Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum



Abstract
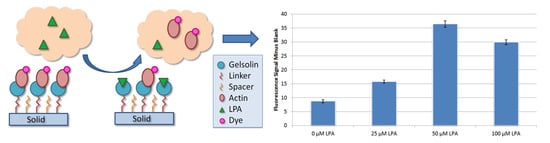
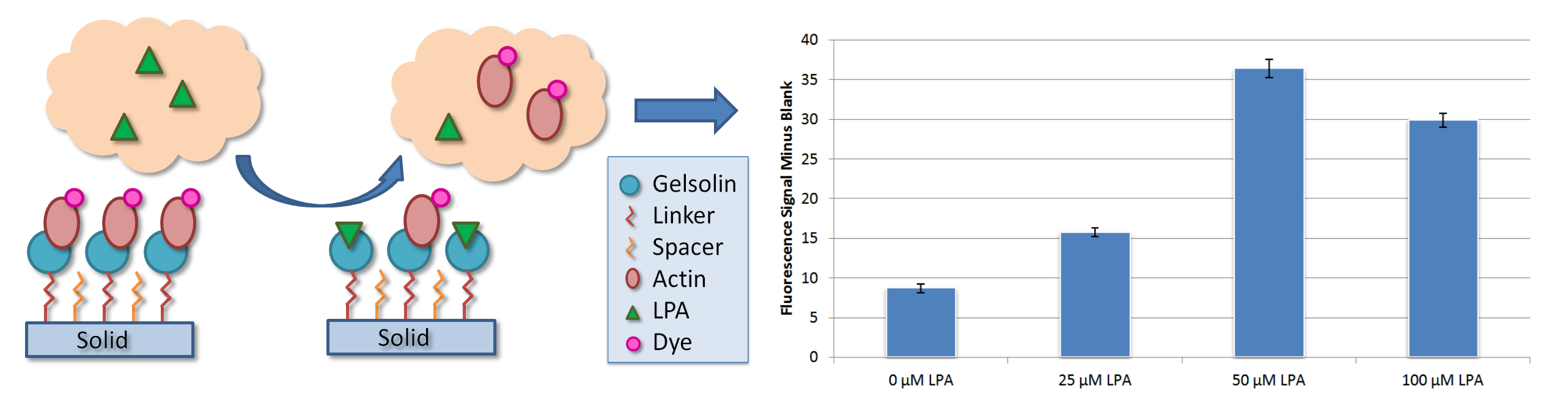
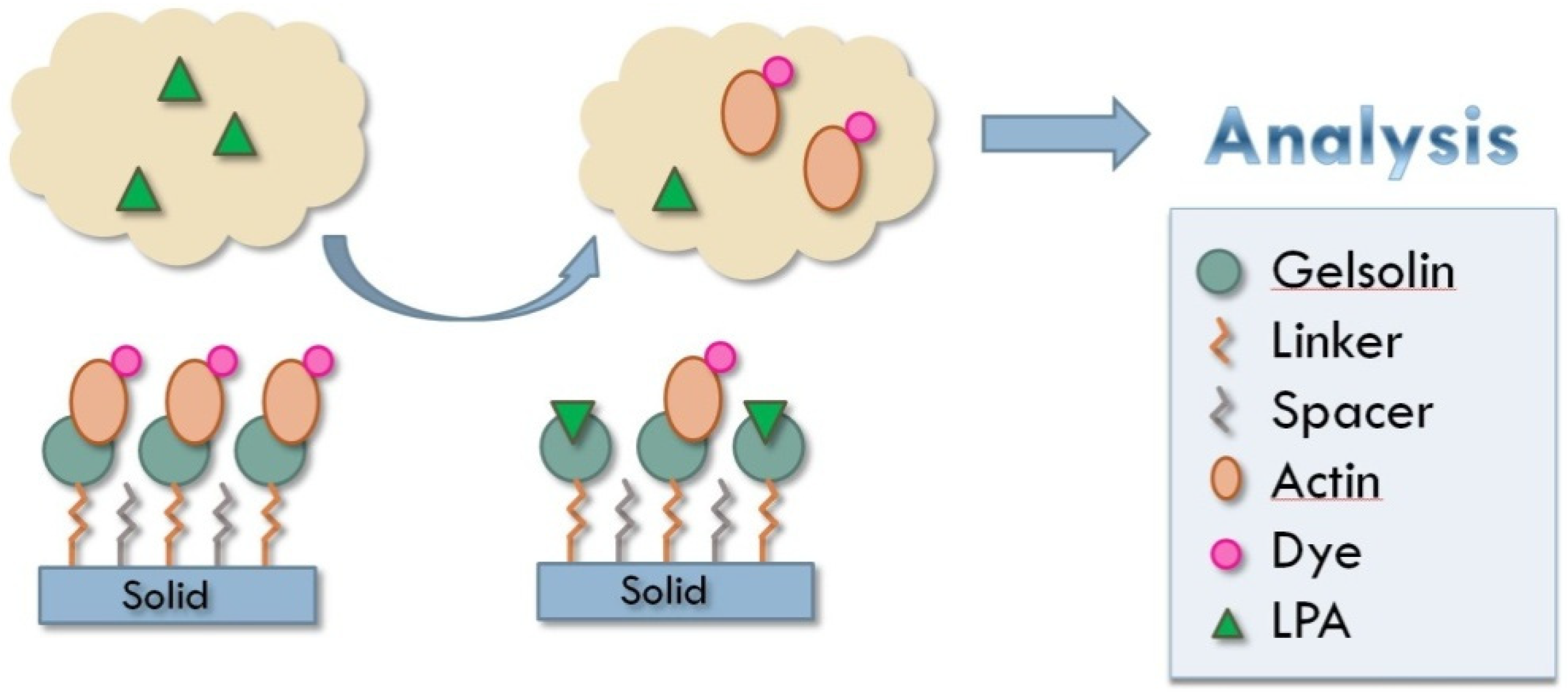
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Expression of Gelsolin Protein
2.3. Modification of Actin Protein
2.4. Non-Denaturing Acrylamide Gel Analysis of Proteins
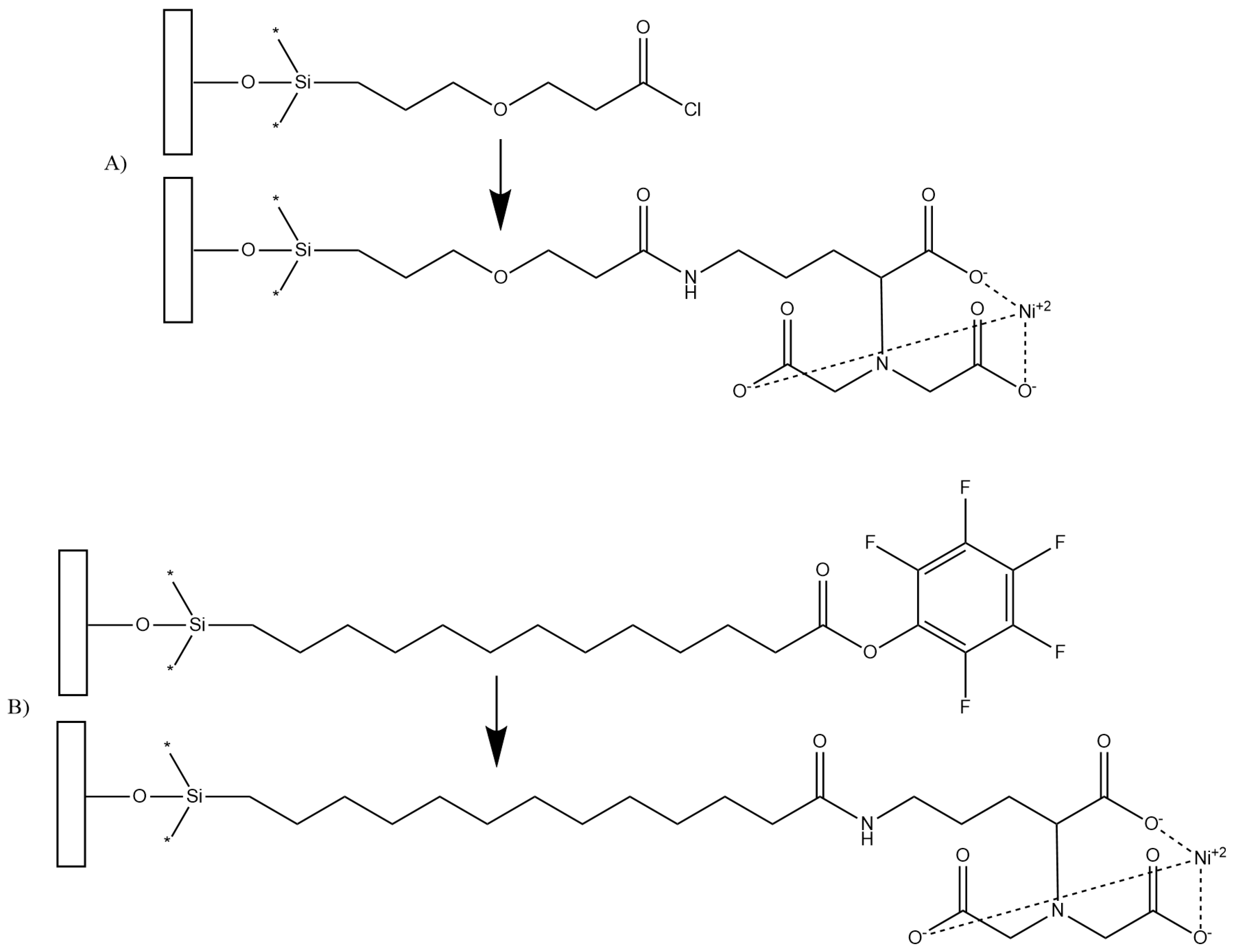
2.5. Cleaning and Surface Modification of Silica Gel
2.6. Fluorescence Measurements
3. Results
4. Discussion
4.1. Actin and Gelsolin Binding by Gel Analysis
4.2. Fluorescence Analysis of Dyed Actin and LPA
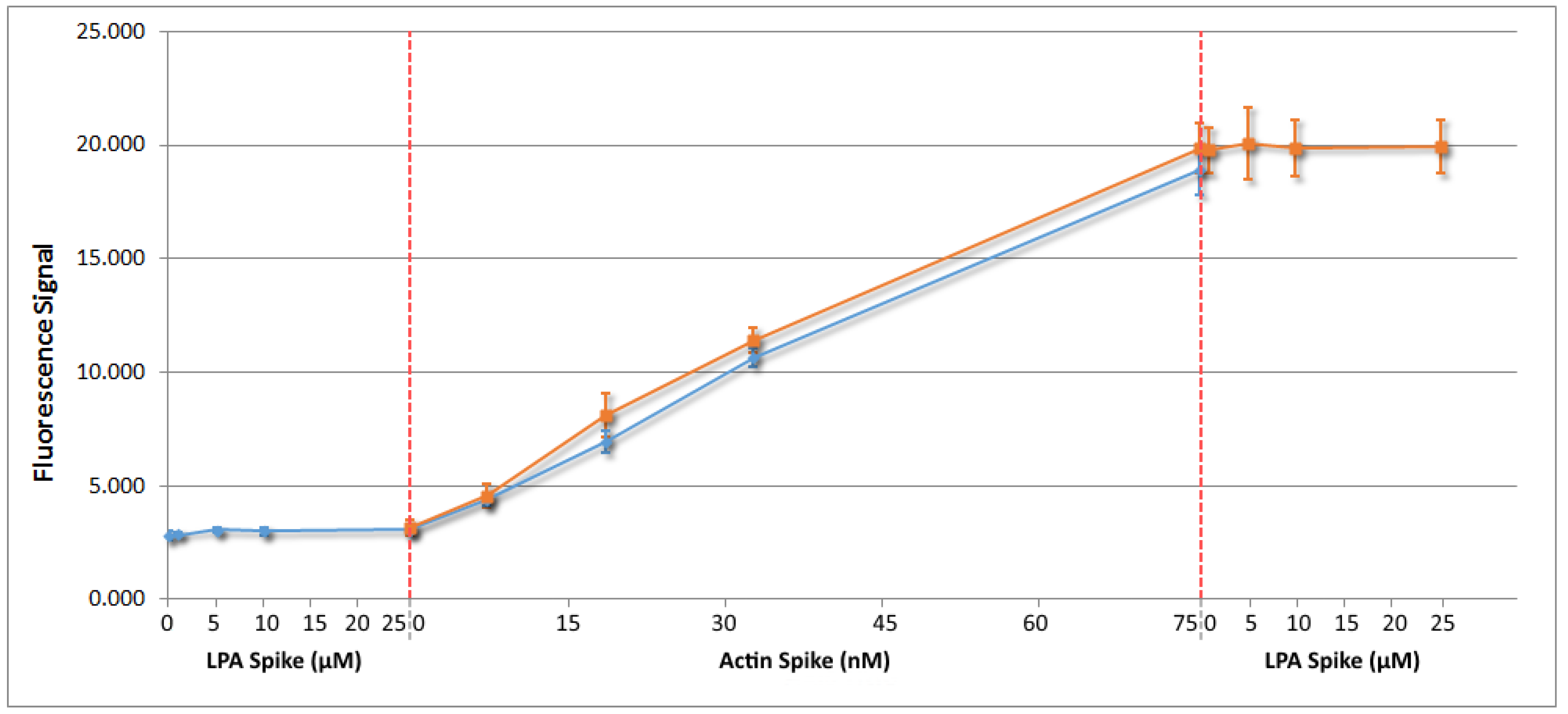
4.3. Initial Test Development with Buffer A Samples
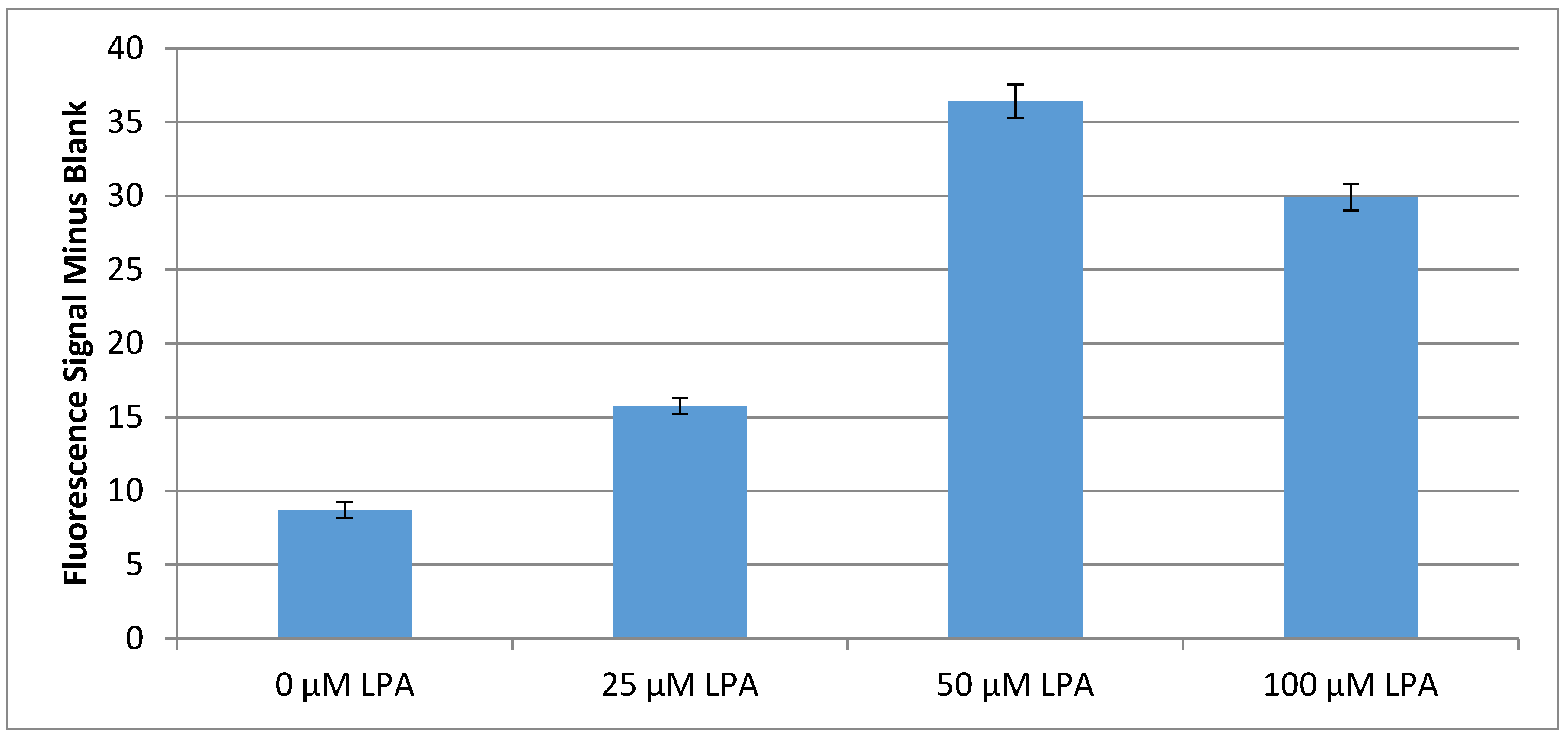
4.4. Serum Based Testing and Conclusions
5. Final Remarks and Future Perspective
“Researchers and funding organizations should focus on the development and assessment of early detection strategies that extend beyond current imaging modalities and biomarkers and that reflect the pathobiology of each ovarian cancer subgroup”.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De La Franier, B.; Thompson, M. Biosensors and Methods for Detection of Lysophosphatidic Acid for Signaling of Ovarian Cancer. U.S. Pantent US 15/572,295, 31 May 2018. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics for hispanics/latinos, 2012. CA Cancer J. Clin. 2012, 62, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Stevens, G.A.; Mathers, C.D.; Beard, J.R. Global mortality trends and patterns in older women. Bull. World Health Organ. 2013, 91, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Sant, M.; Allemani, C.; Santaquilani, M.; Knijn, A.; Marchesi, F.; Capocaccia, R. EUROCARE-4. Survival of cancer patients diagnosed in 1995–1999. Results and commentary. Eur. J. Cancer 2009, 45, 931–991. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Available online: https://www.scienceopen.com/document?vid=19f26dca-3511-48bf-8fab-76242d4b187b (accessed on 14 February 2020).
