Computational Investigation of Tuning the Electron-Donating Ability in Metal-Free Organic Dyes Featuring an Azobenzene Spacer for Dye-Sensitized Solar Cells


Abstract
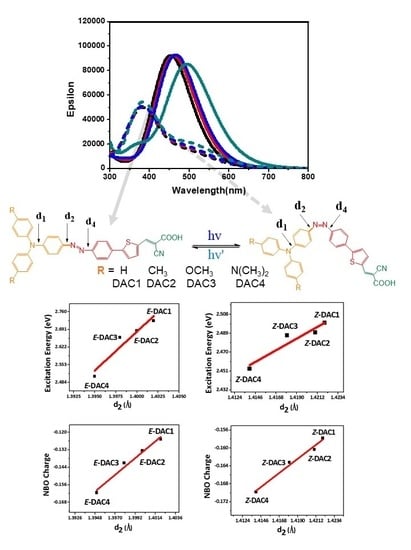
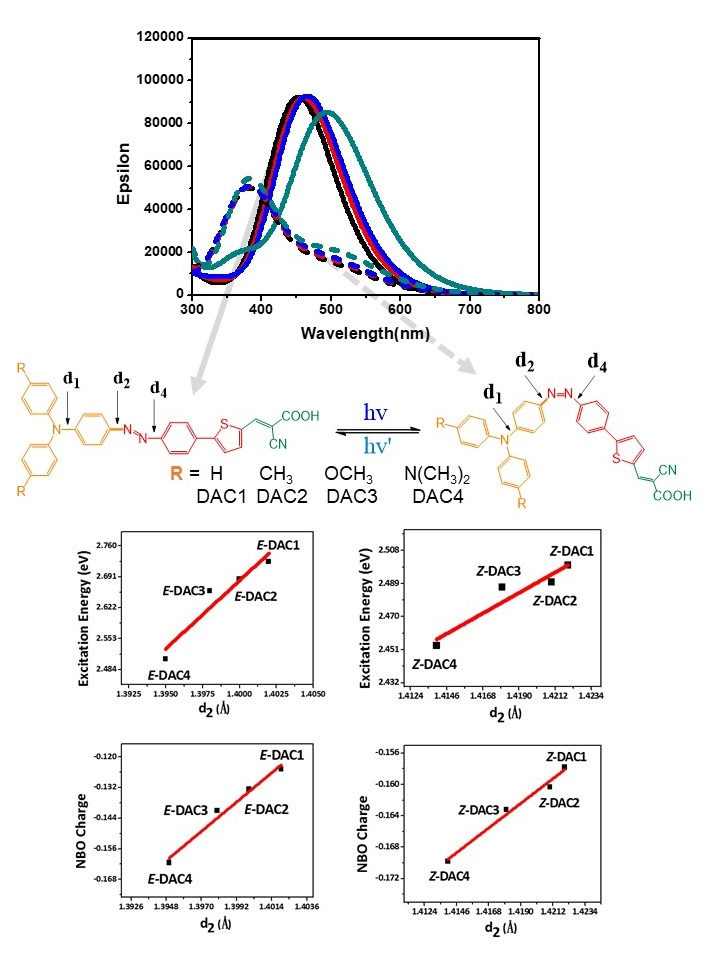
:
1. Introduction
2. Computational Details
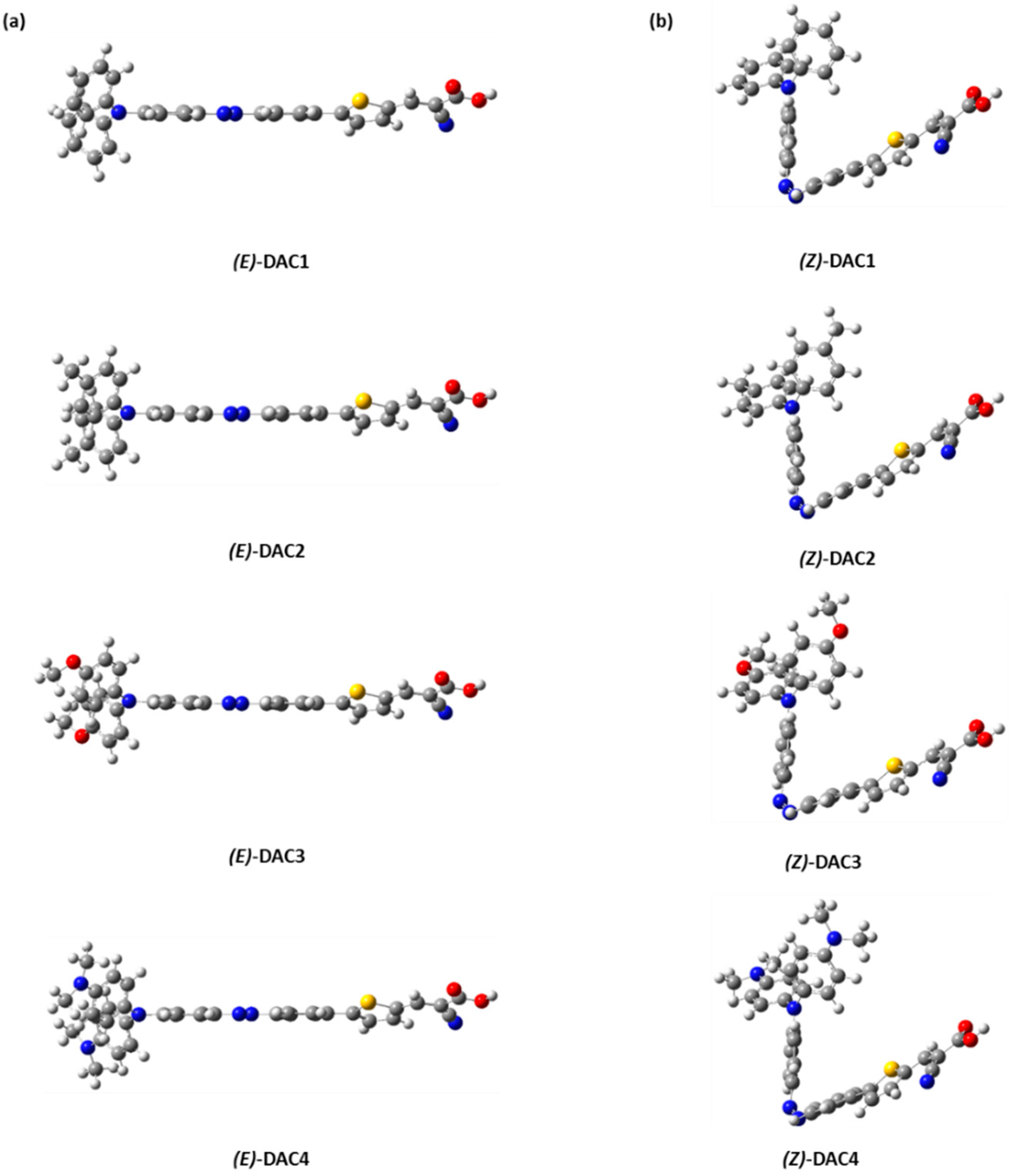
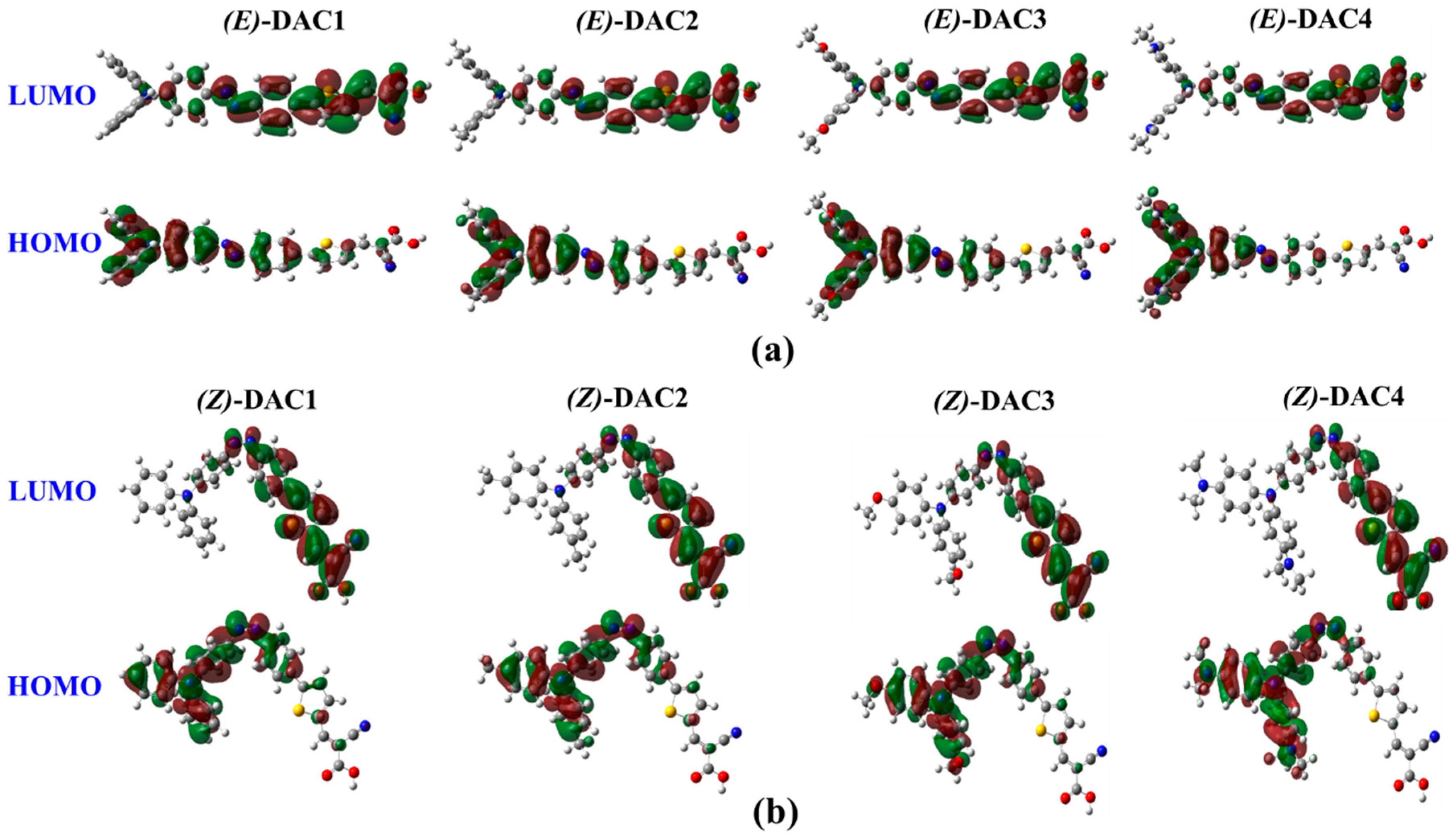
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardin, B.E.; Snaith, H.J.; McGehee, M.D. The renaissance of dye-sensitized solar cells. Nat. Photonics 2012, 6, 162–169. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Numata, Y.; Han, L. Highly efficient dye-sensitized solar cells: Progress and future challenges. Energy Environ. Sci. 2013, 6, 1443–1464. [Google Scholar] [CrossRef]
- Fakharuddin, A.; Jose, R.; Brown, T.M.; Fabregat-Santiago, F.; Bisquert, J. A perspective on the production of dye-sensitized solar modules. Energy Environ. Sci. 2014, 7, 3952–3981. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Chen, J. Arylamine organic dyes for dye-sensitized solar cells. Chem. Soc. Rev. 2013, 42, 3453–3488. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, W. Organic sensitizers from D-π-A to D-A-π-A: Effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances. Chem. Soc. Rev. 2013, 42, 2039–2058. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A. Triphenylamine based dyes for dye sensitized solar cells: A review. Sol. Energy 2016, 123, 127–144. [Google Scholar] [CrossRef]
- Lee, C.-P.; Li, C.-T.; Ho, K.-C. Use of organic materials in dye-sensitized solar cells. Mater. Today 2017, 20, 267–283. [Google Scholar] [CrossRef] [Green Version]
- Martisnovich, N.; Troisi, A. Theoretical studies of dye-sensitized solar cells: From electronic structure to elementary processes. Energy Environ. Sci. 2011, 4, 4473–4495. [Google Scholar] [CrossRef]
