The Stability of a Nanoparticle Diamond Lattice Linked by DNA



Abstract
:1. Introduction
2. Materials and Methods
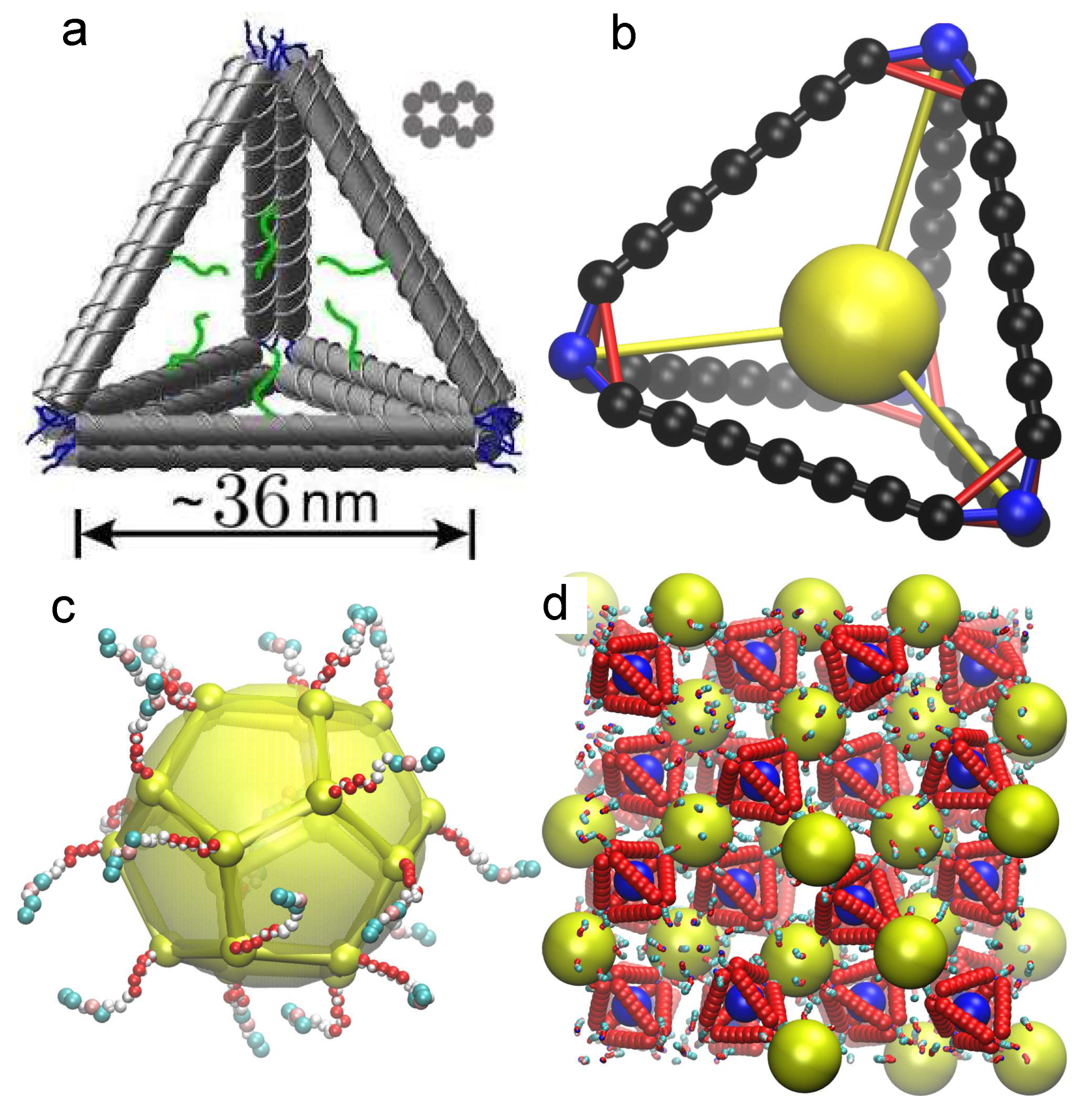
2.1. Tetrahedral Cage Model
2.2. Nanoparticle Model
2.3. DNA Model
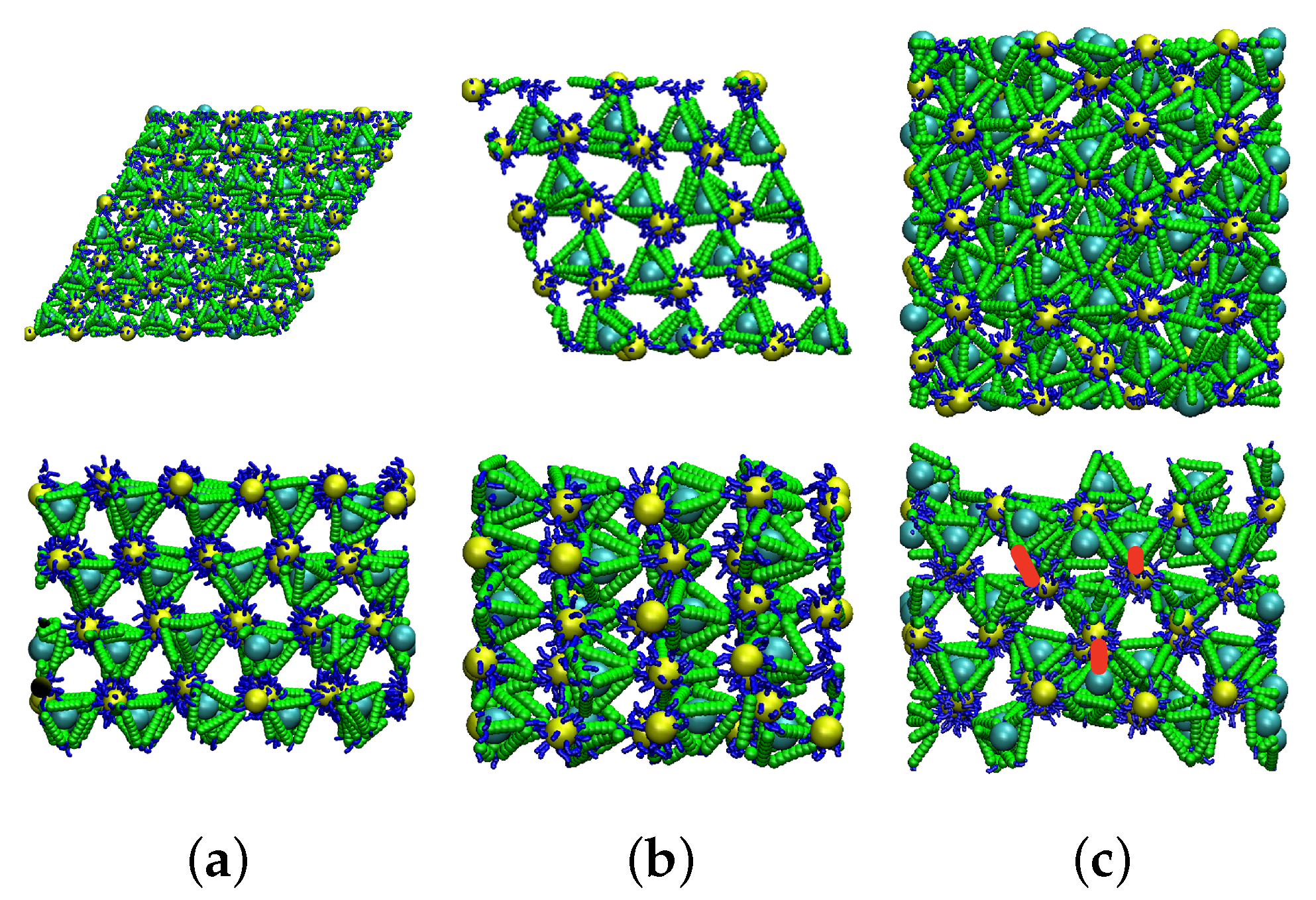
2.4. Simulations and Lattice Preparation
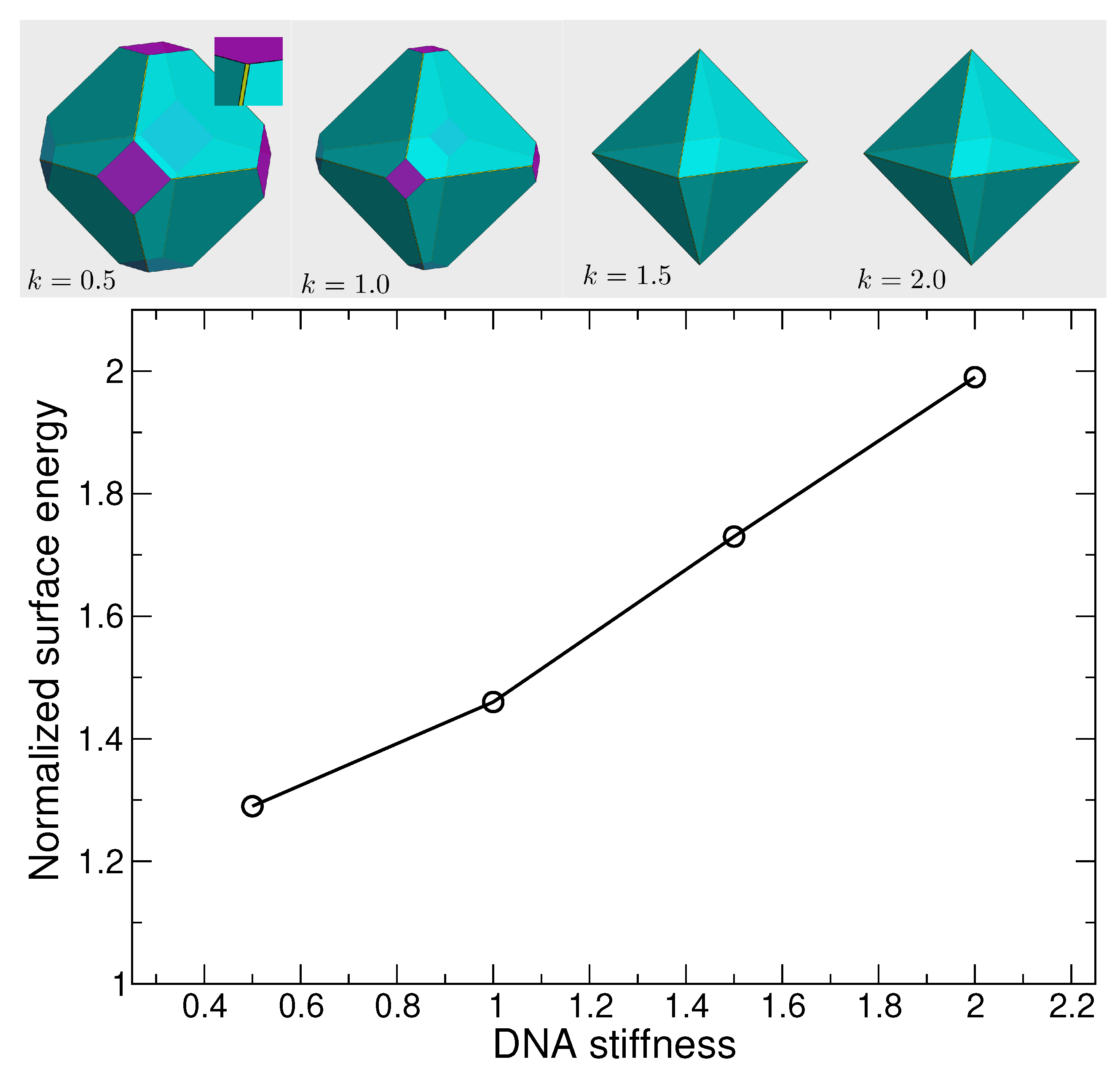
2.5. Surface Energy Calculation
3. Results & Discussion
3.1. Crystallite Shape from the Wulff Construction
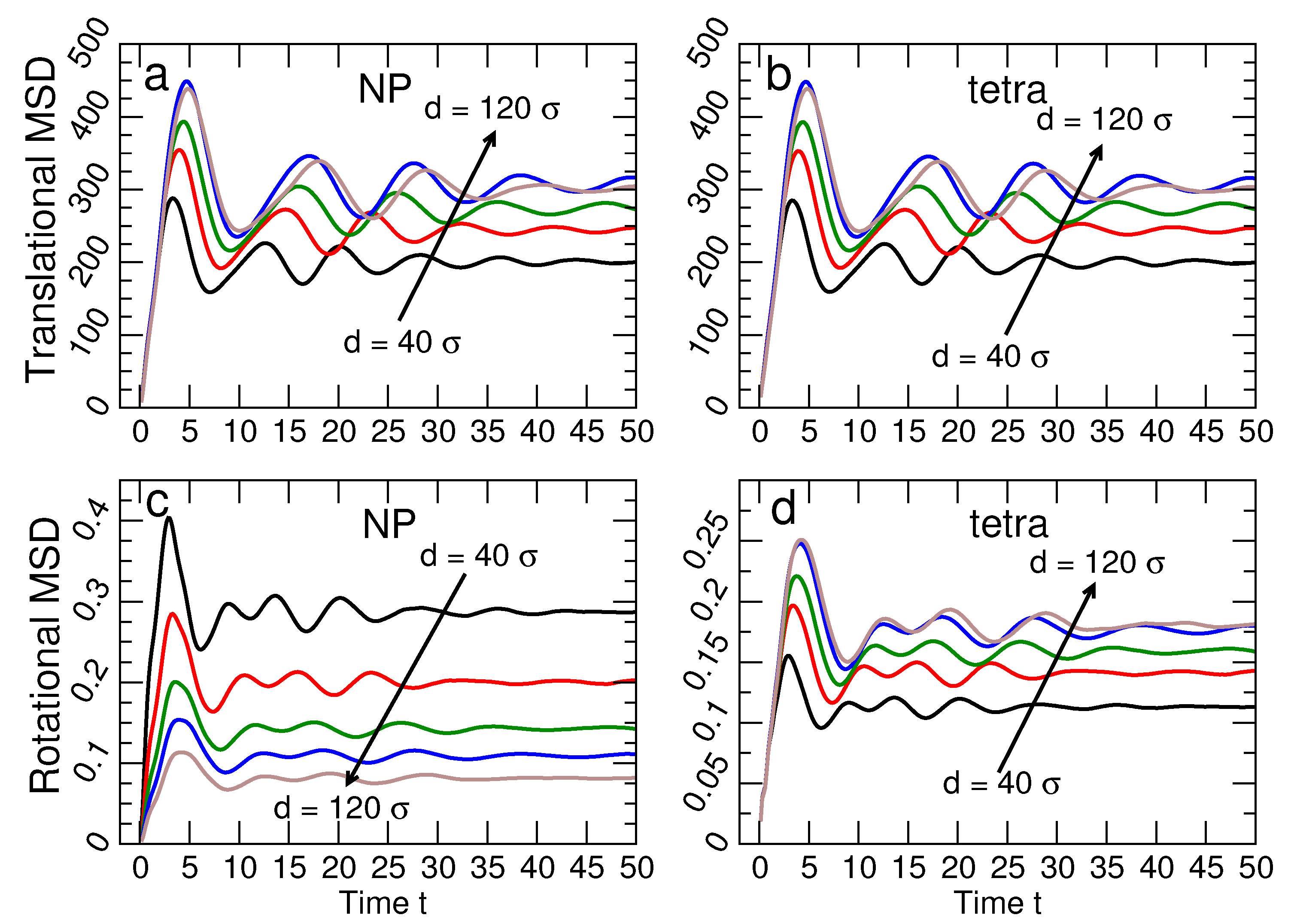
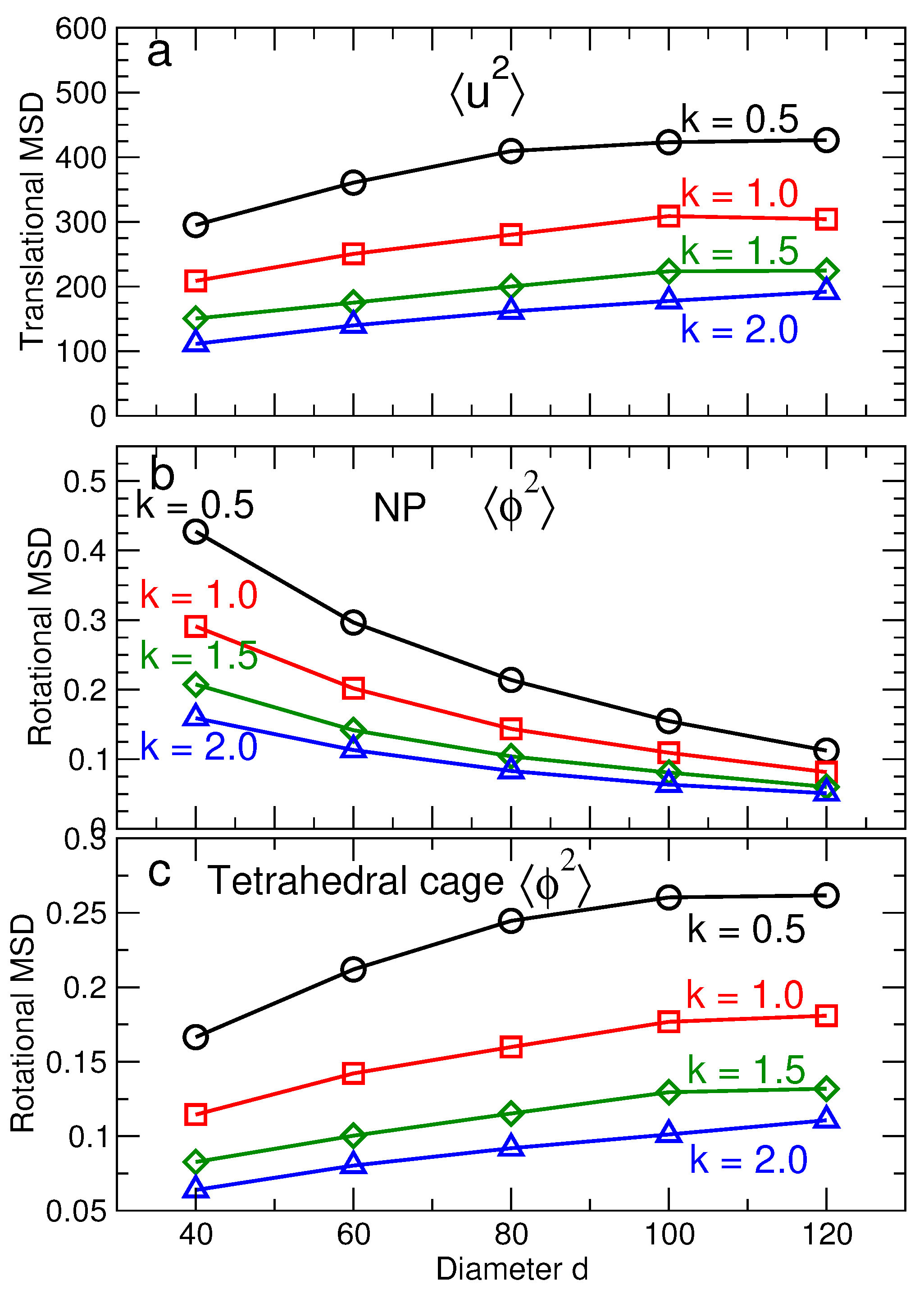
3.2. Lattice Stability
3.2.1. Lattice Structure
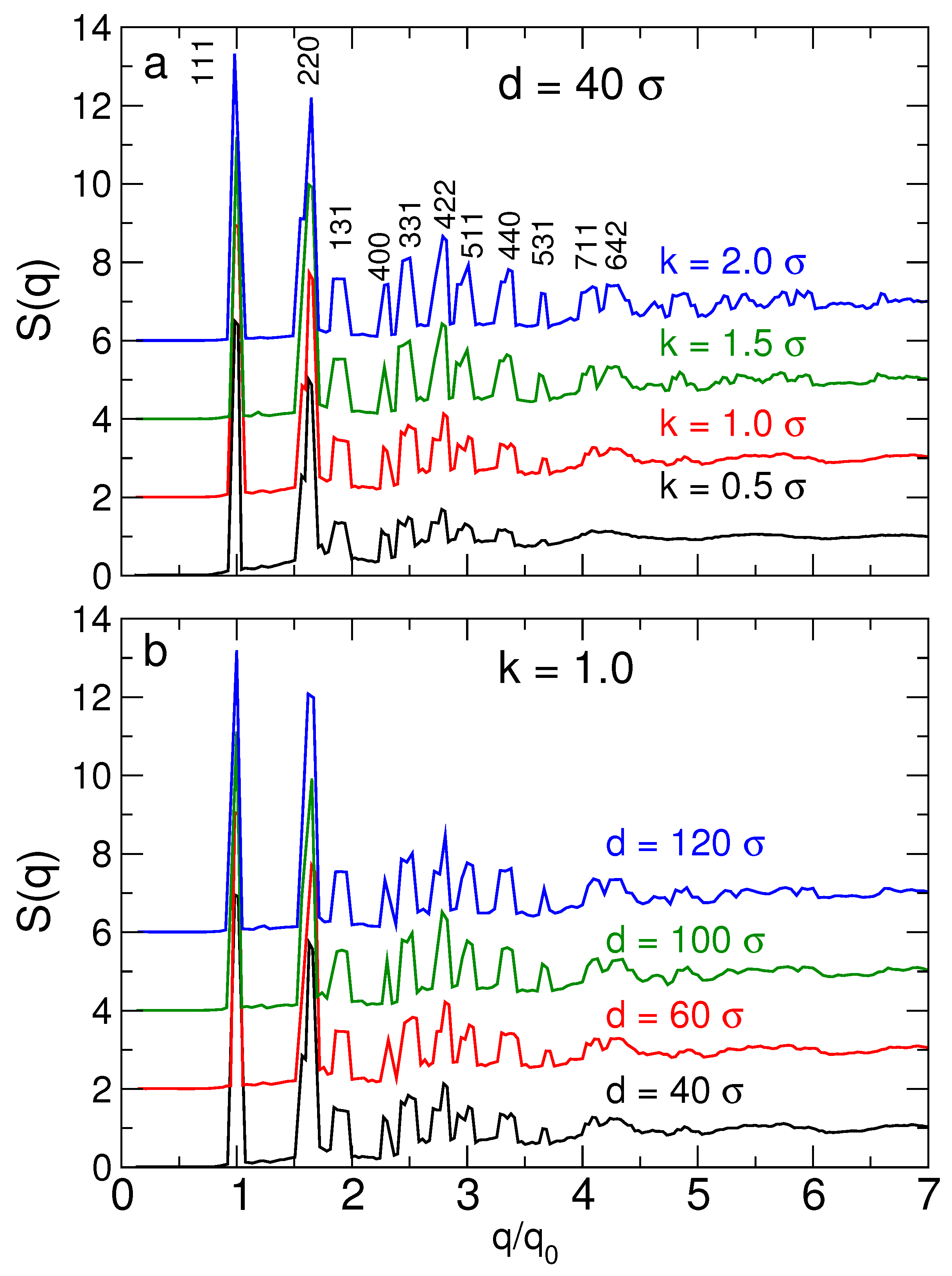
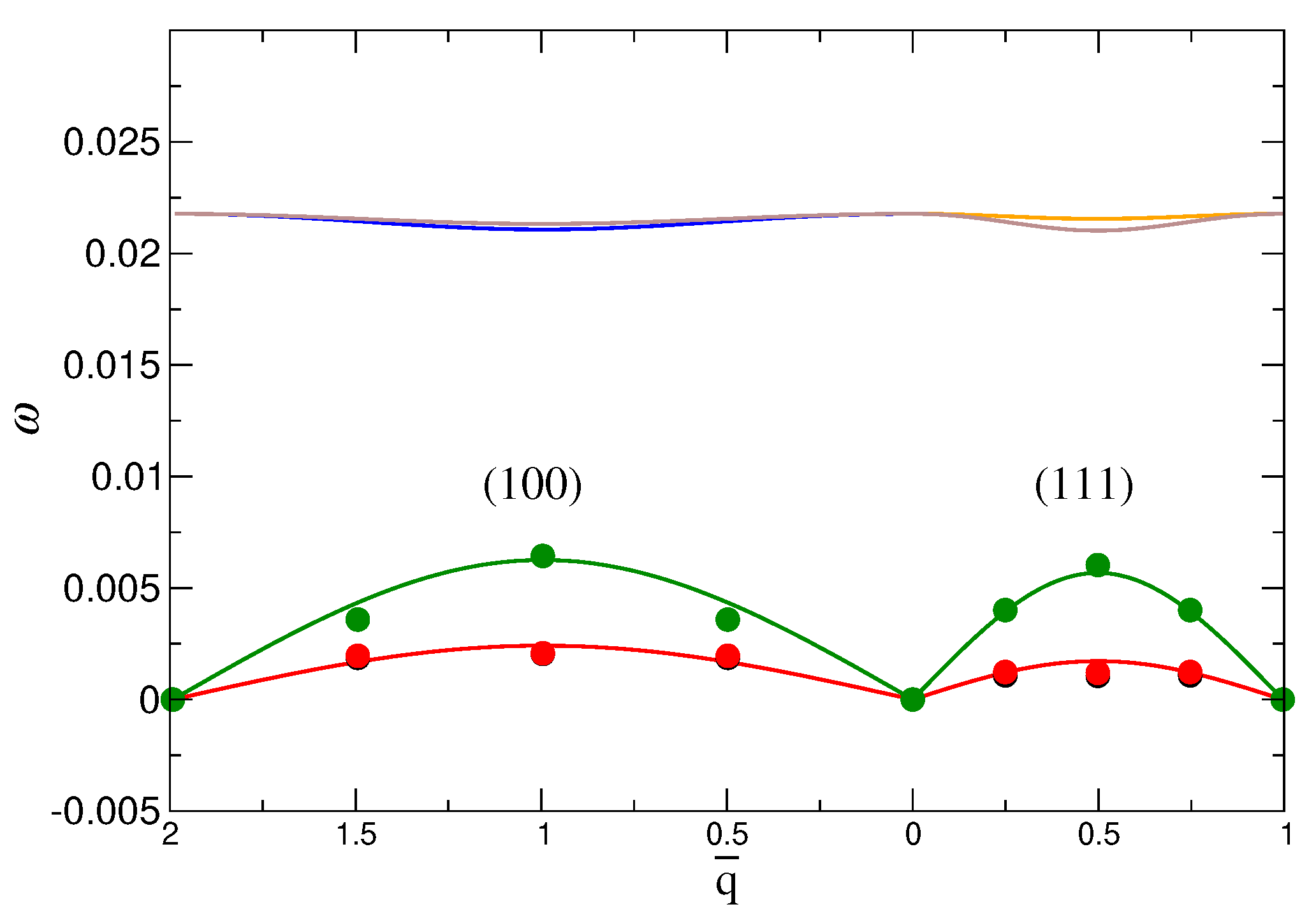
3.2.2. Lattice Vibrational Spectrum
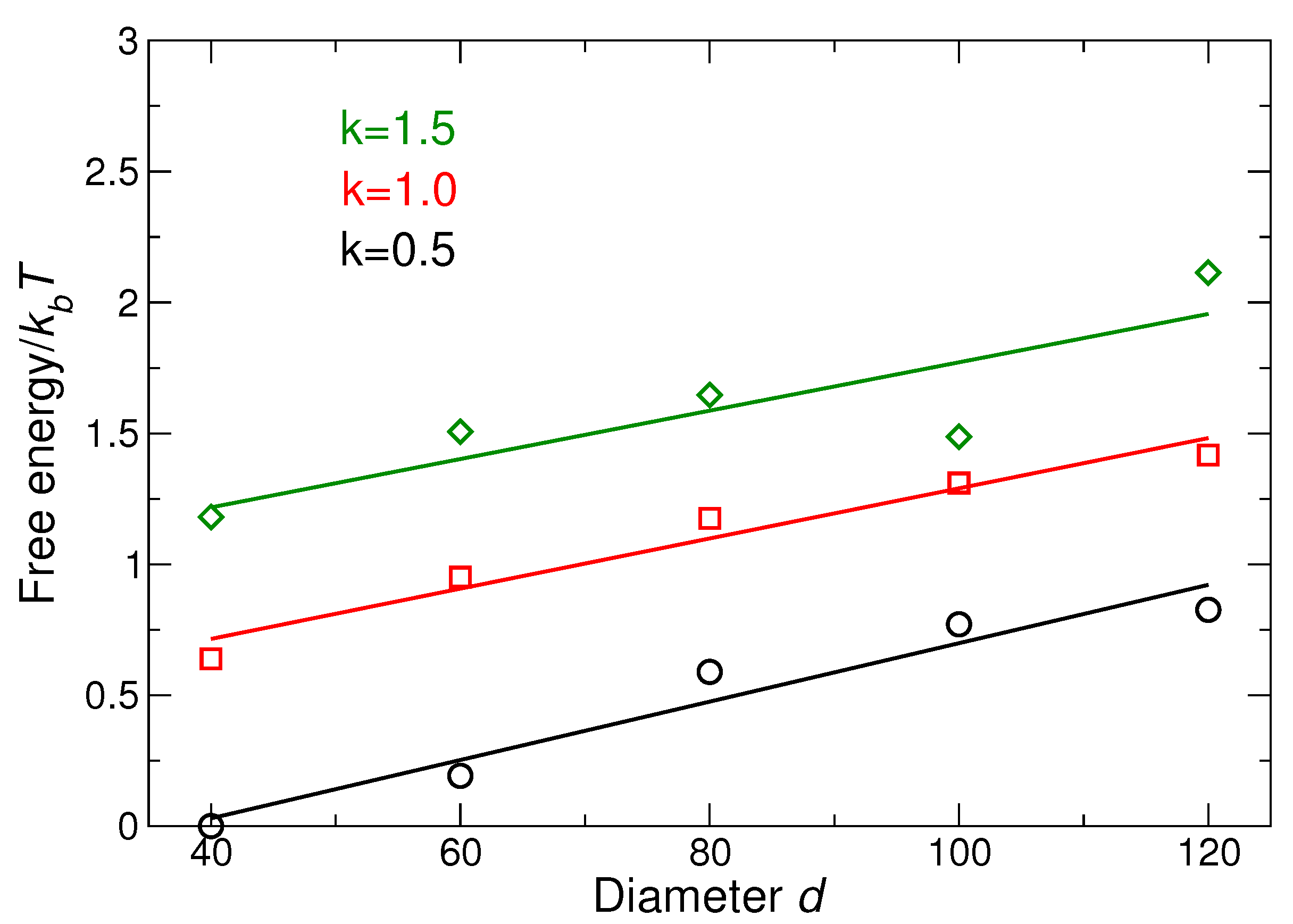
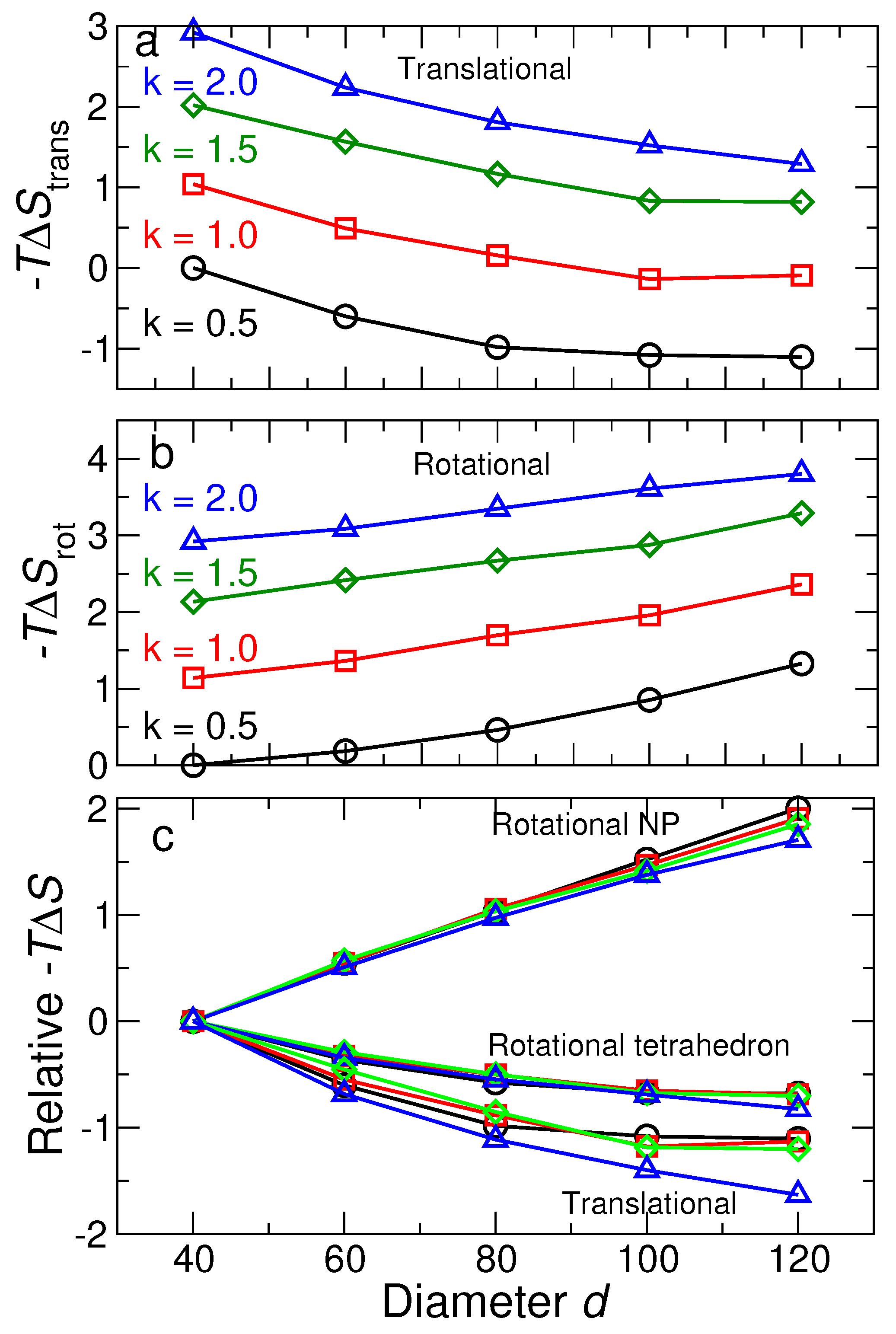
3.2.3. Lattice Free Energy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crocker, J.C. Golden handshake. Nature 2008, 451, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Osakada, Y.; Kawai, K.; Fujitsuka, M.; Majima, T. Charge transfer through DNA nanoscaled assembly programmable with DNA building blocks. Proc. Natl. Acad. Sci. USA 2006, 103, 18072–18076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Seeman, N.C.; Mirkin, C.A. Nanomaterials. Programmable materials and the nature of the DNA bond. Science 2015, 347, 1260901. [Google Scholar] [CrossRef]
- Stebe, K.J.; Lewandowski, E.P.; Ghosh, M. Oriented assembly of metamaterials. Science 2009, 325, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Yablonovitch, E. Photonic band-gap structures. JOSA B 1993, 10, 283–295. [Google Scholar] [CrossRef]
