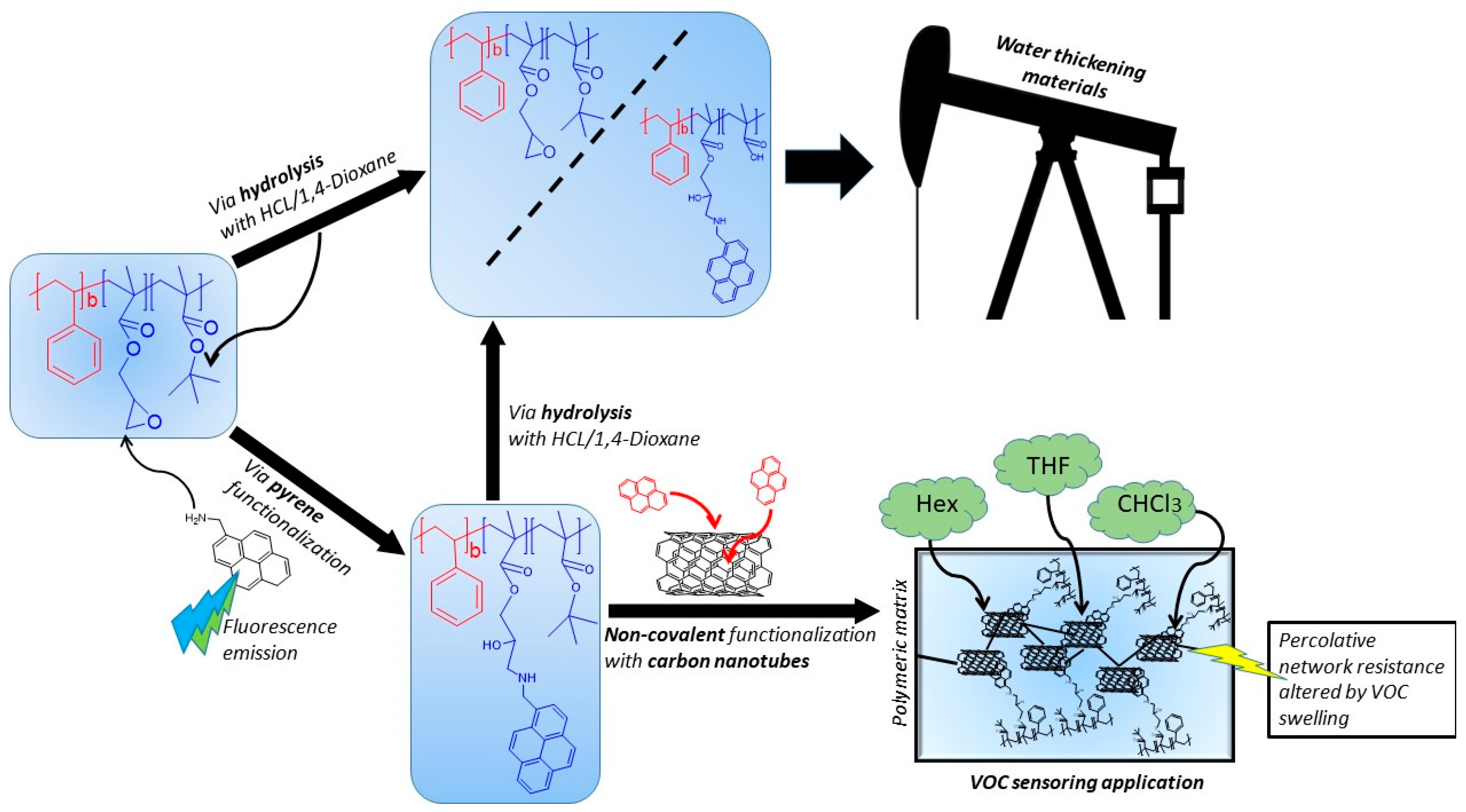
Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis




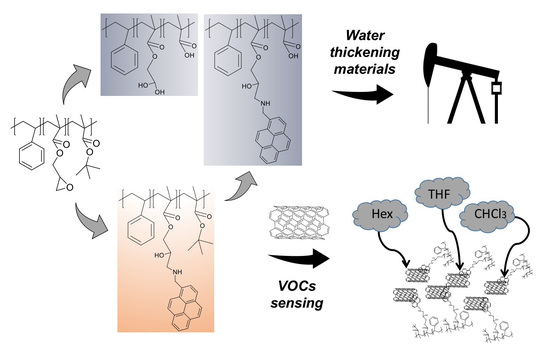
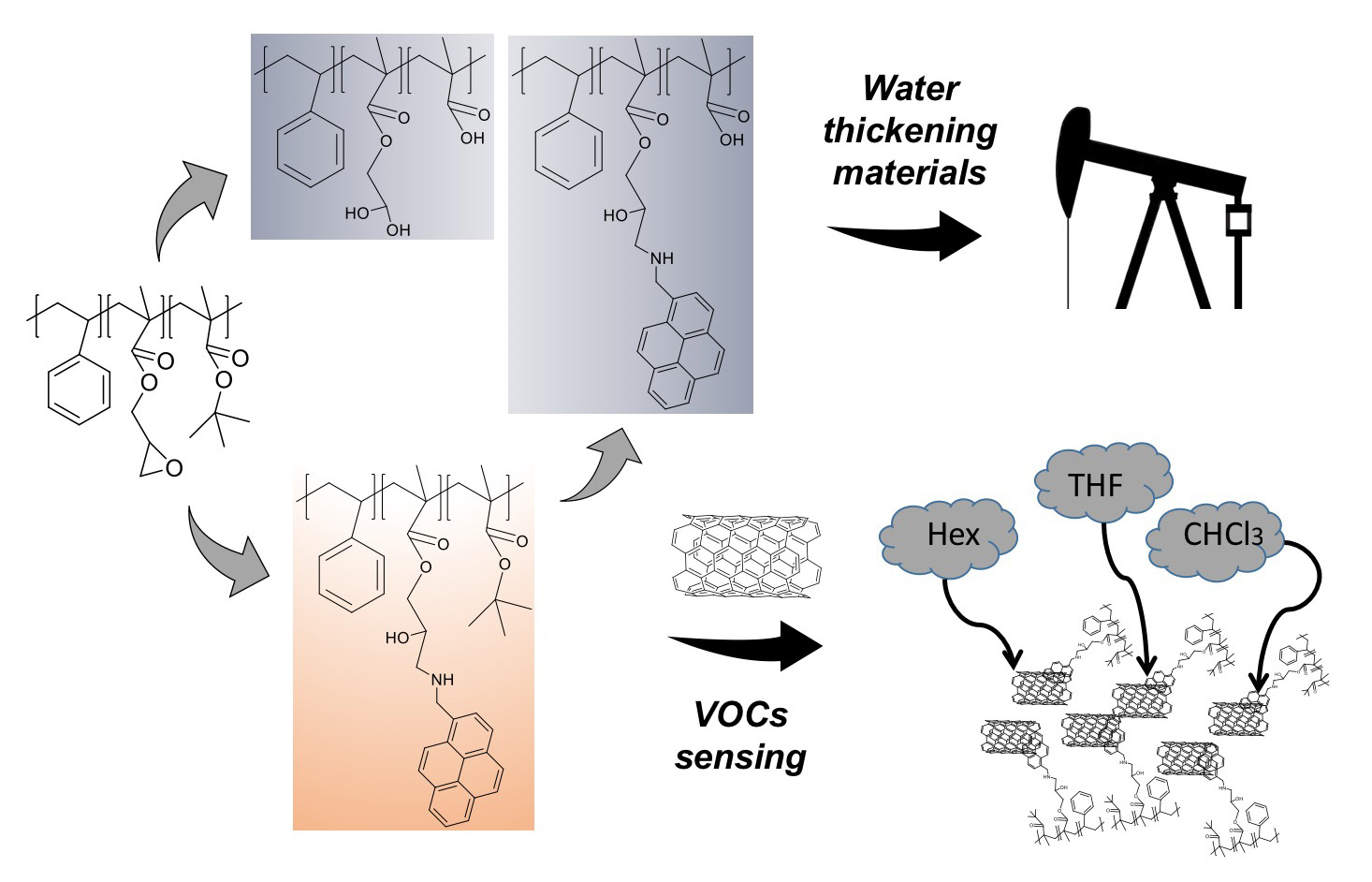
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthetic Procedure, Functionalization, Hydrolysis and Neutralization of Terpolymers
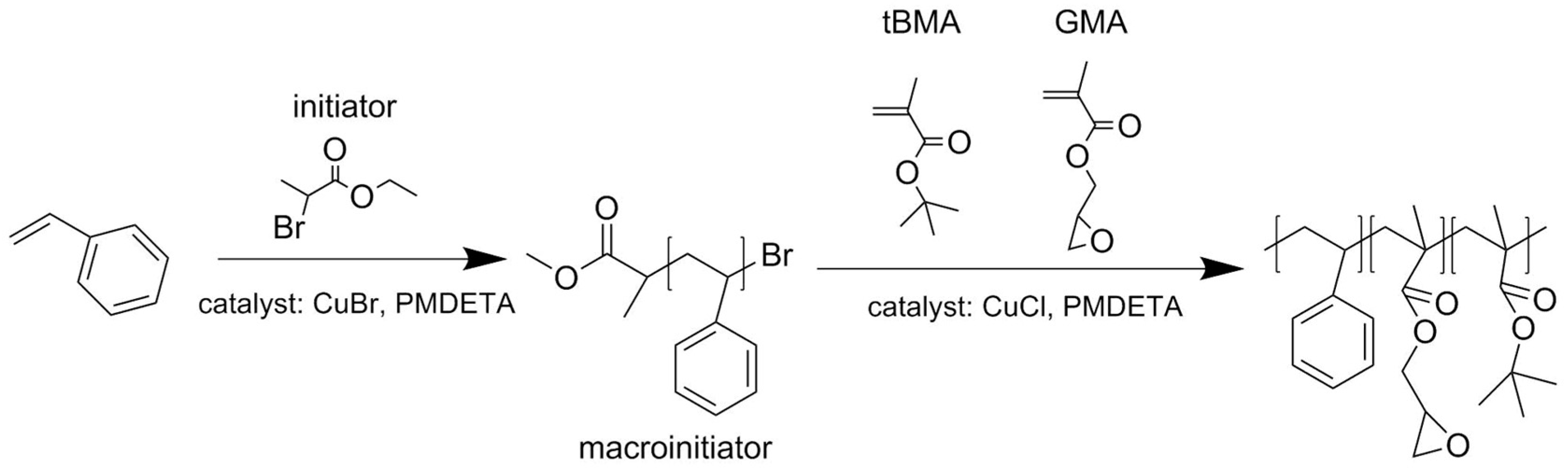
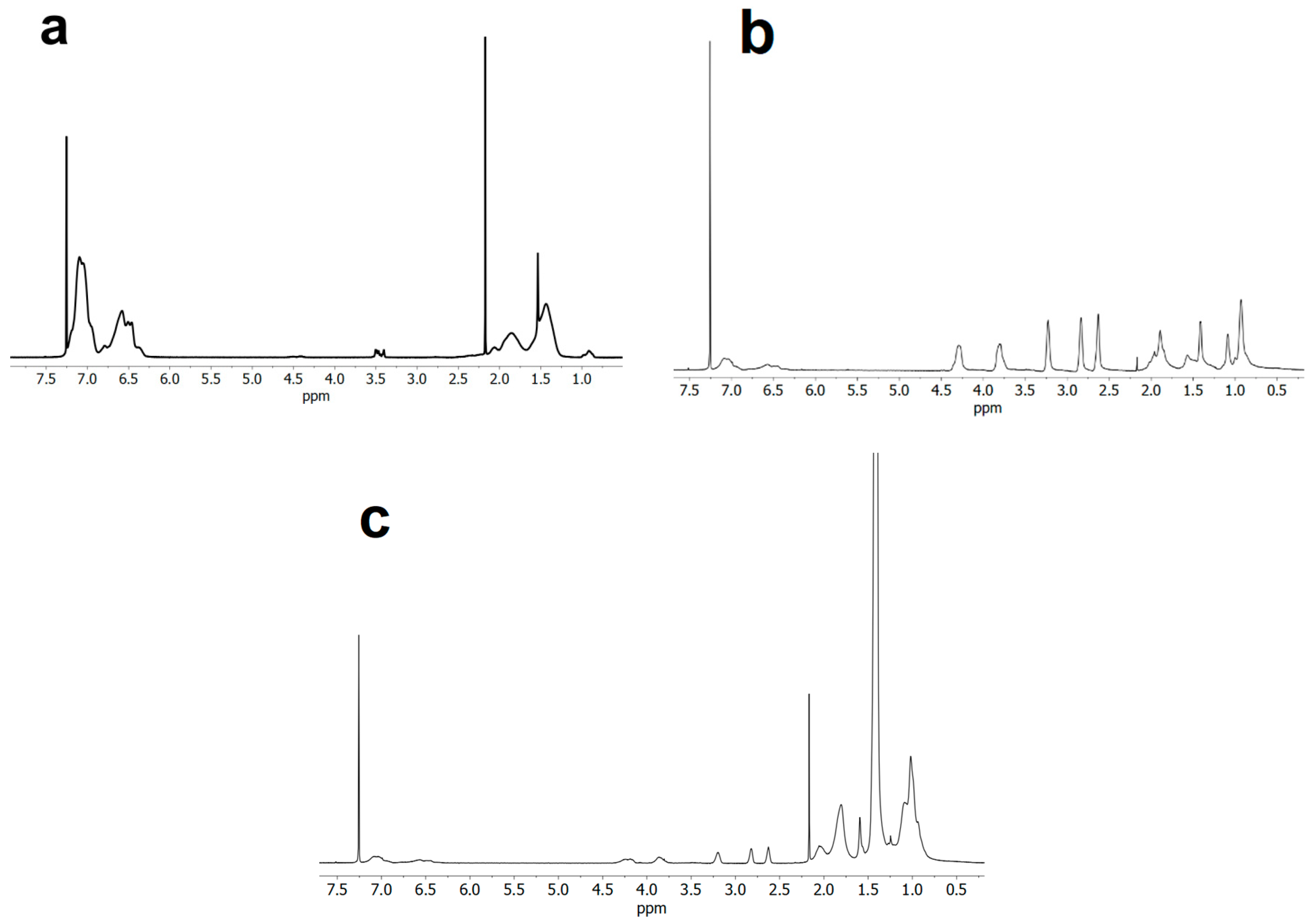
2.2.1. Synthesis of Polystyrene Macroinitiator (PS-Br)
2.2.2. Synthesis of Terpolymer polystyrene-block-(glycidyl methacrylate-co-tert-butyl methacrylate), PS-b-(GMA-r-tBMA)
2.2.3. Kinetic Experiments
2.2.4. Functionalization of PS-b-(GMA-r-tBMA)
2.2.5. Functionalization with 1-AMP
2.2.6. Hydrolysis and Neutralization of PS-b-(GMA-r-tBMA) and TP-AMP
2.3. Nanocomposite and VOC Exposure Setup Preparation
2.4. Characterization and Instruments
3. Results and Discussion
3.1. ATRP Synthesis of PS-b-(tBMA-co-GMA) Terpolymer
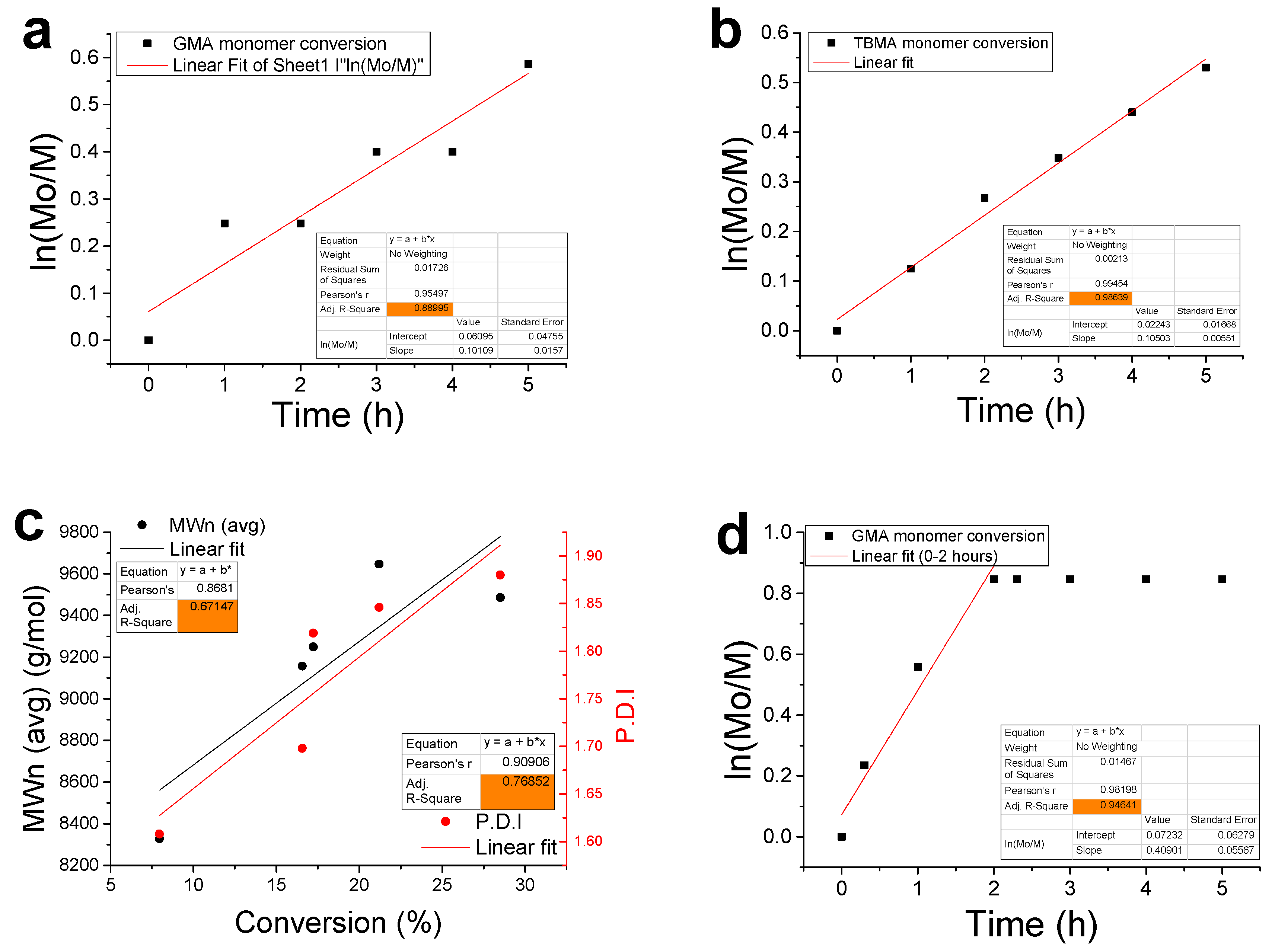
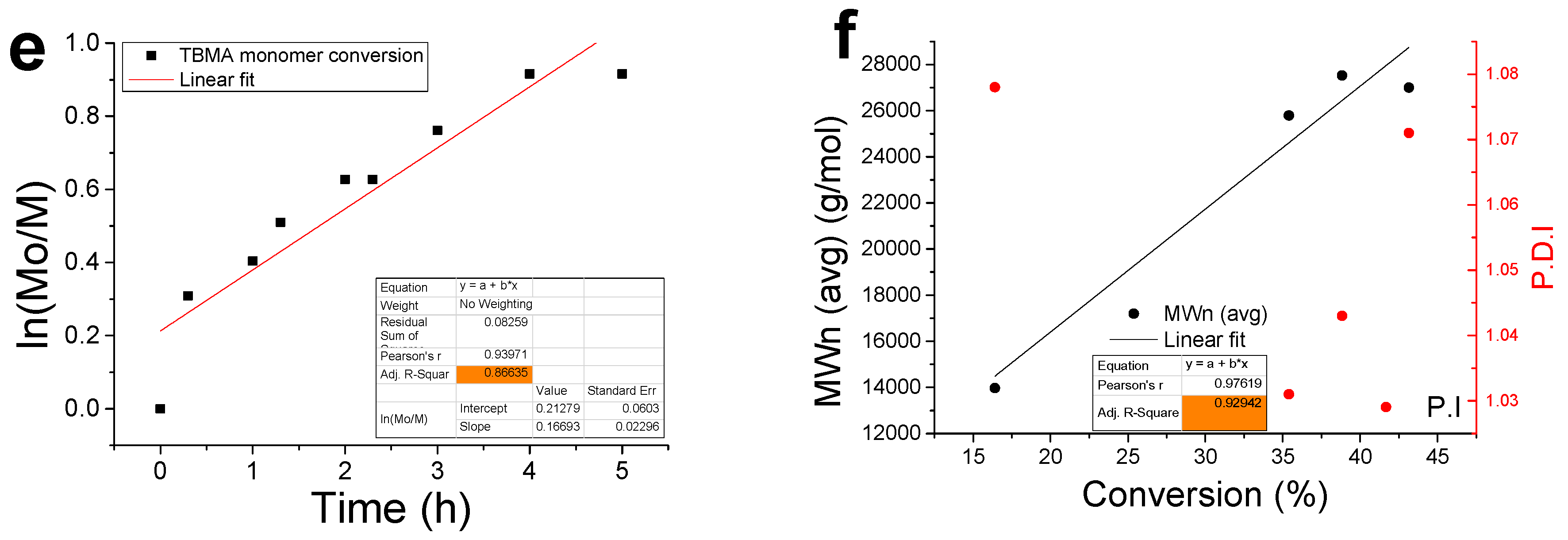
3.2. Kinetic Analysis
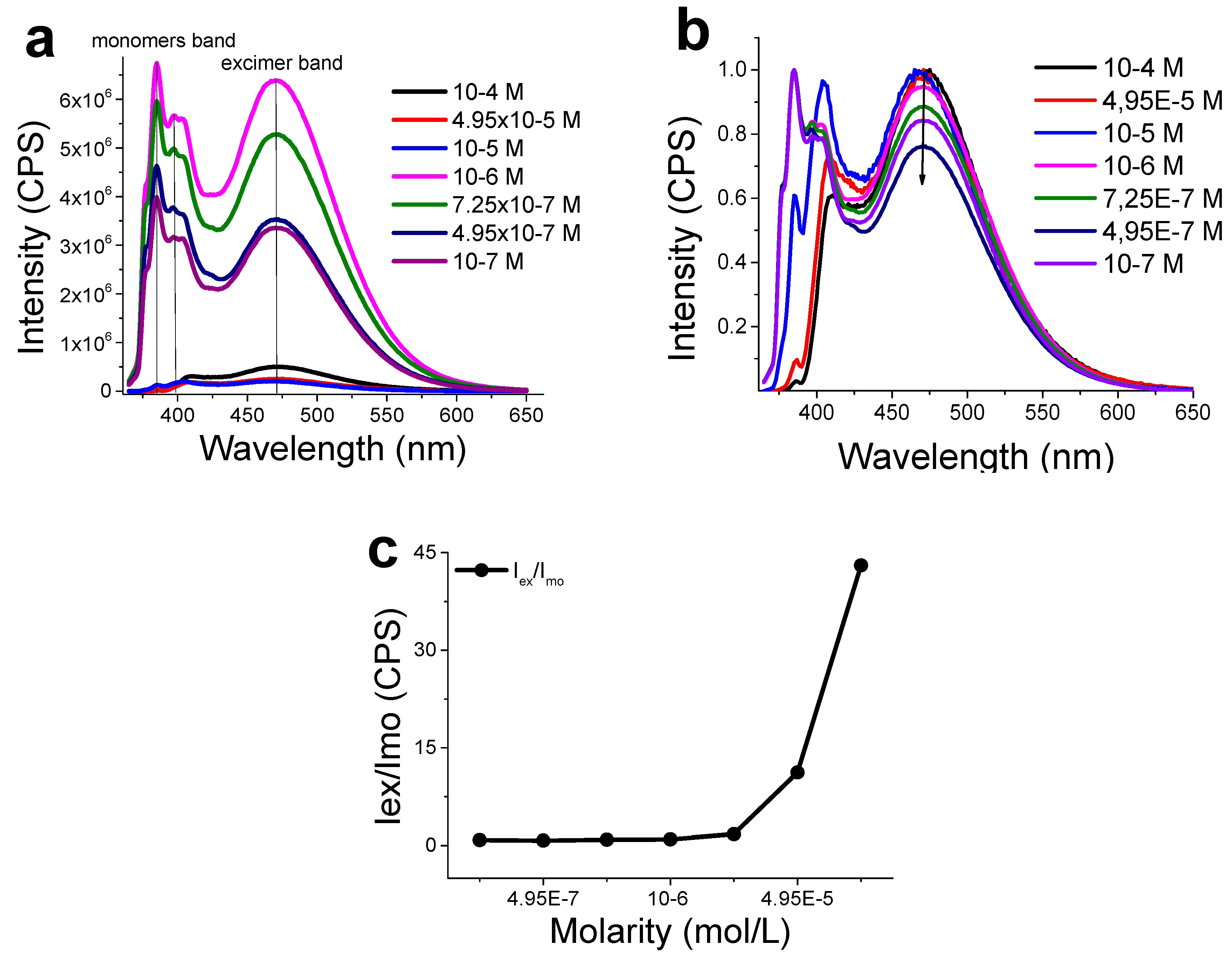
3.3. Functionalization with 1-Pyrenemethylamine (1-AMP)
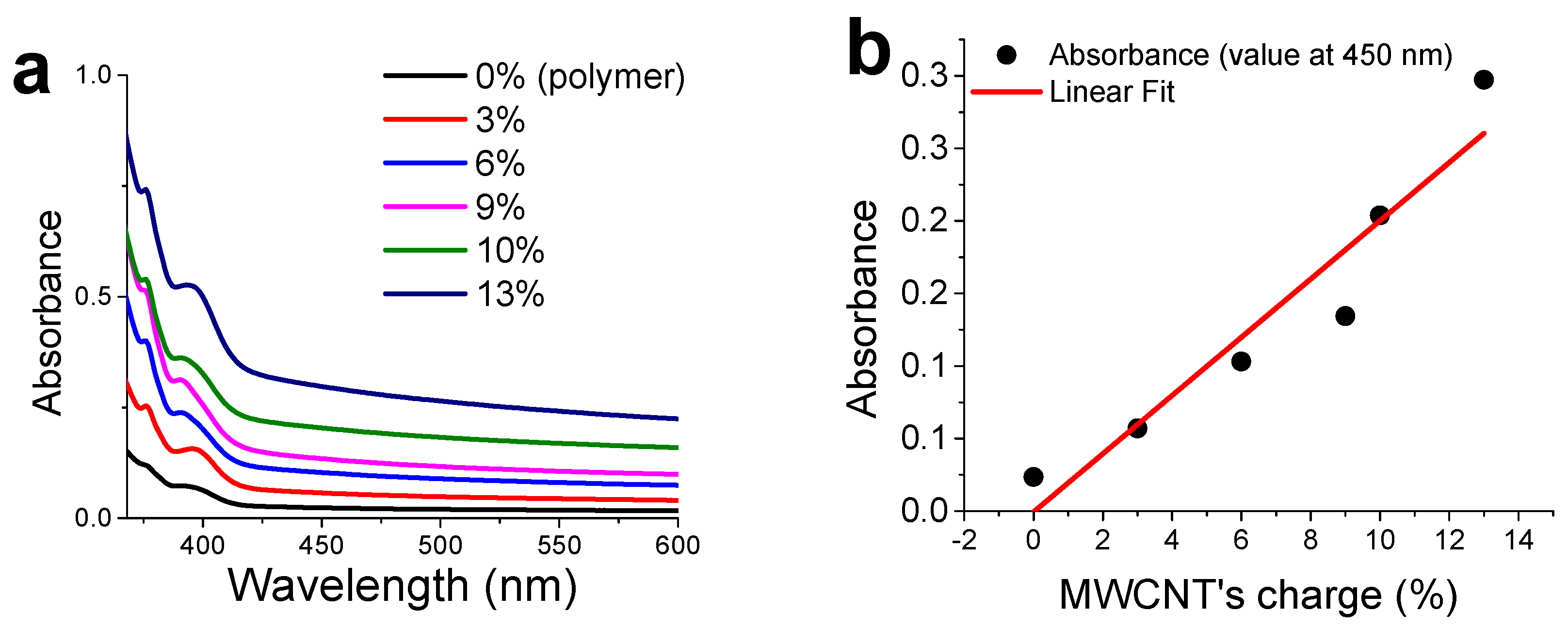
3.4. CNTs Dispersion and Stabilization by AMP-Functionalized Terpolymer
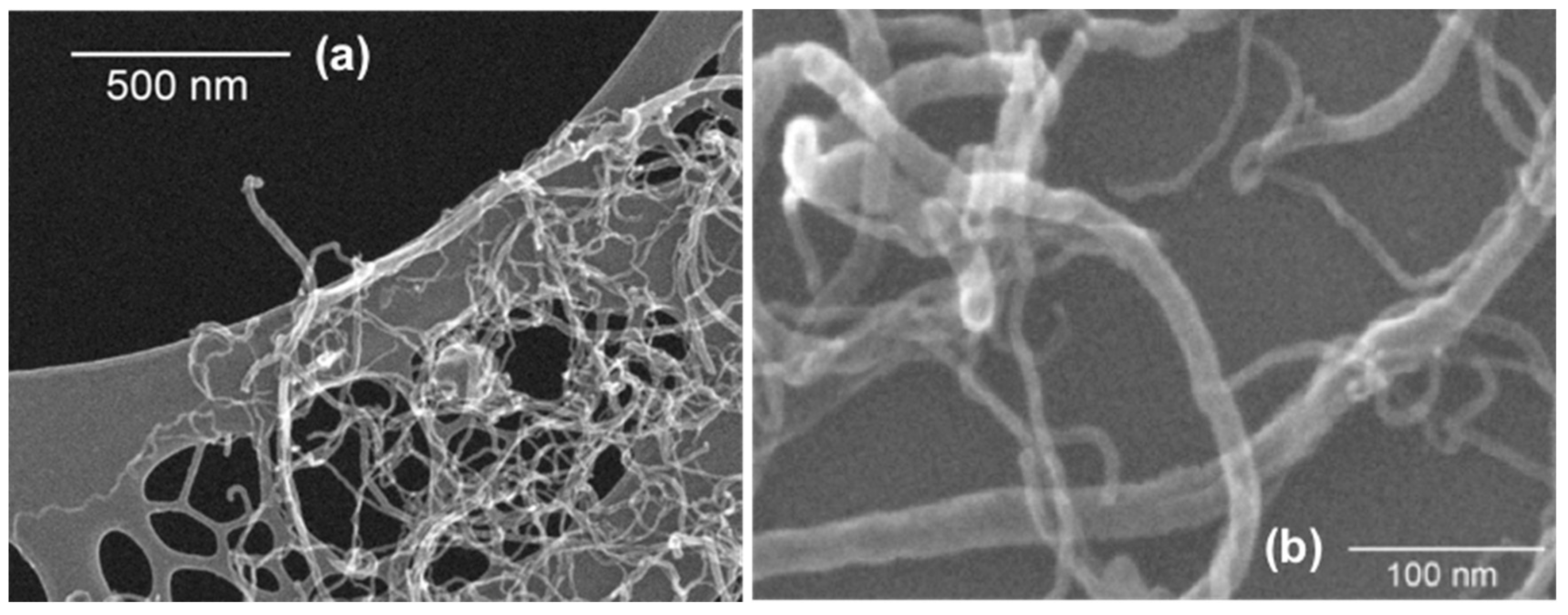
3.5. Scanning Electron Microscopy (SEM) Analysis of CNTs Dispersion
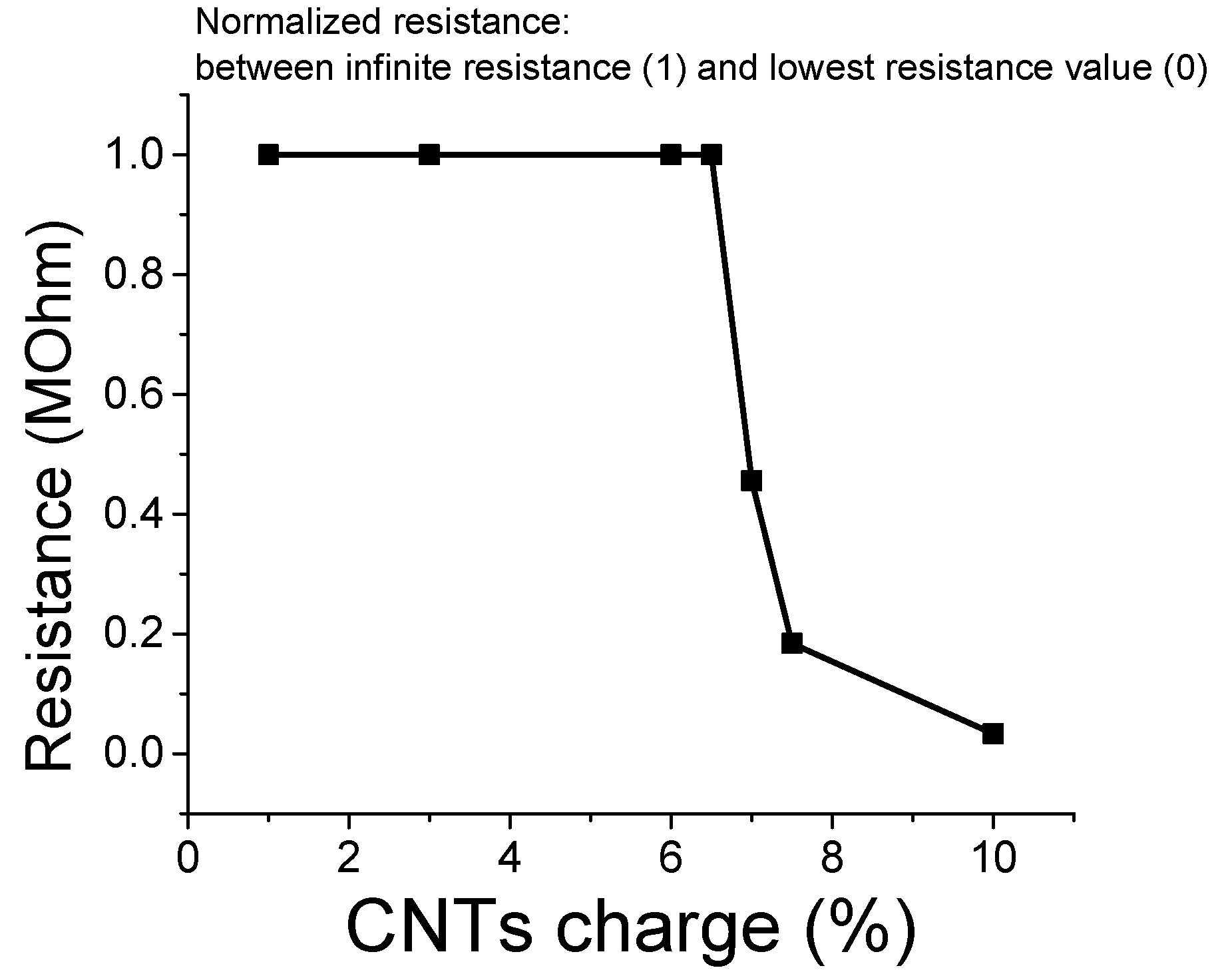
3.6. Percolation Threshold Calculation
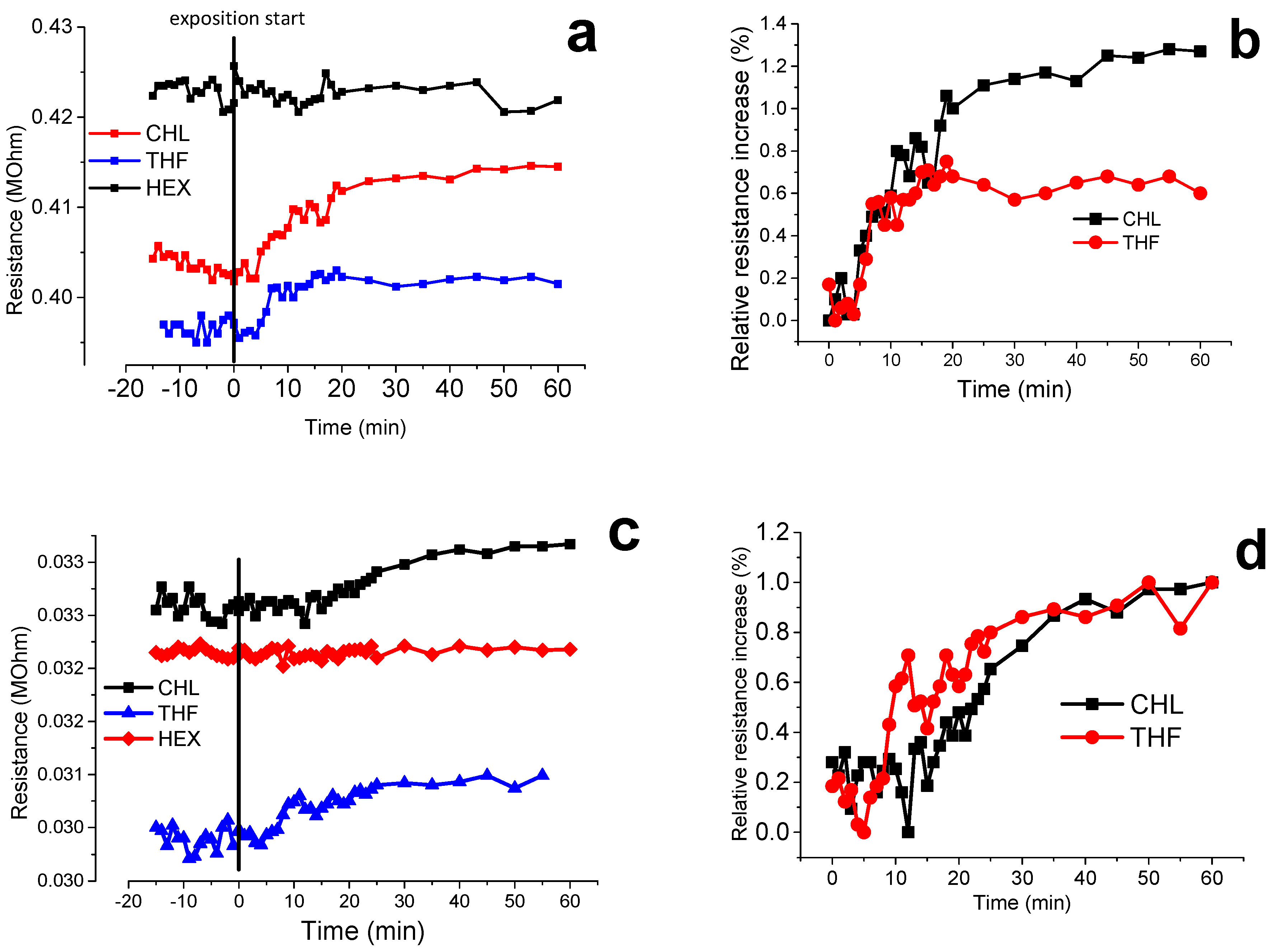
3.7. Volatile Organic Compound (VOCs) Exposure Experiments
3.8. Hydrolysis and Neutralization of TP and AMP-Functionalized Polymers
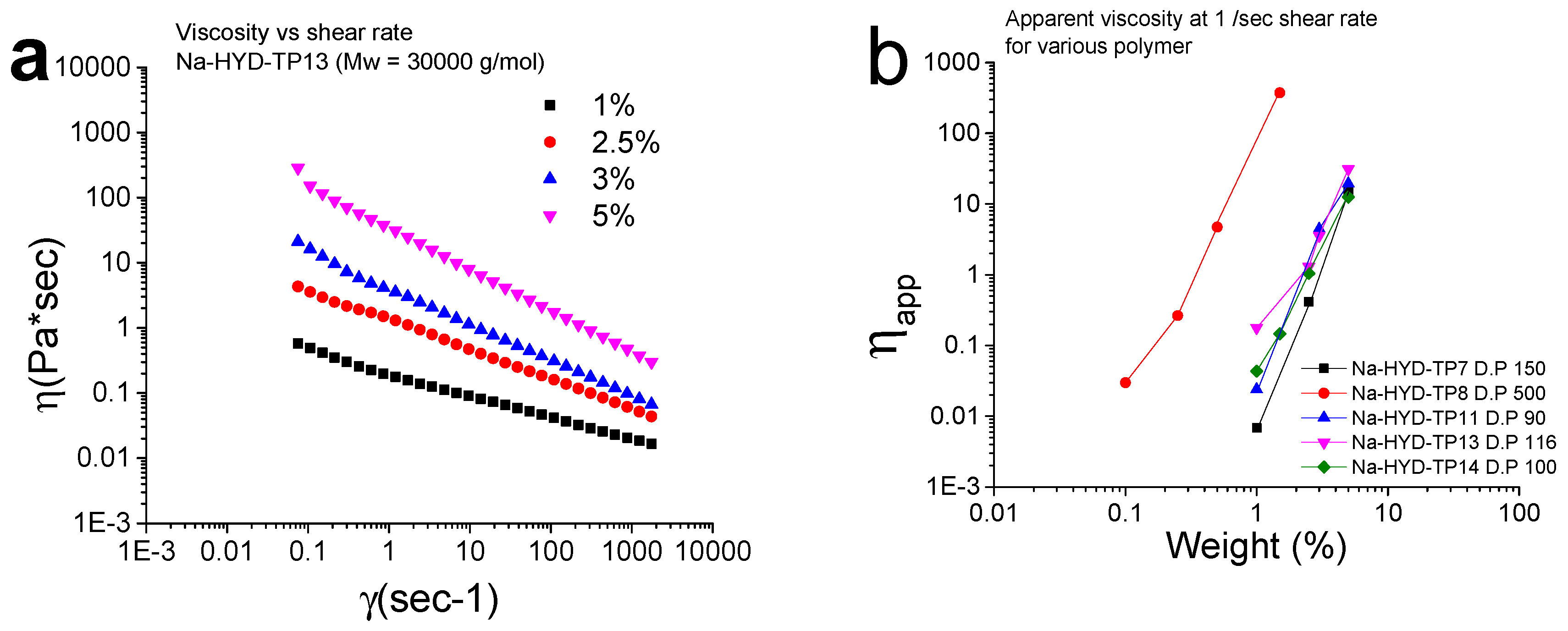
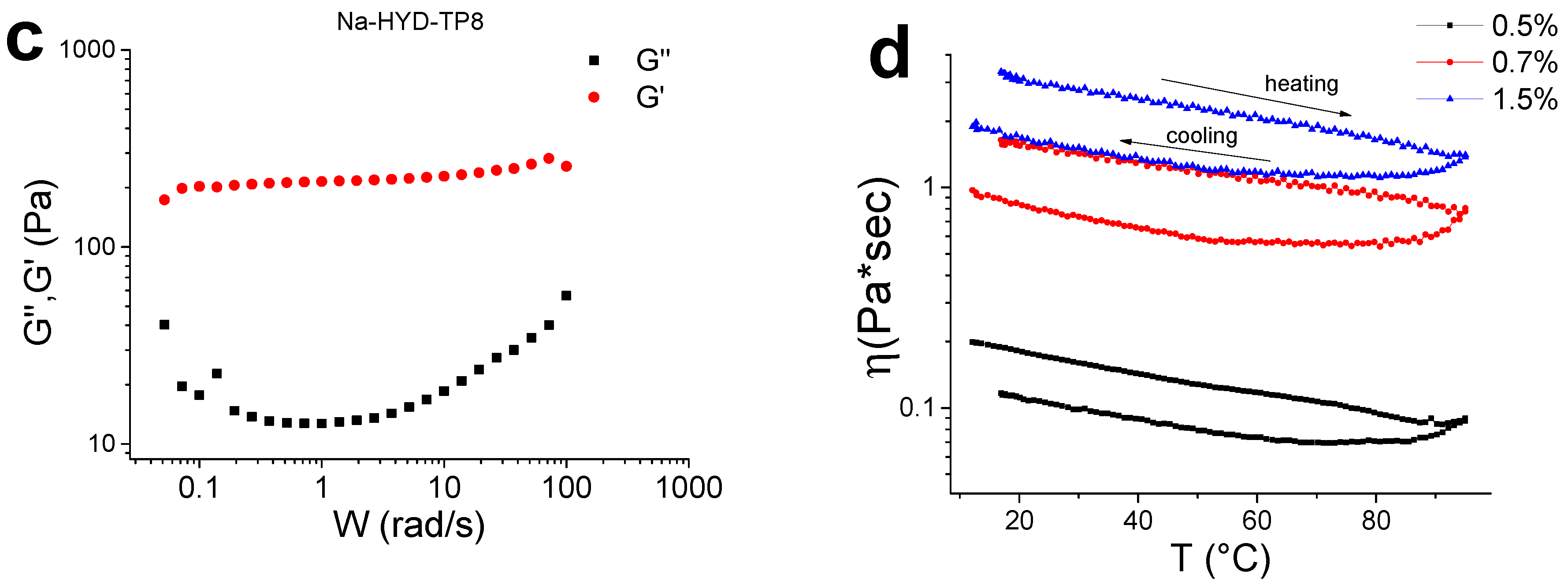
3.9. Rheological Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ganesh, V.A.; Baji, A.; Ramakrishna, S. Smart functional polymers—A new route towards creating a sustainable environment. RSC Adv. 2014, 4, 53352–53364. [Google Scholar] [CrossRef]