- Labat, F.; Bahers, T.L.; Ciofini, I.; Adamo, C. First-principles modeling of dye-sensitized solar cells: Challenges and perspectives. Acc. Chem. Res. 2012, 45, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Preat, J.; Michaux, C.; Jacquemin, D.; Perpete, E.A. Enhanced efficiency of organic dye-sensitized solar cells: Triphenylamine derivatives. J. Phys. Chem. C 2009, 113, 16821–16833. [Google Scholar] [CrossRef]
- Peng, B.; Yang, S.; Li, L.; Cheng, F.; Chen, J. A density functional theory and time-dependent density functional theory investigation on the anchor comparison of triarylamine-based dyes. J. Chem. Phys. 2010, 132, 034305. [Google Scholar] [CrossRef] [PubMed]
- Al-Eid, M.; Lim, S.; Park, K.-W.; Fitzpatrick, B.; Han, C.-H.; Kwak, K.; Hong, J.; Cooke, G. Facile synthesis of metal-free organic dyes featuring a thienylethynyl spacer for dye sensitized solar cell. Dyes Pigment. 2014, 104, 197–203. [Google Scholar] [CrossRef]
- Merino, E.; Ribagorda, M. Control over molecular motion using the cis-trans photoisomerization of the azo group. Beilstein J. Org. Chem. 2012, 8, 1071–1090. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Wysiecka, E.; Lukasik, N.; Biernat, J.F.; Luboch, E. Azo group(s) in selected macrocyclic compounds. J. Incl. Phenom. Macrocycl. Chem. 2018, 90, 189–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cole, J.M. TiO2-assisted photoisomerization of azo dyes using self-assembled monolayers: Case study on para-methyl red towards solar-cell applications. ACS Appl. Mater. Interfaces 2014, 6, 3742–3749. [Google Scholar] [CrossRef] [PubMed]
- Kakiage, K.; Aoyama, Y.; Yamamura, M.; Yano, T.; Unno, M.; Kyomen, T.; Hanaya, M. A novel alkoxysilyl azobenzene dye photosensitizer with alkylamino group for dye-sensitized solar cells. Silicon 2014, 6, 123–127. [Google Scholar] [CrossRef]