- Zou, S.; Schatz, G.C. Theoretical studies of plasmon resonances in one-dimensional nanoparticle chains: narrow lineshapes with tunable widths. Nanotechnology 2006, 17, 2813. [Google Scholar] [CrossRef]
- Soukoulis, C.M.; Wegener, M. Past achievements and future challenges in the development of three-dimensional photonic metamaterials. Nat. Photonics 2011, 5, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Esfandyarpour, M.; Garnett, E.C.; Cui, Y.; McGehee, M.D.; Brongersma, M.L. Metamaterial mirrors in optoelectronic devices. Nat. Nanotechnol. 2014, 9, 542–547. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef]
- Alivisatos, A.P.; Johnsson, K.P.; Peng, X.; Wilson, T.E.; Loweth, C.J.; Bruchez, M.P., Jr.; Schultz, P.G. Organization of ‘nanocrystal molecules’ using DNA. Nature 1996, 382, 609–611. [Google Scholar] [CrossRef]
- Storhoff, J.J.; Mirkin, C.A. Programmed materials synthesis with DNA. Chem. Rev. 1999, 99, 1849–1862. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Macfarlane, R.J.; Young, K.L.; Choi, C.H.J.; Hao, L.; Auyeung, E.; Liu, G.; Zhou, X.; Mirkin, C.A. A general approach to DNA-programmable atom equivalents. Nat. Mater. 2013, 12, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, R.J.; Lee, B.; Jones, M.R.; Harris, N.; Schatz, G.C.; Mirkin, C.A. Nanoparticle superlattice engineering with DNA. Science 2011, 334, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Biancaniello, P.L.; Kim, A.J.; Crocker, J.C. Colloidal interactions and self-assembly using DNA hybridization. Phys. Rev. Lett. 2005, 94, 058302. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lytton-Jean, A.K.; Lee, B.; Weigand, S.; Schatz, G.C.; Mirkin, C.A. DNA-programmable nanoparticle crystallization. Nature 2008, 451, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Nykypanchuk, D.; Maye, M.M.; Van Der Lelie, D.; Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 2008, 451, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Tørring, T.; Voigt, N.V.; Nangreave, J.; Yan, H.; Gothelf, K.V. Advances in DNA-based nanotechnology themed issue Guest editors Eugen Stulz, Guido Clever, Mitsuhiko Shionoya and. Chem. Soc. Rev. 2011, 40, 5621–5928. [Google Scholar]
- Zheng, J.; Birktoft, J.J.; Chen, Y.; Wang, T.; Sha, R.; Constantinou, P.E.; Ginell, S.L.; Mao, C.; Seeman, N.C. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 2009, 461, 74–77. [Google Scholar] [CrossRef]
- Liu, W.; Zhong, H.; Wang, R.; Seeman, N.C. Crystalline Two-Dimensional DNA-Origami Arrays. Angew. Chem. 2011, 123, 278–281. [Google Scholar] [CrossRef]
- Aldaye, F.A.; Palmer, A.L.; Sleiman, H.F. Assembling materials with DNA as the guide. Science 2008, 321, 1795–1799. [Google Scholar] [CrossRef]
- Winfree, E.; Liu, F.; Wenzler, L.A.; Seeman, N.C. Design and self-assembly of two-dimensional DNA crystals. Nature 1998, 394, 539–544. [Google Scholar] [CrossRef]
- Seeman, N.C. Nanomaterials based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, Y.; Wang, T.; Xin, H.L.; Li, H.; Gang, O. Lattice engineering through nanoparticle-DNA frameworks. Nat. Mater. 2016, 15, 654–661. [Google Scholar] [CrossRef]
