Biosensors: Electrochemical Devices—General Concepts and Performance



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
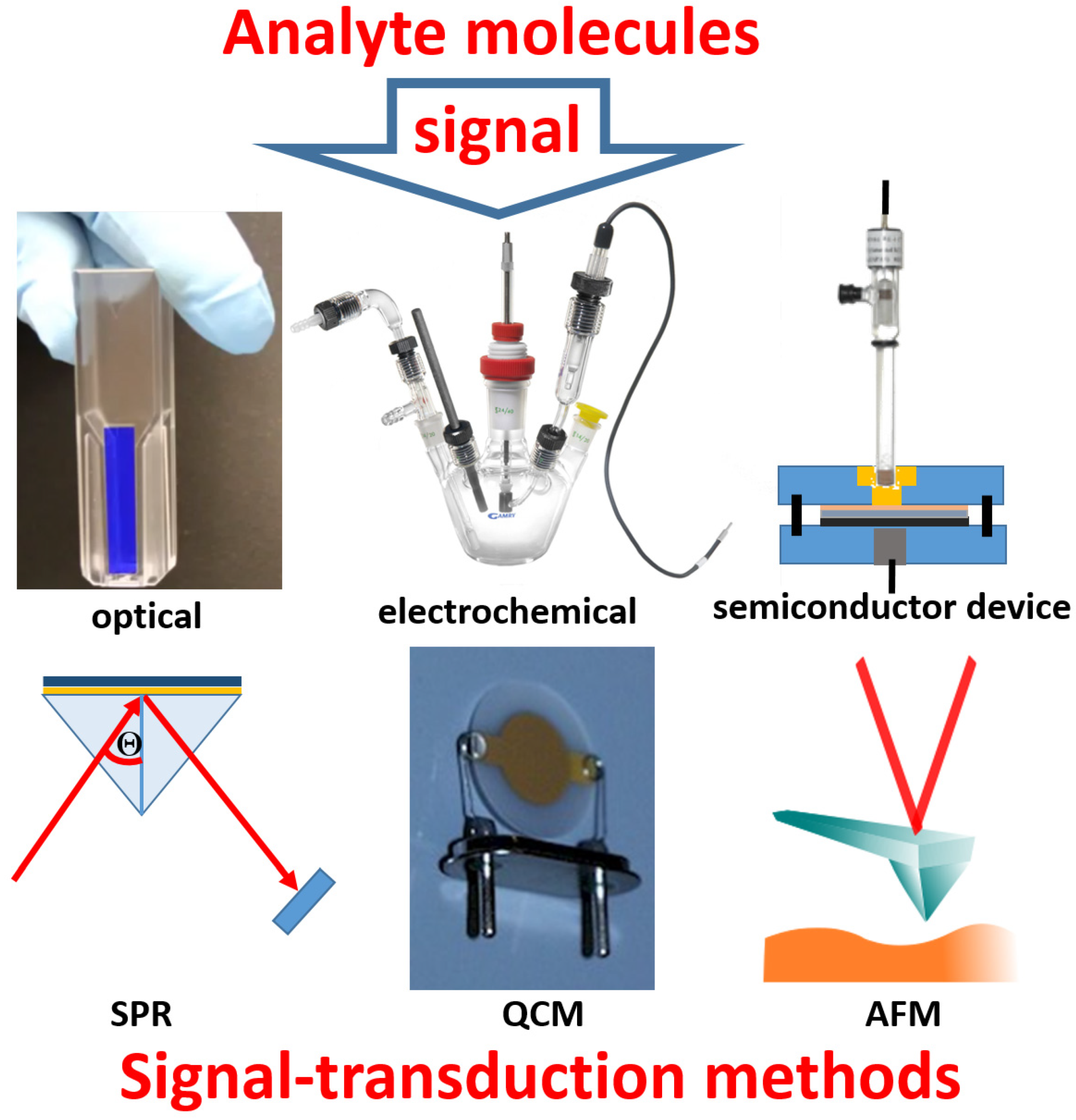
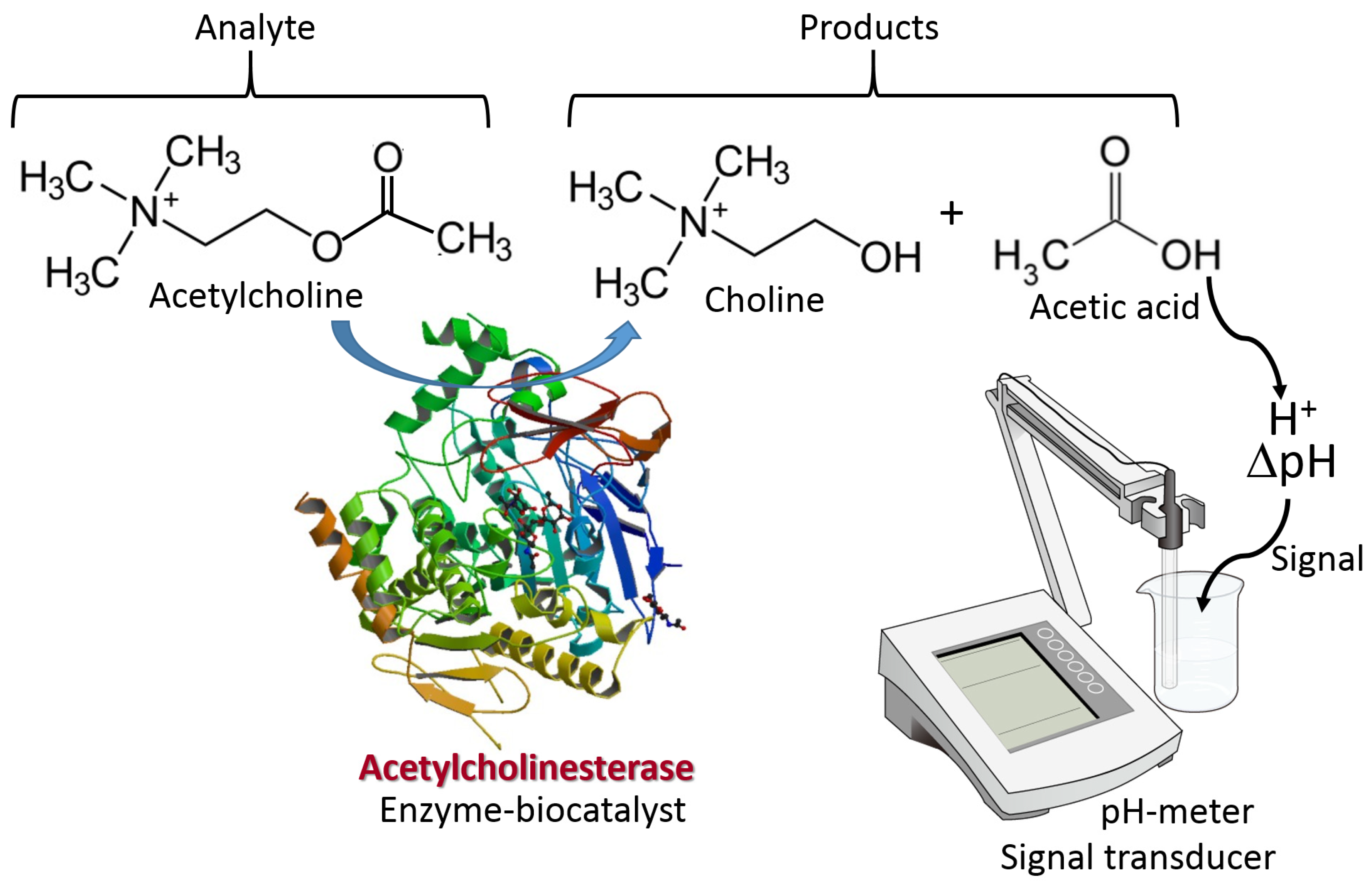
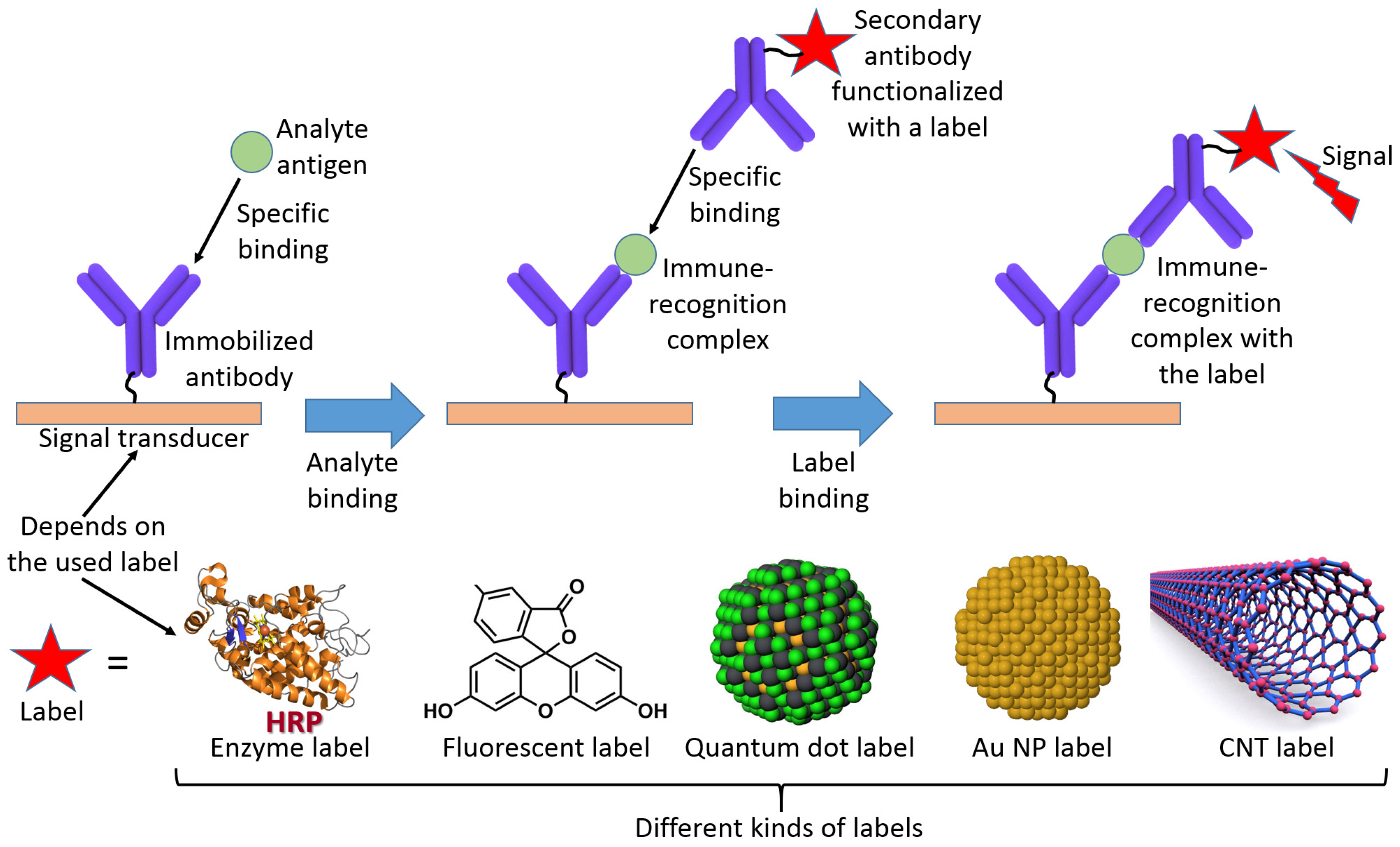