- Ezema, C.G.; Nwanya, A.C.; Ezema, B.E.; Patil, B.H.; Bulakbe, R.N.; Ukoha, P.O.; Lokhande, C.D.; Maaza, M.; Ezema, F.I. Photo-electrochemical studies of chemically deposited nanocrystalline meso-porous n-type TiO2 thin films for dye-sensitized solar cell (DSSC) using simple synthesized azo dye. Appl. Phys. A 2016, 122, 435. [Google Scholar] [CrossRef]
- Chiu, K.Y.; Tran, T.T.H.; Chang, S.H.; Yang, T.-F.; Su, Y.O. A new series of azobenzene-bridged metal-free organic dyes and application on DSSC. Dyes Pigment. 2017, 146, 512–519. [Google Scholar] [CrossRef]
- Ye, Y.; Pang, J.; Zhou, X.; Huang, J. Understanding the torsion effects on optical properties of azobenzene derivatives. Comput. Theor. Chem. 2016, 1076, 17–22. [Google Scholar] [CrossRef]
- Novir, S.B.; Hashemianzadeh, S.M. Quantum chemical investigation of structural and electronic properties of trans- and cis-structures of some azo dyes for dye-sensitized solar cells. Comput. Theor. Chem. 2017, 1102, 87–97. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Naumov, V.A.; Samdal, S.; Naumov, A.V.; Gundersen, S.; Volden, H.V. Molecular Structure of Triphenylamine in the Gas Phase. Russ. J. Gen. Chem. 2005, 75, 1956. [Google Scholar] [CrossRef]
- Preat, J.; Jacquemin, D.; Michaux, C.; Perpète, E.A. Improvement of the efficiency of thiophene-bridged compounds for dye-sensitized solar cells. Chem. Phys. 2010, 376, 56–68. [Google Scholar] [CrossRef]
- Jacquemin, D.; Wathelet, V.; Perpète, E.A.; Adamo, C. Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. [Google Scholar] [CrossRef] [PubMed]