- Alaeian, H.; Dionne, J.A. Plasmon nanoparticle superlattices as optical-frequency magnetic metamaterials. Opt. Express 2012, 20, 15781. [Google Scholar] [CrossRef]
- Jain, A.; Errington, J.R.; Truskett, T.M. Dimensionality and design of isotropic interactions that stabilize honeycomb, square, simple cubic, and diamond lattices. Phys. Rev. X 2014, 4, 031049. [Google Scholar] [CrossRef]
- Rechtsman, M.C.; Stillinger, F.H.; Torquato, S. Synthetic diamond and wurtzite structures self-assemble with isotropic pair interactions. Phys. Rev. E 2007, 75, 031403. [Google Scholar] [CrossRef]
- Liu, W.; Tagawa, M.; Xin, H.L.; Wang, T.; Emamy, H.; Li, H.; Yager, K.G.; Starr, F.W.; Tkachenko, A.V.; Gang, O. Diamond family of nanoparticle superlattices. Science 2016, 351, 582–586. [Google Scholar] [CrossRef]
- Zhang, J.; Luijten, E.; Granick, S. Toward design rules of directional Janus colloidal assembly. Annu. Rev. Phys. Chem. 2015, 66, 581–600. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Breed, D.R.; Manoharan, V.N.; Feng, L.; Hollingsworth, A.D.; Weck, M.; Pine, D.J. Colloids with valence and specific directional bonding. Nature 2012, 491, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jenkins, I.C.; McGinley, J.T.; Sinno, T.; Crocker, J.C. Colloidal crystals with diamond symmetry at optical lengthscales. Nat. Commun. 2017, 8, 14173. [Google Scholar] [CrossRef] [Green Version]