2. The Immobilized Biomolecule Recognition Element and Physical Signal Transducer
3. Immobilization of the Biosensing Component at a Transducer Surface
4. Theoretical Consideration of the Immobilized Biomolecular Systems
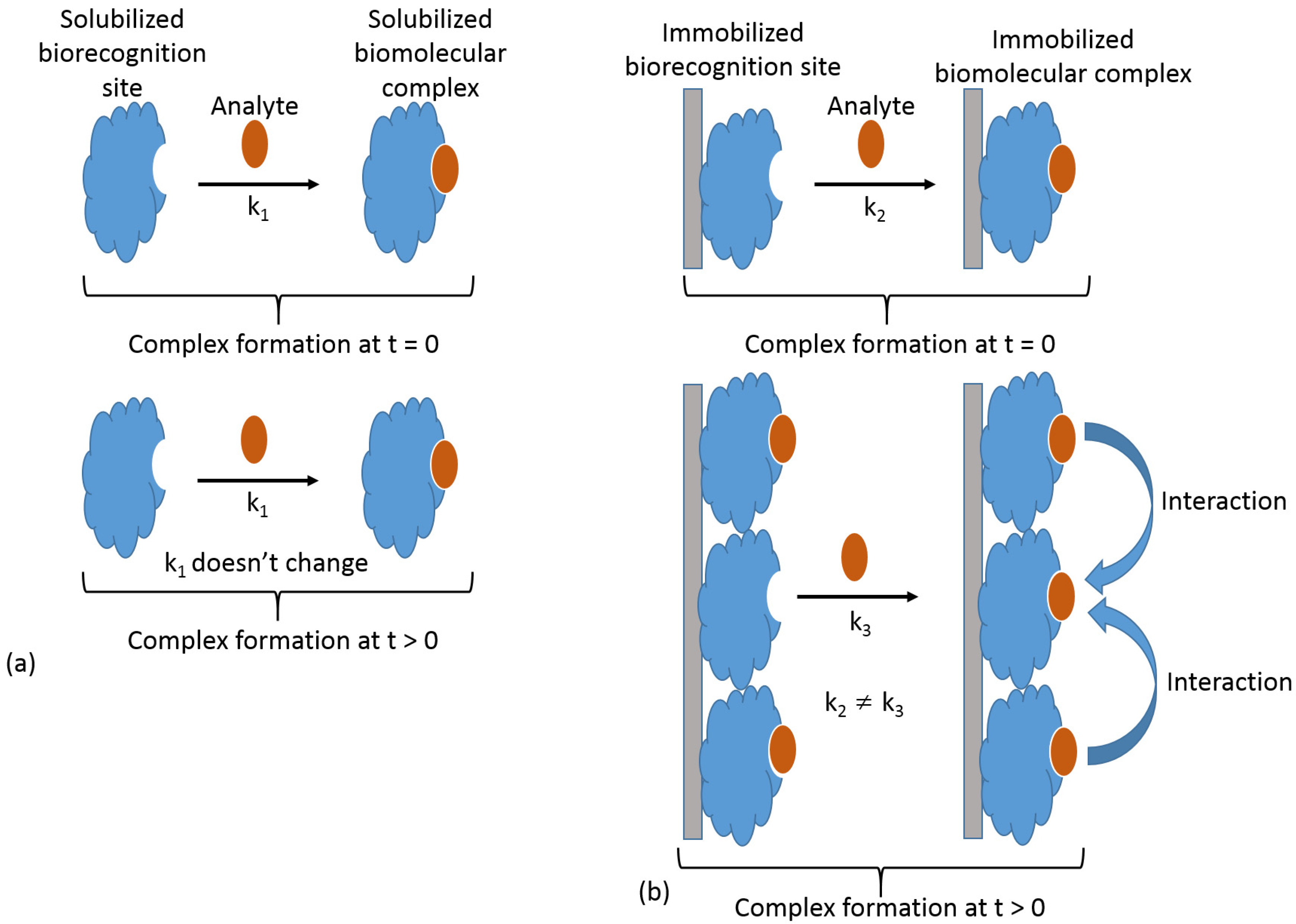