- Berardo, E.; Hu, H.-S.; van Dam, H.J.J.; Shevlin, S.A.; Woodley, S.M.; Kowalski, K.; Zwijnenburg, M.A. Describing Excited State Relaxation and Localization in TiO2 Nanoparticles Using TD-DFT. J. Chem. Theory Comput. 2014, 10, 5538–5548. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Zhao, Z.X.; Zhang, H.X. Theoretical Study of Substituent and Charge Effects on the Thermal Cis→ Trans isomerization of Ortho-fluoroazobenzenes Photoswitches. Org. Electron. 2018, 52, 61–70. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ganesan, P.; Teuscher, J.; Moehl, T.; Kim, Y.J.; Yi, C.; Comte, P.; Pei, K.; Holcombe, T.W.; Nazeeruddin, M.K.; et al. Influence of the donor size in D–π–A organic dyes for dye-sensitized solar cells. J. Am. Chem. Soc. 2014, 136, 5722–5730. [Google Scholar] [CrossRef]
- Boschloo, G.; Hagfeldt, A. Photoinduced absorption spectroscopy of dye-sensitized nanostructured TiO2. Chem. Phys. Lett. 2003, 370, 381–386. [Google Scholar] [CrossRef]
- Tigreros, A.; Ortiz, A.; Insuasty, B. Effect of π-conjugated linkage on photophysical properties: Acetylene linker as the better connection group for highly solvatochromic probes. Dyes Pigment. 2014, 111, 45–51. [Google Scholar] [CrossRef]
- Mo, Y.; Lin, Z.; Wu, W.; Zhang, Q. Bond-distorted orbitals and effects of hybridization and resonance on C–C bond lengths. J. Phys. Chem. 1996, 100, 11569–11572. [Google Scholar] [CrossRef]
- Kreglewski, M. The geometry and inversion-internal rotation potential function of methylamine. J. Mol. Spectrosc. 1989, 113, 10–21. [Google Scholar] [CrossRef]
- Pearson, R.; Lovas, F.J. Microwave spectrum and molecular structure of methylenimine (CH2NH). J. Chem. Phys. 1977, 66, 4149–4156. [Google Scholar] [CrossRef]