- Mane, S. Functional Polymers: A Review. Can. Chem. Trans. 2016, 4, 316–327. [Google Scholar]
- De las Heras Alarcón, C.; Pennadam, S.; Alexander, C. Stimuli responsive polymers for biomedical applications. Chem. Soc. Rev. 2005, 34, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ko, N.R.; Oh, J.K. Recent advances in stimuli-responsive degradable block copolymer micelles: Synthesis and controlled drug delivery applications. Chem. Commun. 2012, 48, 7542–7552. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.; Huck, W.T.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release 2008, 126, 187–204. [Google Scholar] [CrossRef]
- Bae, Y.H.; Okano, T.; Hsu, R.; Kim, S.W. Thermo-sensitive polymers as on-off switches for drug release. Die Makromol. Chemie Rapid Commun. 1987, 8, 481–485. [Google Scholar] [CrossRef]
- Raffa, P.; Wever, D.A.Z.; Picchioni, F.; Broekhuis, A.A. Polymeric surfactants: Synthesis, properties, and links to applications. Chem. Rev. 2015, 115, 8504–8563. [Google Scholar] [CrossRef]
- Grubbs, R.B.; Sun, Z. Shape-changing polymer assemblies. Chem. Soc. Rev. 2013, 42, 7436–7445. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.; Mayadunne, R.T.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition−Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP) Current Status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-polymerization modification reactions of poly(glycidyl methacrylate)s. RSC Adv. 2017, 7, 55874–55884. [Google Scholar] [CrossRef] [Green Version]
- Iwakura, Y.; Kurosaki, T.; Ariga, N.; Ito, T. Copolymerization of Methyl Methacrylate with Glycidyl Methacrylate and the Reaction of the Copolymer with Amines. Die Makromol. Chemie 1966, 97, 128–138. [Google Scholar] [CrossRef]