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, R.; Sant, M.; Coleman, M.P.; Francisci, S.; Baili, P.; Pierannunzio, D.; Trama, A.; Visser, O.; Brenner, H.; Ardanaz, E.; et al. Cancer survival in Europe 1999–2007 by country and age: results of EUROCARE-5—A population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef]
- National Cancer Institute. Cancer of the Ovary (Invasive): 5-Year Relative and Period Survival by Race, Diagnosis Year, Age and Stage at Diagnosis. Available online: http://seer.cancer.gov/csr/1975_2010/browse_csr.php?sectionSEL=21&pageSEL=sect_21_table.08.html (accessed on 17 June 2018).
- Baldwin, L.A.; Huang, B.; Miller, R.W.; Tucker, T.; Goodrich, S.T.; Podzielinski, I.; DeSimone, C.P.; Ueland, F.R.; van Nagell, J.R.; Seamon, L.G. Ten-year relative survival for epithelial ovarian cancer. Obstet. Gynecol. 2012, 120, 612–618. [Google Scholar] [CrossRef]
- Bae, H.S.; Hong, J.H.; Ki, K.D.; Song, J.Y.; Shin, J.W.; Lee, J.M.; Lee, J.K.; Lee, N.W.; Lee, C.; Lee, K.W.; et al. The effect of body mass index on survival in advanced epithelial ovarian cancer. J. Korean Med. Sci. 2014, 29, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Pagès, C.; Simon, M.F.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat 2001, 64, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [Green Version]
- Sedláková, I.; Vávrová, J.; Tošner, J.; Hanousek, L. Lysophosphatidic acid (LPA)—A perspective marker in ovarian cancer. Tumor Biol. 2011, 32, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Sutphen, R.; Xu, Y.; Wilbanks, G.D.; Fiorica, J.; Grendys, E.C.; LaPolla, J.P.; Arango, H.; Hoffman, M.S.; Martino, M.; Wakeley, K.; et al. Lysophospholipids are potential biomarkers of ovarian cancer. Cancer Epidemiol Biomark. Prev. 2004, 13, 1185–1191. [Google Scholar]