- Bouwstra, J.A.; Schouten, A.; Kroon, J. Structural studies of the system trans-azobenzene/trans-stilbene. I. A reinvestigation of the disorder in the crystal structure of trans-azobenzene, C12H10N2. Acta Crystallogr. Sect. C 1983, 39, 1121–1123. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Zhang, T.; Wu, M. Theoretical Study for High-Energy-Density Compounds Derived from Cyclophosphazene. IV. DFT Studies on 1,1-Diamino-3,3,5,5,7,7-hexaazidocyclotetraphosphazene and Its Isomers. Int. J. Mol. Sci. 2009, 10, 3502–3516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhanamoorthi, N.; Lo, C.M.; Jiang, J.C. Molecular Design of Porphyrins for Dye-Sensitized Solar Cells: A DFT/TDDFT Study. J. Phys. Chem. Lett. 2013, 4, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xu, J. Density functional theory study on D-p-A-type organic dyes containing different electron-donors for dye-sensitized solar cells. Bull. Korean Chem. Soc. 2013, 34, 3211–3217. [Google Scholar] [CrossRef]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Campbell, W.M.; Jolley, K.W.; Wagner, P.; Wagner, K.; Walsh, P.J.; Gordon, K.C.; Schmidt-Mende, L.; Nazeeruddin, M.K.; Wang, Q.; Gratzel, M.; et al. Highly Efficient Porphyrin Sensitizers for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2007, 111, 11760–11762. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Li, Y.; Song, P.; Ma, F. An Experimental and Theoretical Investigation of the Electronic Structures and Photoelectrical Properties of Ethyl Red and Carminic Acid for DSSC Application. Materials 2016, 9, 813. [Google Scholar] [CrossRef]
- Angelis, F.D.; Fantacci, S.; Selloni, A.; Gratzel, M.; Nazeeruddin, M.K. Influence of the sensitizer adsorption mode on the open-circuit potential of dye-sensitized solar cells. Nano Lett. 2007, 7, 3189–3195. [Google Scholar] [CrossRef]