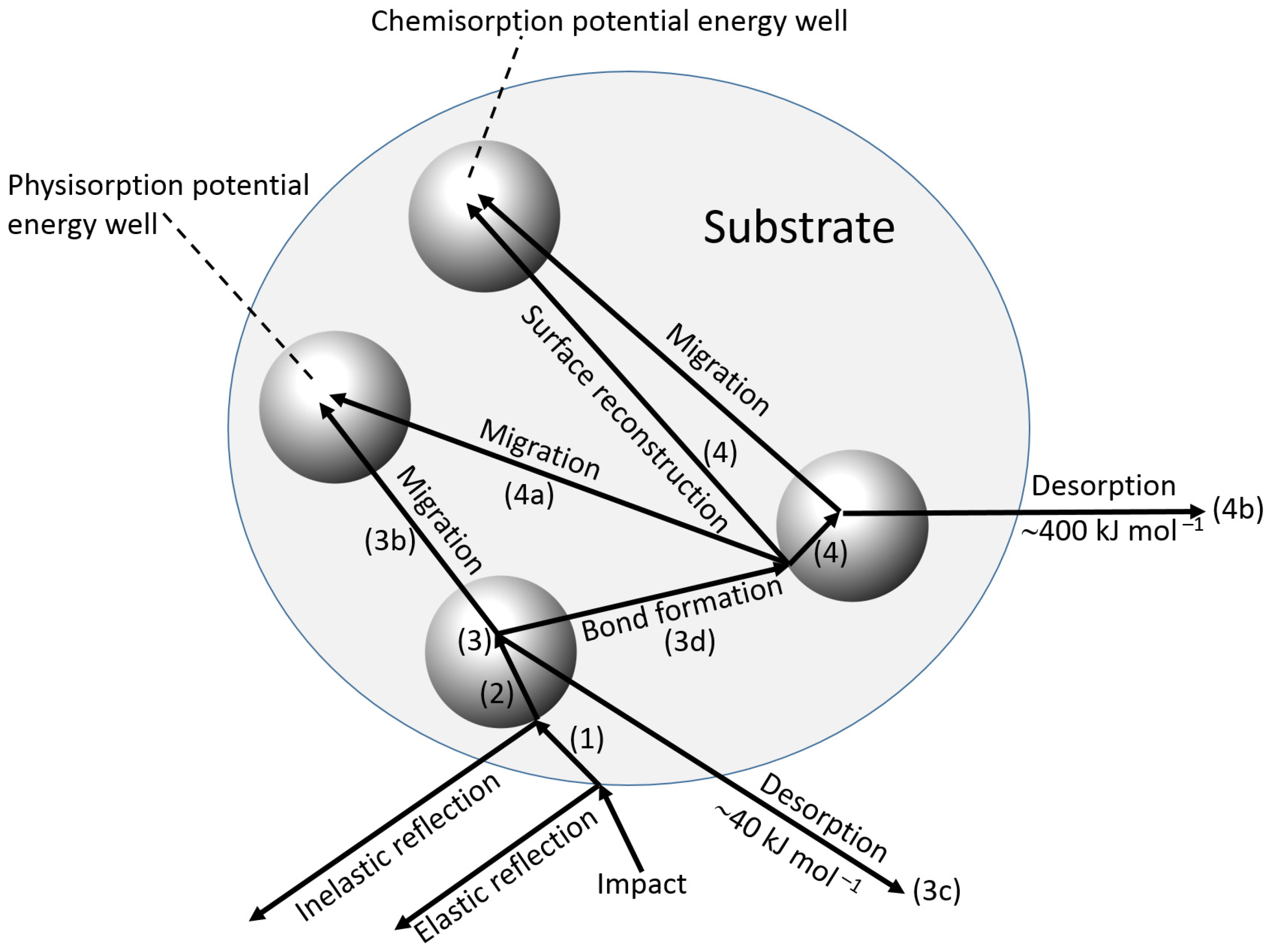
4.1. Reaction at an Interface
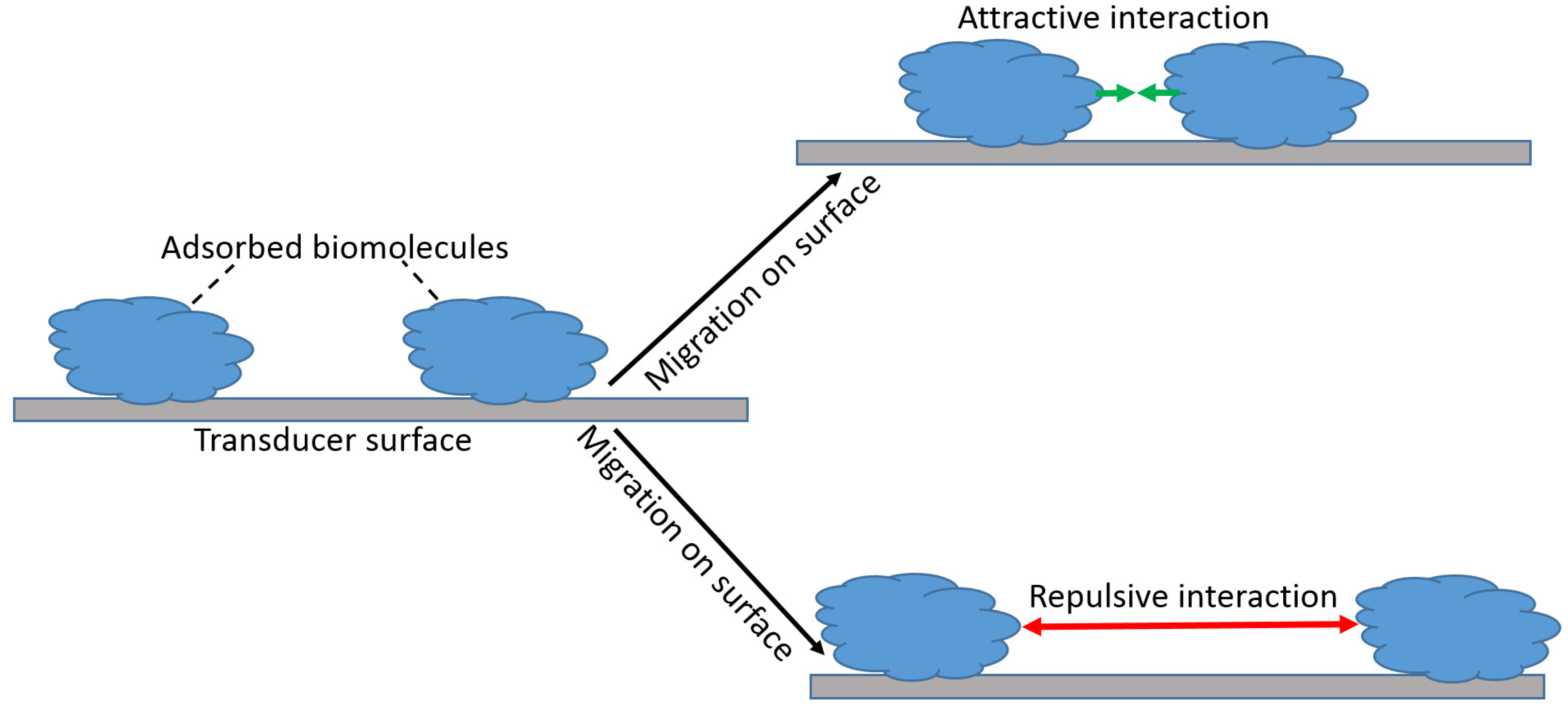
4.2. Range of Forces Affecting Adsorbed Biospecies
- Reflection back into the bulk solution of the adsorbate, with (inelastic) or without (elastic) transfer of energy.
- Transfer of energy (inelastic collision) such that the molecule is unable to “climb” out of the potential well at the surface and is in an excited physisorbed state, which is associated with comparatively weak forces (e.g., van der Waals), and a low enthalpy of adsorption of ca. 40 kJ·mol−1.
- Subsequent possible processes:
- (a)
- Further loss of energy to the surface at the same site.
- (b)
- Migration over the surface with loss of energy at other sites.
- (c)
- Desorption with a gain in energy from the adsorbent.
- (d)
- Transfer to the chemisorbed state, either at the initial site or after migration.
- 4.
- Once chemisorption has occurred, further possibilities exist.
- (a)
- Migration of the chemisorbed species.