- Kalsin, A.M.; Fialkowski, M.; Paszewski, M.; Smoukov, S.K.; Bishop, K.J.; Grzybowski, B.A. Electrostatic self-assembly of binary nanoparticle crystals with a diamond-like lattice. Science 2006, 312, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Julin, S.; Korpi, A.; Shen, B.; Liljeström, V.; Ikkala, O.; Keller, A.; Linko, V.; Kostiainen, M.A. DNA origami directed 3D nanoparticle superlattice via electrostatic assembly. Nanoscale 2019, 11, 4546–4551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hartl, C.; Frank, K.; Heuer-Jungemann, A.; Fischer, S.; Nickels, P.C.; Nickel, B.; Liedl, T. 3D DNA origami crystals. Adv. Mater. 2018, 30, 1800273. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, P.F.; Engel, M.; Glotzer, S.C. Crystalline assemblies and densest packings of a family of truncated tetrahedra and the role of directional entropic forces. ACS Nano 2011, 6, 609–614. [Google Scholar] [CrossRef]
- Travesset, A. Binary nanoparticle superlattices of soft-particle systems. Proc. Natl. Acad. Sci. USA 2015, 112, 9563–9567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulff, G. On the question of speed of growth and dissolution of crystal surfaces. Z. Kristallogr. 1901, 34, 449–530. [Google Scholar]
- Auyeung, E.; Li, T.I.; Senesi, A.J.; Schmucker, A.L.; Pals, B.C.; de La Cruz, M.O.; Mirkin, C.A. DNA-mediated nanoparticle crystallization into Wulff polyhedra. Nature 2014, 505, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Li, T.I.; Olvera de la Cruz, M. Surface energy fluctuation effects in single crystals of DNA-functionalized nanoparticles. J. Chem. Phys. 2015, 143, 243156. [Google Scholar] [CrossRef]