- Georgiev, A.; Bubev, E.; Dimov, D.; Yancheva, D.; Zhivkov, I.; Krajcovic, J.; Vala, M.; Weiter, M.; Machkova, M. Synthesis, Structure, Spectral properties and DFT Quantum Chemical Calculations of 4-aminoazobenzene Dyes. Effect of Intramolecular Hydrogen Bonding on photoisomerization. Spectrochim. Acta Part A 2017, 175, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-R.; Liu, L.; Zhe, J.-W.; Jin, N.-Z.; Ma, Y.; Yuan, L.-H.; Zhang, M.-L.; Wu, Y.-Z.; Liu, Z.-J.; Chen, H.-S. The Role of the Conjugate Bridge in Electronic Structures and Related Properties of Tetrahydroquinoline for Dye Sensitized Solar Cells. Int. J. Mol. Sci. 2013, 14, 5461–5481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, D.; Park, K.W.; Kim, J.; Hong, J.; Kwak, K. DFT computational investigation of tuning the electron donating ability in metal-free organic dyes featuring a thienylethynyl spacer for dye sensitized solar cells. Comput. Theor. Chem. 2016, 1081, 30–37. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (E)-DAC1 | (E)-DAC2 | (E)-DAC3 | (E)-DAC4 | (Z)-DAC1 | (Z)-DAC2 | (Z)-DAC3 | (Z)-DAC4 | |
|---|---|---|---|---|---|---|---|---|
| d1(C5-N3) | 1.404 | 1.401 | 1.397 | 1.392 | 1.405 | 1.403 | 1.397 | 1.392 |
| d2(C1-N1) | 1.402 | 1.400 | 1.398 | 1.395 | 1.422 | 1.421 | 1.418 | 1.414 |
| d3(N1=N2) | 1.261 | 1.262 | 1.263 | 1.264 | 1.247 | 1.248 | 1.249 | 1.251 |
| d4(C2-N2) | 1.412 | 1.411 | 1.410 | 1.409 | 1.426 | 1.425 | 1.424 | 1.422 |
| d5(C6-C7) | 1.461 | 1.461 | 1.461 | 1.461 | 1.461 | 1.461 | 1.461 | 1.461 |
| A1(C1N1N2C2) | 179.351 | 179.371 | 179.219 | 179.652 | −12.214 | −12.376 | −12.417 | −12.694 |
| A2(C3C1N1N2) | −2.036 | −1.903 | −1.512 | −1.765 | −37.821 | −36.679 | −34.342 | −30.862 |
| A3(N1N2C2C4) | −1.562 | −1.322 | −0.804 | −0.722 | −54.091 | −54.507 | −55.525 | −56.612 |
| Dyes | qDonor | qπ-spacer | qAcceptor | ∆qD−A |
|---|---|---|---|---|
| (E)-DAC1 | 0.2827 | −0.1249 | −0.1578 | 0.4405 |