- Höhne, S.; Uhlmann, P. Synthesis of functional block copolymers and terpolymers containing polyglycidyl methacrylate blocks. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 675–684. [Google Scholar] [CrossRef]
- Kalal, J.; Švec, F.; Maroušek, V. Reactions of epoxide groups of glycidyl methacrylate copolymers. J. Polym. Sci. Polym. Symp. 2007, 47, 155–166. [Google Scholar] [CrossRef]
- Durmaz, H.; Dag, A.; Tunca, U.; Hizal, G. Synthesis and characterization of pyrene bearing amphiphilic miktoarm star polymer and its noncovalent interactions with multiwalled carbon nanotubes. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2406–2414. [Google Scholar] [CrossRef]
- Gao, H.; Jones, M.-C.; Tewari, P.; Ranger, M.; Leroux, J.-C. Star-shaped alkylated poly(glycerol methacrylate) reverse micelles: Synthesis and evaluation of their solubilizing properties in dichloromethane. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2425–2435. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Z.; Zhang, J.; Li, Z.; Gao, Y.; Wang, C.; Zhang, H.; Yang, B. Multifunctional nanoparticles/silica microsphere assemblies using polyglycidyl methacrylate shells as supports. J. Colloid Interface Sci. 2009, 339, 83–90. [Google Scholar] [CrossRef]
- Dong, X.; Zheng, Y.; Huang, Y.; Chen, X.; Jing, X. Synthesis and characterization of multifunctional poly(glycidyl methacrylate) microspheres and their use in cell separation. Anal. Biochem. 2010, 405, 207–212. [Google Scholar] [CrossRef]
- Kocak, G.; Solmaz, G.; Dikmen, Z.; Bütün, V. Preparation of Cross-Linked Micelles from Glycidyl Methacrylate Based Block Copolymers and Their Usages as Nanoreactors in the Preparation of Gold Nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 514–526. [Google Scholar] [CrossRef]
- Bains, G.; Patel, A.B.; Narayanaswami, V. Pyrene: A Probe to Study Protein Conformation and Conformational Changes. Molecules 2011, 16, 7909–7935. [Google Scholar] [CrossRef] [PubMed]