- Stekolnikov, A.A.; Bechstedt, F. Shape of free and constrained group-IV crystallites: Influence of surface energies. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72, 125326. [Google Scholar] [CrossRef]
- Yang, X.; Fujiwara, K.; Maeda, K.; Nozawa, J.; Koizumi, H.; Uda, S. Crystal growth and equilibrium crystal shapes of silicon in the melt. Prog. Photovolt. Res. Appl. 2014, 22, 574–580. [Google Scholar] [CrossRef]
- Eaglesham, D.; White, A.; Feldman, L.; Moriya, N.; Jacobson, D. Equilibrium shape of Si. Phys. Rev. Lett. 1993, 70, 1643. [Google Scholar] [CrossRef]
- Chi, C.; Vargas-Lara, F.; Tkachenko, A.V.; Starr, F.W.; Gang, O. Internal structure of nanoparticle dimers linked by DNA. ACS Nano 2012, 6, 6793–6802. [Google Scholar] [CrossRef] [PubMed]
- Starr, F.W.; Sciortino, F. Model for assembly and gelation of four-armed DNA dendrimers. J. Phys. Condens. Matter 2006, 18, L347. [Google Scholar] [CrossRef] [PubMed]
- Lara, F.V.; Starr, F.W. Stability of DNA-linked nanoparticle crystals I: Effect of linker sequence and length. Soft Matter 2011, 7, 2085–2093. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Saito, Y. Wulff polyhedra derived from morse potentials and crystal habits of bcc and fcc metal particles. J. Cryst. Growth 1981, 53, 273–279. [Google Scholar] [CrossRef]
- Zucker, R.V.; Chatain, D.; Dahmen, U.; Hagège, S.; Carter, W.C. New software tools for the calculation and display of isolated and attached interfacial-energy minimizing particle shapes. J. Mater. Sci. 2012, 47, 8290–8302. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Cristal Lattices; Oxford University Press: Oxford, UK, 1954. [Google Scholar]