| (E)-DAC2 | 0.2931 | −0.1328 | −0.1603 | 0.4534 |
| (E)-DAC3 | 0.3044 | −0.1411 | −0.1632 | 0.4676 |
| (E)-DAC4 | 0.3313 | −0.1615 | −0.1698 | 0.5011 |
| (Z)-DAC1 | 0.2128 | −0.0529 | −0.1598 | 0.3726 |
| (Z)-DAC2 | 0.2234 | −0.0607 | −0.1626 | 0.386 |
| (Z)-DAC3 | 0.2384 | −0.0726 | −0.1657 | 0.4041 |
| (Z)-DAC4 | 0.2677 | −0.0936 | −0.1741 | 0.4418 |
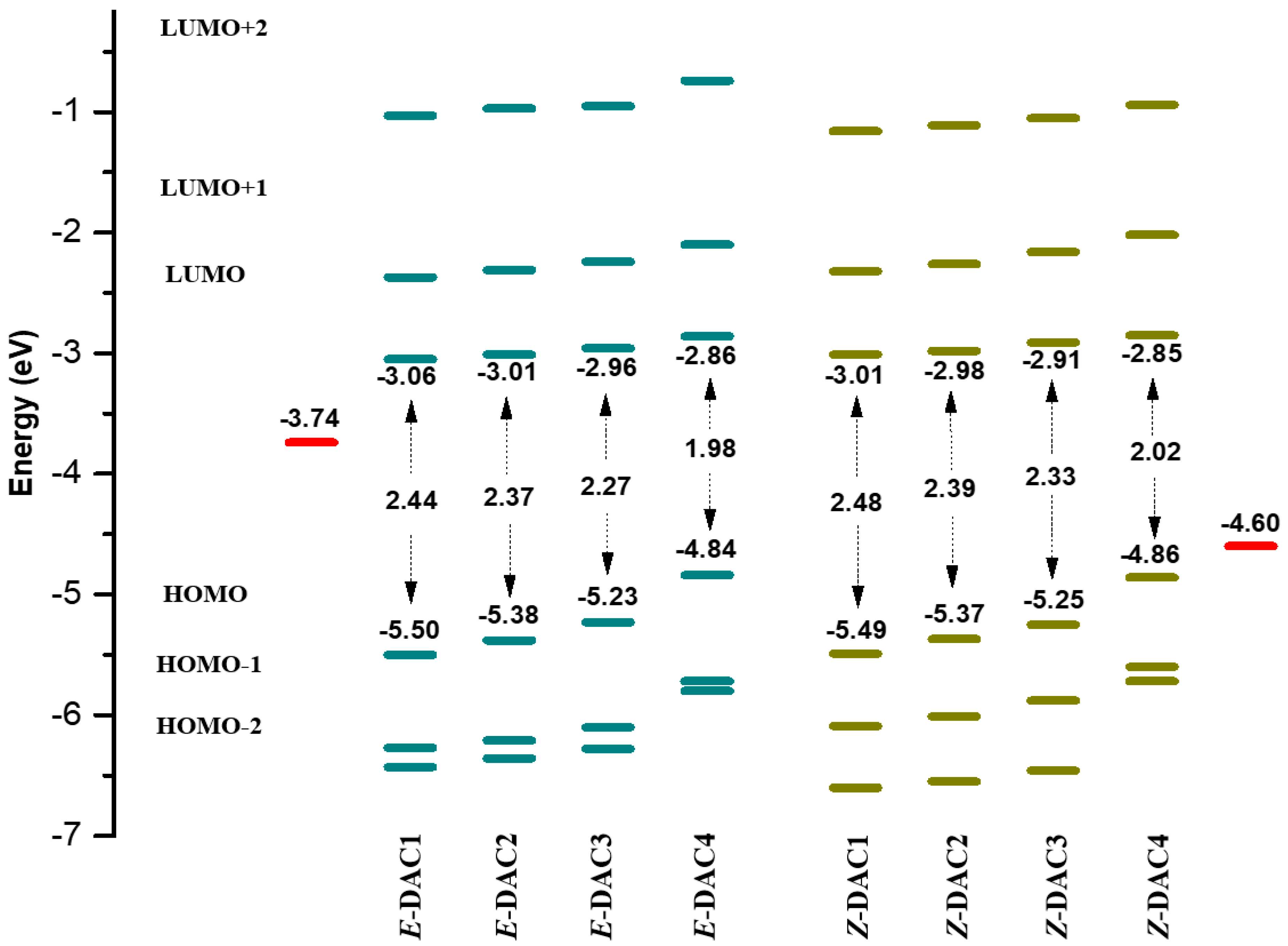
| Dyes | HOMO | LUMO | Egap | IP | EA | η | Dipole, µ (Debye) | ∆E (Ecis − Etrans) |
|---|---|---|---|---|---|---|---|---|
| (E)-DAC1 | −5.50 | −3.06 | 2.44 | 5.32 | 3.26 | 1.03 | 8.564 | 0.71 |
| (Z)-DAC1 | −5.49 | −3.01 | 2.48 | 5.30 | 3.16 | 1.07 | 6.524 | |
| (E)-DAC2 | −5.38 | −3.01 | 2.37 | 5.22 | 3.25 | 0.99 | 9.765 | 0.71 |
| (Z)-DAC2 | −5.37 | −2.98 | 2.39 | 5.20 | 3.15 | 1.03 | 7.319 | |
| (E)-DAC3 | −5.23 | −2.96 | 2.27 | 5.11 | 3.24 | 0.94 | 12.228 | 0.72 |
| (Z)-DAC3 | −5.25 | −2.91 | 2.34 | 5.11 | 3.14 | 0.99 | 9.497 | |
| (E)-DAC4 | −4.84 | −2.86 | 1.98 | 4.74 | 3.22 | 0.76 | 13.981 | 0.71 |
| (Z)-DAC4 | −4.86 | −2.85 | 2.01 | 4.74 | 3.13 | 0.81 | 11.036 |
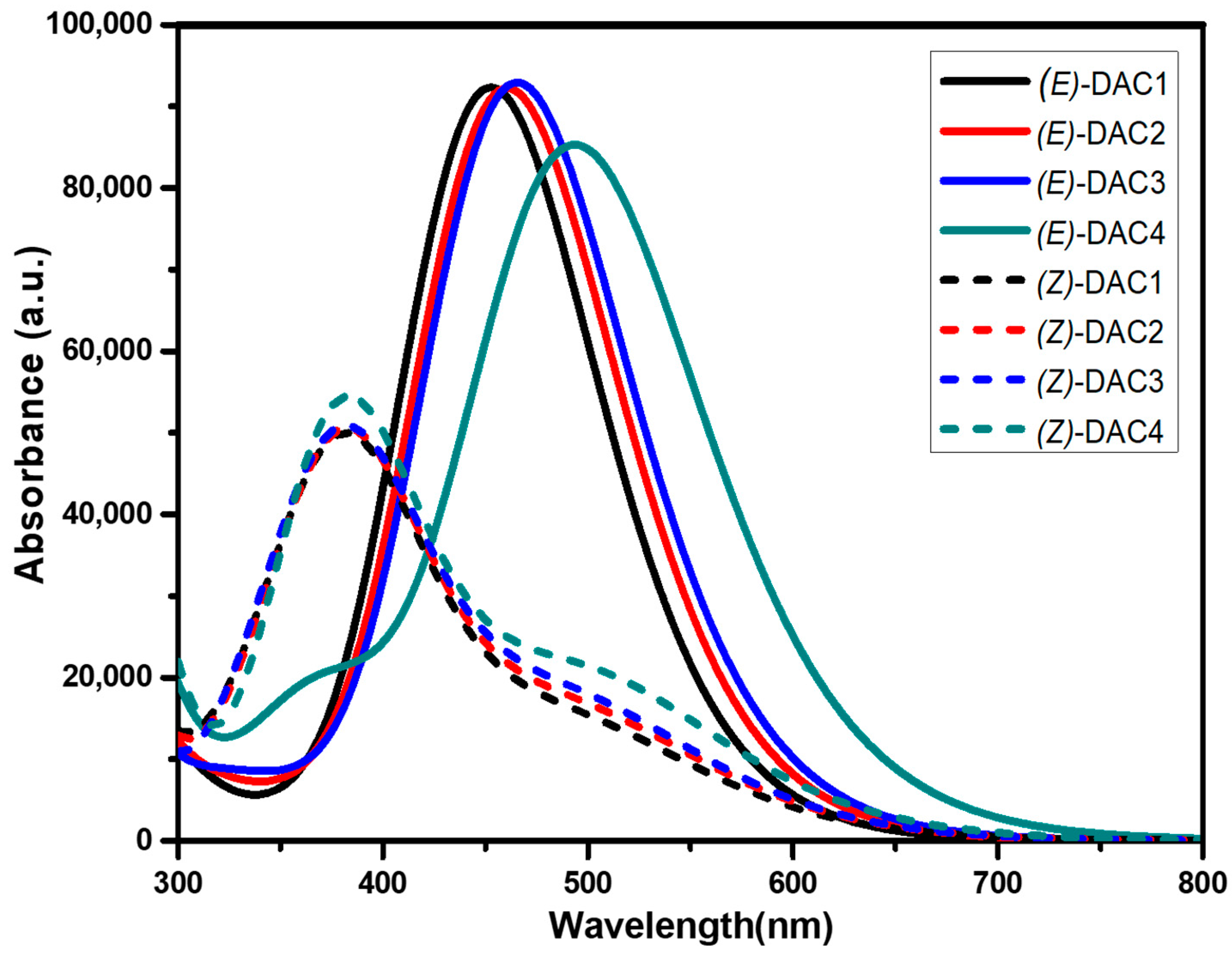
| Dyes | Excited State | Eex (λmax) | f | LHE | Transition Assignment | |||
|---|---|---|---|---|---|---|---|---|
| (E)-DAC1 | π → π* | 2.725 (455.05) | 1.721 | 0.981 | H−0→L+0 44% | H−1→L+0 14% | H−0→L+1 14% | |
| (E)-DAC2 | π → π* | 2.685 (461.75) | 2.167 | 0.993 | H−0→L+0 55% | H−1→L+0 17% | H−0→L+1 19% | |
| (E)-DAC3 | π → π* | 2.660 (466.21) | 2.253 | 0.994 | H−0→L+0 55% | H−1→L+0 19% | H−0→L+1 19% | |
| (E)-DAC4 | π → π* | 2.507 (494.51) | 2.091 | 0.992 | H−0→L+0 54% | H−1→L+0 20% | H−0→L+1 18% | |
| (Z)-DAC1 | n → π* | 2.450 (496.05) | 0.330 | 0.532 | H−0→L+0 17% | H−1→L+0 11% | H−0→L+1 27% | H−2→L+1 21% |
| π → π* | 3.156 (392.87) | 0.907 | 0.876 | H−0→L+0 22% | H−1→L+0 36% | H−1→L+1 10% | ||