- Bauhofer, W.; Kovacs, J.Z. A review and analysis of electrical percolation in carbon nanotube polymer composites. Compos. Sci. Technol. 2009, 69, 1486–1498. [Google Scholar] [CrossRef] [Green Version]
- Petrov, P.; Stassin, F.; Pagnoulle, C.; Jérôme, R. Noncovalent functionalization of multi-walled carbon nanotubes by pyrene containing polymers. Chem. Commun. 2003, 0, 2904–2905. [Google Scholar] [CrossRef]
- Meuer, S.; Braun, L.; Schilling, T.; Zentel, R. α-Pyrene polymer functionalized multiwalled carbon nanotubes: Solubility, stability and depletion phenomena. Polymer (Guildf) 2009, 50, 154–160. [Google Scholar] [CrossRef]
- Bahun, G.J.; Wang, C.; Adronov, A. Solubilizing single-walled carbon nanotubes with pyrene-functionalized block copolymers. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 1941–1951. [Google Scholar] [CrossRef]
- Parikh, K.; Cattanach, K.; Rao, R.; Suh, D.-S.; Wu, A.; Manohar, S.K. Flexible vapour sensors using single walled carbon nanotubes. Sensors Actuators B Chem. 2006, 113, 55–63. [Google Scholar] [CrossRef]
- Zhao, J.; Buldum, A.; Han, J.; Lu, J.P. Gas molecule adsorption in carbon nanotubes and nanotube bundles. Nanotechnology 2002, 13, 195. [Google Scholar] [CrossRef]
- Kong, J.; Chapline, M.G.; Dai, H. Functionalized Carbon Nanotubes for Molecular Hydrogen Sensors. Adv. Mater. 2011, 13, 1384–1386. [Google Scholar] [CrossRef]
- Raffa, P.; Brandenburg, P.; Wever, D.A.Z.; Broekhuis, A.A.; Picchioni, F. Polystyrene-poly(sodium methacrylate) amphiphilic block copolymers by ATRP: Effect of structure, pH, and ionic strength on rheology of aqueous solutions. Macromolecules 2013, 46, 7106–7111. [Google Scholar] [CrossRef]