- Meleh, M.; Pozlep, B.; Mlakar, A.; Meden-Vrtovec, H.; Zupancic-Kralj, L. Determination of serum lysophosphatidic acid as a potential biomarker for ovarian cancer. J. Chromatogr. B. 2007, 858, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Xu, Y. Determination of lysophosphatidic acids by capillary electrophoresis with indirect ultraviolet detection. J. Chromatogr. B. 2001, 753, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Xu, Y. Isolation and quantitation of plasma lysophosphatidic acids by solid-phase extraction and capillary electrophoresis. J. Liq. Chromatogr. Relat. Technol. 2002, 25, 843–855. [Google Scholar] [CrossRef]
- Tokumura, A.; Harada, K.; Yoshioka, Y.; Tsukatani, H.; Handa, Y. Analyses of lysophosphatidic acids by gas chromatography mass spectrometry without hydrolytic pretreatment. Biol. Mass Spectrom 1984, 11, 167–171. [Google Scholar] [CrossRef]
- Sugiura, T.; Nakane, S.; Kishimoto, S.; Waku, K.; Yoshioka, Y.; Tokumura, A. Lysophosphatidic acid, a growth factor-like lipid, in the saliva. J. Lipid Res. 2002, 43, 2049–2055. [Google Scholar] [CrossRef] [Green Version]
- Bese, T.; Barbaros, M.; Baykara, E.; Guralp, O.; Cengiz, S.; Demirkiran, F.; Sanioglu, C.; Arvas, M. Comparison of total plasma lysophosphatidic acid and serum CA-125 as a tumor marker in the diagnosis and follow-up of patients with epithelial ovarian cancer. J. Gynecol. Oncol. 2010, 21, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.J.; Schwartz, B.; Washington, M.; Kennedy, A.; Webster, K.; Belinson, J.; Xu, Y. Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: Comparison of the lysophospholipid contents in malignant vs. nonmalignantascitic fluids. Anal. Biochem. 2001, 290, 302–313. [Google Scholar] [CrossRef]