- Sólyom, J. Fundamentals of the Physics of Solids: Volume 1: Structure and Dynamics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007; Volume 1. [Google Scholar]
- Lax, M. Symmetry Principles in Solid State and Molecular Physics; John Wiley and Sons: New York, NY, USA, 1974. [Google Scholar]
- Smith, H.M. The theory of the vibrations and the Raman spectrum of the diamond lattice. Philos. Trans. R. Soc. Lond. A 1948, 241, 105–145. [Google Scholar] [CrossRef]
- Musgrave, M.; Pople, J.A. A general valence force field for diamond. Proc. R. Soc. Lond. A 1962, 268, 474–484. [Google Scholar]
- Keim, P.; Maret, G.; Herz, U.; von Grünberg, H.H. Harmonic lattice behavior of two-dimensional colloidal crystals. Phys. Rev. Lett. 2004, 92, 215504. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.T. Phonon dispersion measured directly from molecular dynamics simulations. Comput. Phys. Commun. 2011, 182, 2201–2207. [Google Scholar] [CrossRef]
- Padovan-Merhar, O.; Lara, F.V.; Starr, F.W. Stability of DNA-linked nanoparticle crystals: Effect of number of strands, core size, and rigidity of strand attachment. J. Chem. Phys. 2011, 134, 244701. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Mittal, J. Insights into DNA-mediated interparticle interactions from a coarse-grained model. J. Chem. Phys. 2014, 141, 184901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallenburg, F.; Sciortino, F. Liquids more stable than crystals in particles with limited valence and flexible bonds. Nat. Phys. 2013, 9, 554–558. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Type | Bond | ||||