- Raffa, P.; Stuart, M.C.A.; Broekhuis, A.A.; Picchioni, F. The effect of hydrophilic and hydrophobic block length on the rheology of amphiphilic diblock Polystyrene-b-Poly(sodium methacrylate) copolymers prepared by ATRP. J. Colloid Interface Sci. 2014, 428, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Meijerink, M.; van Mastrigt, F.; Franken, L.E.; Stuart, M.C.A.; Picchioni, F.; Raffa, P. Triblock copolymers of styrene and sodium methacrylate as smart materials: Synthesis and rheological characterization. Pure Appl. Chem. 2017, 89, 1641–1658. [Google Scholar] [CrossRef]
- Raffa, P.; Broekhuis, A.A.; Picchioni, F. Polymeric surfactants for enhanced oil recovery: A review. J. Pet. Sci. Eng. 2016, 145, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Tsarevsky, N.V.; Jakubowski, W. Atom transfer radical polymerization of functional monomers employing Cu-based catalysts at low concentration: Polymerization of glycidyl methacrylate. J. Polym. Sci. Pol. Chem. 2011, 49, 918–925. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Polymer molecular weight analysis by1H NMR spectroscopy. J. Chem. Educ. 2011, 88, 1098–1104. [Google Scholar] [CrossRef]
- Wang, T.-L.; Liu, Y.-Z.; Jeng, B.-C.; Cai, Y.-C. The Effect of Initiators and Reaction Conditions on the Polymer Syntheses by Atom Transfer Radical Polymerization. J. Polym. Res. 2005, 12, 67–75. [Google Scholar] [CrossRef]
- Chakraborti, A.K.; Rudrawar, S.; Kondaskar, A. An efficient synthesis of 2-amino alcohols by silica gel catalysed opening of epoxide rings by amines. Org. Biomol. Chem. 2004, 2, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Haldar, S.; Chattopadhyay, A. Organization and dynamics in micellar structural transition monitored by pyrene fluorescence. Biochem. Biophys. Res. Commun. 2009, 390, 728–732. [Google Scholar] [CrossRef]
- Numata, Y.; Nirasawa, T.; Suzuka, I. Excited states of pyrene excimer observed by photodissociation spectroscopy in a supersonic jet. J. Photochem. Photobiol. A Chem. 2010, 209, 27–31. [Google Scholar] [CrossRef]