- Shan, L.; Jaffe, K.; Li, S.; Davis, L. Quantitative determination of lysophosphatidic acid by LC/ESI/MS/MS employing a reversed phase HPLC column. J. Chromatogr. B 2008, 864, 22–28. [Google Scholar] [CrossRef]
- Yoon, H.R.; Kim, H.; Cho, S.H. Quantitative analysis of acyl-lysophosphatidic acid in plasma using negative ionization tandem mass spectrometry. J. Chromatogr. B. 2003, 788, 85–92. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, Y. Measurement of endogenous lysophosphatidic acid by ESI-MS/MS in plasma samples requires pre-separation of lysophosphatidylcholine. J. Chromatogr. B. 2009, 877, 3739–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.L.; Umstot, E.S.; Desiderio, D.M.; Tigyi, G.J. Quantitative analysis of lysophosphatidic acid in human blood fractions. Ann. N. Y. Acad. Sci. 2000, 905, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.L.; Desiderio, D.M.; Miller, D.D.; Tolley, B.; Tigyi, G.J. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal. Biochem. 2001, 292, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Schmitz, G.; Liebisch, G. High-throughput analysis of sphingosine 1-phosphate, sphinganine 1-phosphate, and lysophosphatidic acid in plasma samples by liquid chromatography-tandem mass spectrometry. Clin. Chem. 2009, 55, 1218–1222. [Google Scholar] [CrossRef]
- Bollinger, J.G.; Ii, H.; Sadilek, M.; Gelb, M.H. Improved method for the quantification of lysophospholipids including enol ether species by liquid chromatography-tandem mass spectrometry. J. Lipid Res. 2010, 51, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sibrian-Vazquez, M.; Escobedo, J.O.; Lowry, M.; Wang, L.; Chu, Y.H.; Moore, R.G.; Strongin, R.M. Simple enrichment and analysis of plasma lysophosphatidic acids. Analyst 2013, 138, 6852–6859. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Stauter, E.C.; Stith, B.J. Quantification of phosphatidic acid and lysophosphatidic acid by HPLC with evaporative light-scattering detection. J. Lipid Res. 2003, 44, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Kawasaki, A.; Takahashi, M.; Yamada, A.; Koike, T. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of phosphorylated compounds using a novel phosphate capture molecule. Rapid Commun. Mass Spectrom. 2003, 17, 2075–2081. [Google Scholar] [CrossRef]
- Morishige, J.; Urikura, M.; Takagi, H.; Hirano, K.; Koike, T.; Tanaka, T.; Satouchi, K. A clean-up technology for the simultaneous determination of lysophosphatidic acid and sphingosine-1-phosphate by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry using a phosphate-capture molecule, Phos-tag. Rapid Commun. Mass Spectrom 2010, 24, 1075–1084. [Google Scholar] [CrossRef]
- Tanaka, T.; Tsutsui, H.; Hirano, K.; Koike, T.; Tokumura, A.; Satouchi, K. Quantitative analysis of lysophosphatidic acid by time-of-flight mass spectrometry using a phosphate-capture molecule. J. Lipid Res. 2004, 45, 2145–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Lee, H.; Azuma, T.; Stossel, T.P.; Turck, C.W.; Karliner, J.S. Gelsolin binding and cellular presentation of lysophosphatidic acid. J. Biol. Chem. 2000, 275, 14573–14578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mintzer, E.; Sargsyan, H.; Bittman, R. Lysophosphatidic acid and lipopolysaccharide bind to the PIP 2-binding domain of gelsolin. Biochim. Biophys. Acta (Bba) Biomembr. 2006, 1758, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, K.; Herr, D.; Mutoh, T.; Chun, J. Lysophosphatidic acid (LPA) and its receptors. Curr. Opin. Pharmacol. 