| (Z)-DAC2 | n → π* | 2.490 (497.98) | 0.382 | 0.585 | H−0→L+0 16% | H−1→L+0 14% | H−0→L+1 25% | H−1→L+1 14% |
| π → π* | 3.121 (397.31) | 0.785 | 0.836 | H−0→L+0 25% | H−1→L+0 27% | H−0→L+1 13% | ||
| (Z)-DAC3 | n → π* | 2.487 (498.55) | 0.358 | 0.561 | H−0→L+0 17% | H−1→L+0 13% | H−0→L+1 26% | H−1→L+1 11% |
| π → π* | 3.134 (395.60) | 0.823 | 0.850 | H−0→L+0 24% | H−1→L+0 30% | H−0→L+1 12% | ||
| (Z)-DAC4 | n → π* | 2.453 (505.38) | 0.457 | 0.651 | H−0→L+0 17% | H−0→L+1 20% | H−2→L+0 17% | H−2→L+1 22% |
| π → π* | 3.234 (383.38) | 0.890 | 0.871 | H−0→L+0 11% | H−0→L+1 43% | H−3→L+0 27% | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashid, M.A.M.; Hayati, D.; Kwak, K.; Hong, J. Computational Investigation of Tuning the Electron-Donating Ability in Metal-Free Organic Dyes Featuring an Azobenzene Spacer for Dye-Sensitized Solar Cells. Nanomaterials 2019, 9, 119. https://doi.org/10.3390/nano9010119
Rashid MAM, Hayati D, Kwak K, Hong J. Computational Investigation of Tuning the Electron-Donating Ability in Metal-Free Organic Dyes Featuring an Azobenzene Spacer for Dye-Sensitized Solar Cells. Nanomaterials. 2019; 9(1):119. https://doi.org/10.3390/nano9010119
Chicago/Turabian StyleRashid, Md Al Mamunur, Dini Hayati, Kyungwon Kwak, and Jongin Hong. 2019. "Computational Investigation of Tuning the Electron-Donating Ability in Metal-Free Organic Dyes Featuring an Azobenzene Spacer for Dye-Sensitized Solar Cells" Nanomaterials 9, no. 1: 119. https://doi.org/10.3390/nano9010119
APA StyleRashid, M. A. M., Hayati, D., Kwak, K., & Hong, J. (2019). Computational Investigation of Tuning the Electron-Donating Ability in Metal-Free Organic Dyes Featuring an Azobenzene Spacer for Dye-Sensitized Solar Cells. Nanomaterials, 9(1), 119. https://doi.org/10.3390/nano9010119