breanne, Jeoung Soo Lee, and Angela A. Alexander-Bryant. 2019. "Vectors for Glioblastoma Gene Therapy: Viral & Non-Viral Delivery Strategies" Nanomaterials 9, no. 1: 105. https://doi.org/10.3390/nano9010105
APA StyleCaffery, B., Lee, J. S., & Alexander-Bryant, A. A. (2019). Vectors for Glioblastoma Gene Therapy: Viral & Non-Viral Delivery Strategies. Nanomaterials, 9(1), 105. https://doi.org/10.3390/nano9010105