2009, 9, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Ma, Q.; Wang, H.; Chumnarnsilpa, S.; Lee, W.L.; Larsson, M.; Kannan, B.; Hernandez-Valladares, M.; Burtnick, L.D.; Robinson, R.C. Ca2+ binding by domain 2 plays a critical role in the activation and stabilization of gelsolin. Proc. Natl. Acad. Sci. USA 2009, 106, 13713–13718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X. Identification of a polyphosphoinositide-binding sequence in an actin monomer-binding domain of gelsolin. J. Biol. Chem. 1992, 267, 14616–14621. [Google Scholar]
- McLaughlin, P.J.; Gooch, J.T.; Mannherz, H.G.; Weeds, A.G. Structure of gelsolin segment 1-actin complex and the mechanism of filament severing. Nature 1993, 364, 685–692. [Google Scholar] [CrossRef]
- Xian, W.; Janmey, P.A. Dissecting the gelsolin--polyphosphoinositide interaction and engineering of a polyphosphoinositide-sensitive gelsolin C-terminal half protein. J. Mol. Biol. 2002, 322, 755–771. [Google Scholar] [CrossRef]
- Bryan, J. Gelsolin has three actin-binding sites. J. Cell Biol. 1988, 106, 1553–1562. [Google Scholar] [CrossRef]
- Schoepper, B.; Wegner, A. Rate constants and equilibrium constants for binding of actin to the 1: 1 gelsolin-actin complex. Eur. J. Biochem. 1991, 202, 1127–1131. [Google Scholar] [CrossRef]
- Gremm, D.; Wegner, A. Co-operative binding of Ca2+ ions to the regulatory binding sites of gelsolin. Eur. J. Biochem. 1999, 262, 330–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meerschaert, K.; De Corte, V.; De Ville, Y.; Vandekerckhove, J.; Gettemans, J. Gelsolin and functionally similar actin-binding proteins are regulated by lysophosphatidic acid. EMBO J. 1998, 17, 5923–5932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Franier, B.; Jankowski, A.; Thompson, M. Functionalizable self-assembled trichlorosilyl-based monolayer for application in biosensor technology. Appl. Surf. Sci. 2017, 414, 435–441. [Google Scholar] [CrossRef]