- Kathiravan, A.; Sundaravel, K.; Jaccob, M.; Dhinagaran, G.; Rameshkumar, A.; Arul Ananth, D.; Sivasudha, T. Pyrene Schiff Base: Aggregation Induced Emission, and Antimicrobial Properties. J. Phys. Chem. B 2014, 118, 13573–13581. [Google Scholar] [CrossRef]
- De Halleux, V.; Mamdouh, W.; De Feyter, S.; De Schryver, F.; Levin, J.; Geerts, Y.H. Emission properties of a highly fluorescent pyrene dye in solution and in the liquid state. J. Photochem. Photobiol. A Chem. 2006, 178, 251–257. [Google Scholar] [CrossRef]
- Bains, G.K.; Kim, S.H.; Sorin, E.J.; Narayanaswami, V. The extent of pyrene excimer fluorescence emission is a reflector of distance and flexibility: Analysis of the segment linking the LDL receptor-binding and tetramerization domains of apolipoprotein E3. Biochemistry 2012, 51, 6207–6219. [Google Scholar] [CrossRef]
- Saltiel, C.; Manickavasagam, S.; Mengüc, M.P.; Andrews, R. Light-scattering and dispersion behavior of multiwalled carbon nanotubes. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2005, 22, 1546–1554. [Google Scholar] [CrossRef]
- Malhofer, A.; Rother, M.; Zakharko, Y.; Graf, A.; Schießl, S.P.; Zaumseil, J. Direct visualization of percolation paths in carbon nanotube/polymer composites. Org. Electron. Phys. Mater. Appl. 2017, 45, 151–158. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Winey, K.I. Polymer nanocomposites containing carbon nanotubes. Macromolecules 2006, 39, 5194–5205. [Google Scholar] [CrossRef]
- Hsu, J.C.; Cao, W.; Yang, F.; Yang, T.J.; Lee, S. Absorption behavior of poly(methyl methacrylate)-multiwalled carbon nanotube composites: Effects of UV irradiation. Phys. Chem. Chem. Phys. 2017, 19, 7359–7369. [Google Scholar] [CrossRef]
- Vayer, M.; Vital, A.; Sinturel, C. New insights into polymer-solvent affinity in thin films. Eur. Polym. J. 2017, 93, 132–139. [Google Scholar] [CrossRef]