|---|---|---|---|---|---|
| Tetrahedral cage | Edge beads, first neighbor | 240 | 1.5 | 8 | 6.36 |
| Edge beads, second neighbor | 240 | 1.5 | 8 | 13.67 | |
| Vertex to center | 240 | 1.5 | 8 | 36.52 | |
| Vertex to edge | 30 | 1.5 | 1 | 15.08 | |
| Edge to edge | 30 | 1.5 | 1 | 9.15 | |
| Nanoparticle | Center to vertex | 30 | 1.5 | 1 | |
| Vertex to vertex | 30 | 1.5 | 1 |
| Particle Type | Diameter () |
|---|---|
| Tetrahedral cage, edge particles | 9.9 |
| Tetrahedral cage, central particle | 40 |
| NP, central particle | ranging between 40 to 120 |
| DNA backbone | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emamy, H.; Gang, O.; Starr, F.W. The Stability of a Nanoparticle Diamond Lattice Linked by DNA. Nanomaterials 2019, 9, 661. https://doi.org/10.3390/nano9050661
Emamy H, Gang O, Starr FW. The Stability of a Nanoparticle Diamond Lattice Linked by DNA. Nanomaterials. 2019; 9(5):661. https://doi.org/10.3390/nano9050661
Chicago/Turabian StyleEmamy, Hamed, Oleg Gang, and Francis W. Starr. 2019. "The Stability of a Nanoparticle Diamond Lattice Linked by DNA" Nanomaterials 9, no. 5: 661. https://doi.org/10.3390/nano9050661
APA StyleEmamy, H., Gang, O., & Starr, F. W. (2019). The Stability of a Nanoparticle Diamond Lattice Linked by DNA. Nanomaterials, 9(5), 661. https://doi.org/10.3390/nano9050661