- Sheikh, S.; Blaszykowski, C.; Thompson, M. Sacrificial BSA to block non-specific adsorption on organosilane adlayers in ultra-high frequency acoustic wave sensing. Surf. Interface Anal. 2013, 45, 1781–1784. [Google Scholar] [CrossRef]
- Hochuli, E.; Dobeli, H.; Schacher, A. New metal chelate adsorbent selective for proteins and peptides containing neighboring histidine residues. J. Chromatogr. 1987, 411, 177–184. [Google Scholar] [CrossRef]
- Safer, D. An electrophoretic procedure for detecting proteins that bind actin monomers. Anal. Biochemistry. 1989, 178, 32–37. [Google Scholar] [CrossRef]
- Avci, C.; Sheikh, S.; Blaszykowski, C.; Thompson, M. Critical role of surface hydration on the dynamics of serum adsorption studied with monoethylene glycol adlayers on gold. Chem. Commun. 2013, 49, 466–468. [Google Scholar] [CrossRef]
- Pawlowska, N.M.; Fritzsche, H.; Blaszykowski, C.; Sheikh, S.; Vezvaie, M.; Thompson, M. Probing the hydration of ultrathin antifouling organosilane adlayers using neutron reflectometry. Langmuir 2014, 30, 1199–1203. [Google Scholar] [CrossRef]
- Pál-Gábor, H.; Gombos, L.; Micsonai, A.; Kovács, E.; Petrik, É.; Kovács, J.; Gráf, L.; Fidy, J.; Naiki, H.; Goto, Y.; et al. Mechanism of lysophosphatidic acid-induced amyloid fibril formation of β2-microglobulin in vitro under physiological conditions. Biochemistry 2009, 48, 5689–5699. [Google Scholar] [CrossRef]
- Chiu, C.-S.; Guo, S. Quantitative surface acoustic wave Detection based on colloidal gold nanoparticles and their bioconjugates. Anal. Chem. 2008, 80, 3318–3326. [Google Scholar] [CrossRef]