- Paoletti, C.; He, M.; Salvo, P.; Melai, B.; Calisi, N.; Mannini, M.; Cortigiani, B.; Bellagambi, F.G.; Swager, T.M.; Di Francesco, F.; et al. Room temperature amine sensors enabled by sidewall functionalization of single-walled carbon nanotubes. RSC Adv. 2018, 8, 5578–5585. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, B.; Holtzer, M. Structural Examination of The Cross-Linking Reaction Mechanism of Polyacrylate Binding Agents. Arch. Metall. Mater. 2009, 54, 427–437. [Google Scholar]
- Kimerling, A.S.; Rochefort, W.E.; Bhatia, S.R. Rheology of Block Polyelectrolyte Solutions and Gels: A Review. Ind. Eng. Chem. Res. 2006, 45, 6885–6889. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | [Sty]:[I]:[C]:[L] | Sty (mL) | Solvent (mL) | Mn 1 (g/mol) | Time (h) | Yield (%) | Sty Unit | PDI |
|---|---|---|---|---|---|---|---|---|
| PS1 | 30:1:1:1 | 20 | Bulk | 3700 | 1.5 | 55 | 33 | 1.1 |
| PS2 | 30:1:1:1 | 40 | 20 (toluene) | 2500 | 3 | 45 | 21 | 1.1 |
| Polymer | Molar Ratio 1 | Solvent (Anisole) Volume % | Time (h) | T (°C) | Mn (GPC) g/mol | Mn (NMR) g/mol | PDI 2 | Sty-GMA-TBMA Units |
|---|---|---|---|---|---|---|---|---|
| TP1 | 1:1:1:270:30 | 25 | 18 | 90 | 26,400 | 23,000 | 1.2 | 33-27-239 |
| TP2 | 1:1:1:270:30 | 25 | 4 | 90 | 18,700 | 41,300 | 1.6 | 33-23-155 |
| TP3 | 1:1:1:210:90 | 25 | 2 | 90 | 11,800 | 45,500 | 1.85 | 33-99-197 |
| TP4 | 1:1:1:210:90 | 25 | 1 | 90 | 14,700 | 17,200 | 1.59 | 33-52-45 |
| TP5 | 1:1:1:210:90 | 25 | 0.5 | 90 | 11,300 | 19,650 | 1.47 | 33-21-39 |
| TP6 | 1:1:1:270:30 | 50 | 48 | 30 | 12,900 | 20,200 | 1.36 | 33-109-9 |
| TP7 | 1:1:1:270:30 | 50 | 5 | 60 | 35,100 | 29,700 | 1.09 | 33-33-152 |
| TP8 | 1:1:1:270:30 | 50 | 15 | 60 | 31,900 | 118,000 | 1.13 | 33-57-531 |
| TP9 | 1:1:1:210:90 | 50 | 5 | 60 | 29,600 | 12,400 | 1.25 | 33-37-26 |
| TP10 | 1:1:1:255:45 | 50 | 5 | 60 | 24,400 | 27,150 | 1.1 | 33-23-144 |