- Ovarian Cancers: Evolving Paradigms in Research and Care. Available online: http://nationalacademies.org/HMD/reports/2016/state-of-ovarian-cancer (accessed on 17 June 2018).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Condition | 25 μM LPA Signal | Buffer A | Overall | Imidazole |
|---|---|---|---|---|---|
| Signal | Signal | Signal | |||
| Linker: Spacer Ratio | 1PFP:1HTS | 0.27 ± 0.07 | 0.23 ± 0.06 | 0.04 ± 0.07 | 1.4 ± 0.2 |
| 2PFP:0HTS | 0.32 ± 0.06 | 0.17 ± 0.06 | 0.15 ± 0.06 | 0.8 ± 0.2 | |
| 2PFP:1HTS | 0.9 ± 0.3 | 0.5 ± 0.2 | 0.4 ± 0.3 | 3.0 ± 0.3 | |
| 1PFP:2HTS | 0.61 ± 0.04 | 0.40 ± 0.05 | 0.21 ± 0.05 | 4.3 ± 0.4 | |
| Silica Pore Size | 60Å | 3.0 ± 0.5 | 4.2 ± 0.5 | −1.2 ± 0.5 | 9.8 ± 0.6 |
| 150Å | 0.9 ± 0.2 | 0.6 ± 0.2 | 0.3 ± 0.2 | 4.3 ± 0.4 | |
| Plasma cleaning time | 1 min | 0.7 ± 0.2 | 1.1 ± 0.2 | −0.4 ± 0.5 | 4.1±0.4 |
| 10 min | 1.0 ± 0.2 | 0.7 ± 0.2 | 0.3 ± 0.2 | 3.2 ± 0.2 | |
| Linker | PFP-TTTA | 0.0 ± 0.2 | 0.3 ± 0.2 | −0.3 ± 0.2 | 3.2 ± 0.2 |
| MEG-Cl | 2.2 ± 0.3 | 1.2 ± 0.2 | 1.0 ± 0.3 | 2.3 ± 0.5 | |
| Protein | Gelsolin | 0.2 ± 0.2 | 0.6 ± 0.3 | −0.5 ± 0.3 | 1.3 ± 0.3 |
| Gelsolin 1–3 | 0.9 ± 0.1 | 0.6 ± 0.2 | 0.3 ± 0.2 | 2.7 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De La Franier, B.; Thompson, M. Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum. Biosensors 2020, 10, 13. https://doi.org/10.3390/bios10020013
De La Franier B, Thompson M. Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum. Biosensors. 2020; 10(2):13. https://doi.org/10.3390/bios10020013
Chicago/Turabian StyleDe La Franier, Brian, and Michael Thompson. 2020. "Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum" Biosensors 10, no. 2: 13. https://doi.org/10.3390/bios10020013
APA StyleDe La Franier, B., & Thompson, M. (2020). Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum. Biosensors, 10(2), 13. https://doi.org/10.3390/bios10020013