| TP11 | 1:1:1:105:45 | 50 | 10 | 60 | 15,400 | 18,700 | 1.67 | 33-31-85 |
| TP12 | 1:1:1:210:90 | 50 | 8 | 60 | 30,700 | 48,900 | 1.15 | 33-133-196 |
| TP13 | 1:1:1:210:90 | 50 | 8 | 60 | 32,500 | 27,450 | 1.12 | 33-63-116 |
| TP14 | 1:1:1:210:90 | 50 | 5 | 60 | 30,900 | 24,500 | 1.14 | 33-49-106 |
| Sample | GMA Molar Amount in the Polymer | AMP: Polymer (Molar Ratio) | SiO2 w/w on Polymer | Reaction Time (h) |
|---|---|---|---|---|
| TP9-PYR(0) | 36% | 1.0 | 0% | 48 1 |
| TP9-PYR (1) | 36% | 2.5 | 0% | 48 1 |
| TP7-PYR (2) | 16% | 1.5 | 10% | 48 |
| TP9-PYR(3) | 39% | 1.5 | 5% | 24 |
| TP11-PYR(4) | 24% | 1.0 | 10% | 24 |
| TP11-PYR(5) | 24% | 1.0 | 7.5% | 48 |
| TP9-PYR(6) | 39% | 1.0 | 7.5% | 48 |
| TP13-PYR(7) | 31% | 1.0 | 7.5% | 48 |
| TP13-PYR(8) | 31% | 1.0 | 7.5% | 48 |
| TP14-PYR(9) | 28% | 1.0 | 5% | 48 |
| Sample | CNTs Feed (%) | Average Residue (%) | CNTs Average Effective Charge (%) |
|---|---|---|---|
| TP9-PYR(6) | 0% | 7.3 | 0 |
| D3 | 3% | 9.8 | 2.4 |
| D6 | 6% | 13.0 | 5.6 |
| D75 | 7.5% | 13.9 | 6.5 |
| D8 | 8% | 14.9 | 7.5 |
| D9 | 9% | 16.0 | 8.5 |
| D10 | 10% | 15.9 | 8.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Sacco, F.; Pucci, A.; Raffa, P. Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis. Nanomaterials 2019, 9, 458. https://doi.org/10.3390/nano9030458
Di Sacco F, Pucci A, Raffa P. Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis. Nanomaterials. 2019; 9(3):458. https://doi.org/10.3390/nano9030458
Chicago/Turabian StyleDi Sacco, Federico, Andrea Pucci, and Patrizio Raffa. 2019. "Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis" Nanomaterials 9, no. 3: 458. https://doi.org/10.3390/nano9030458
APA StyleDi Sacco, F., Pucci, A., & Raffa, P. (2019). Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis. Nanomaterials, 9(3), 458. https://doi.org/10.3390/nano9030458