- (b)
- Desorption from the chemisorbed state.
- (c)
- Further chemisorption giving multiple attachments.
- 5.
- Surface reaction may take place between the incoming molecule and another species already adsorbed, but not directly involve the substrate.
5. Methods of Immobilization
5.1. Physical Adsorption
5.2. Physical Retention in Polymer Matrices
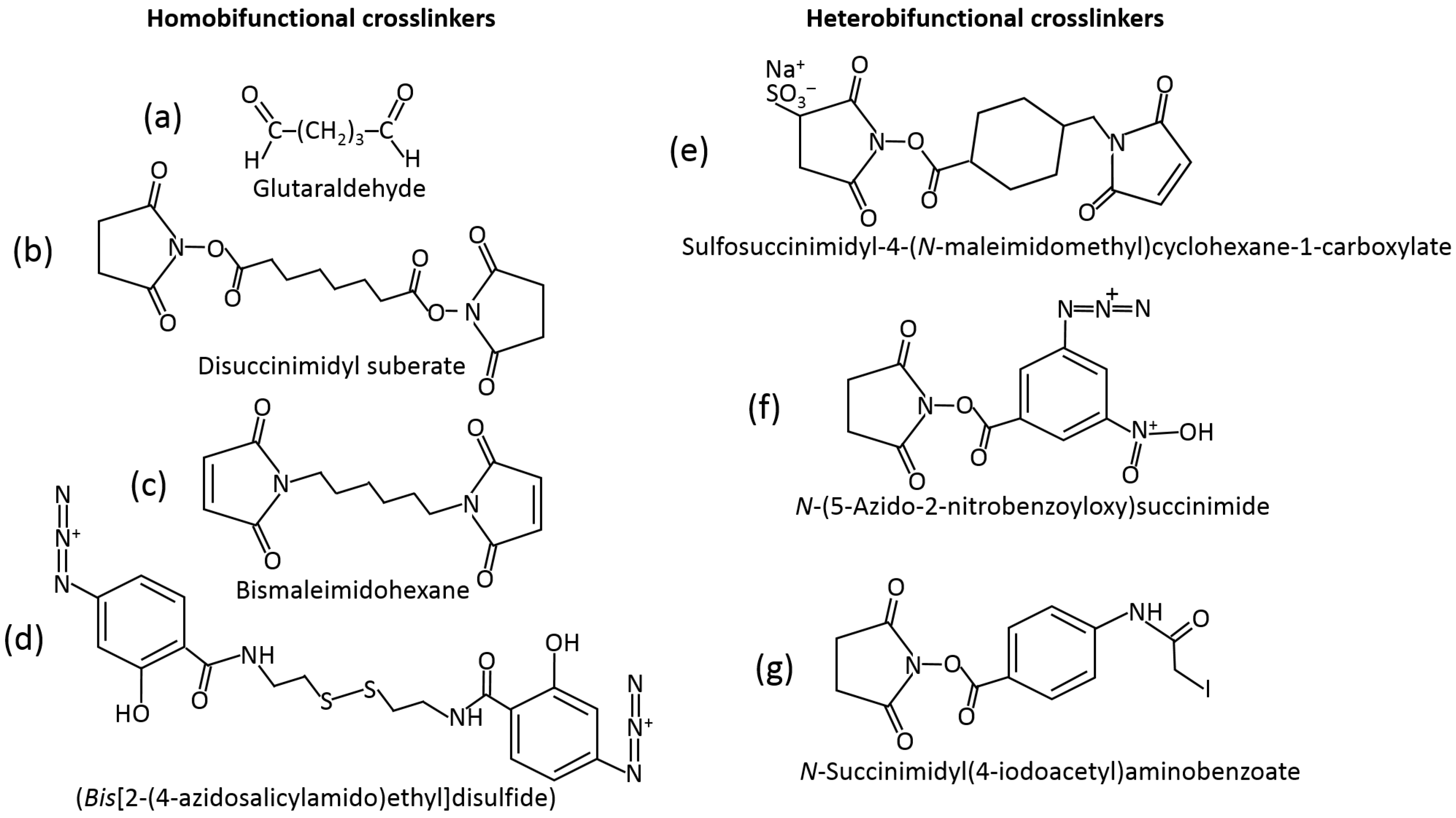
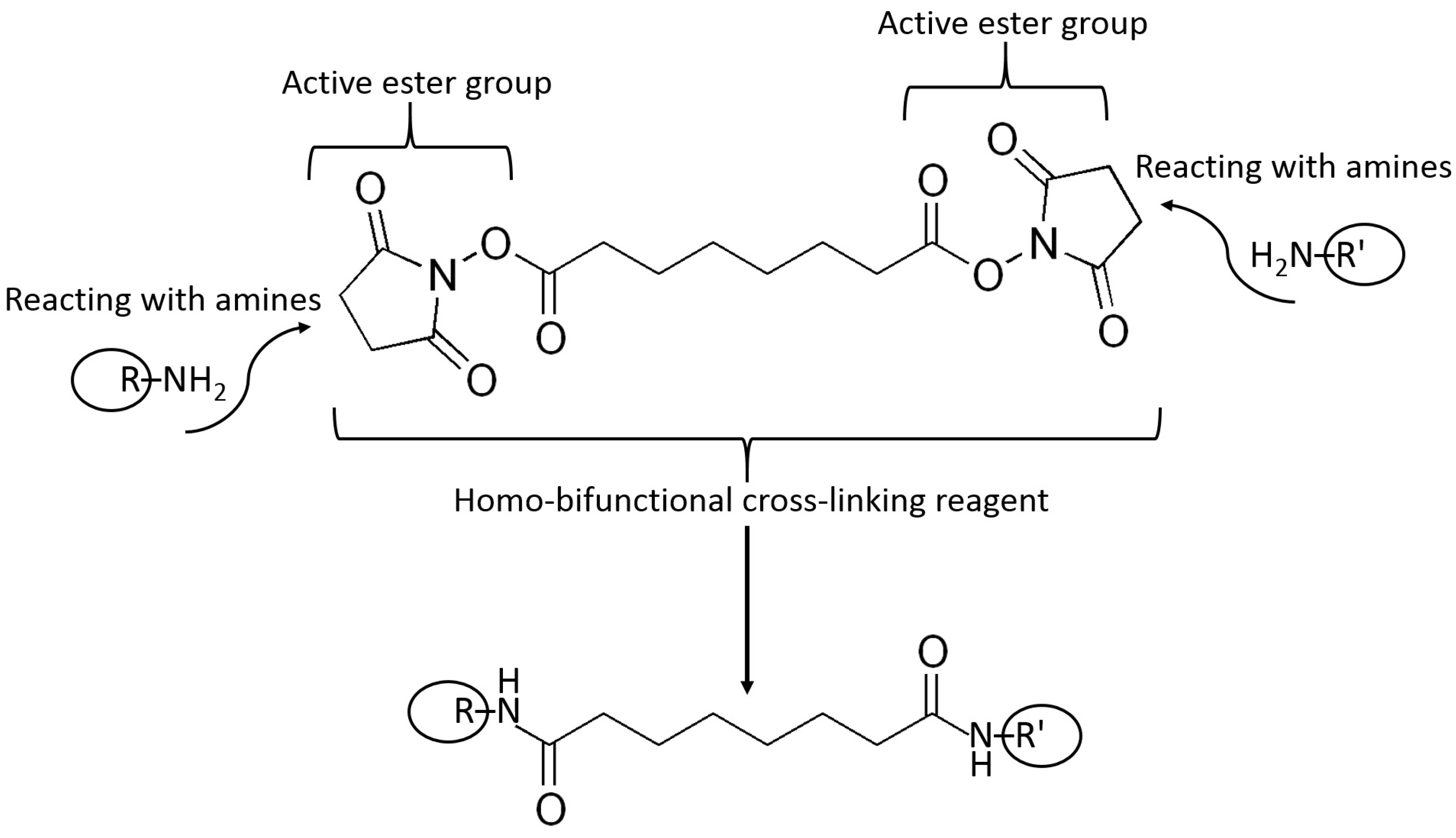
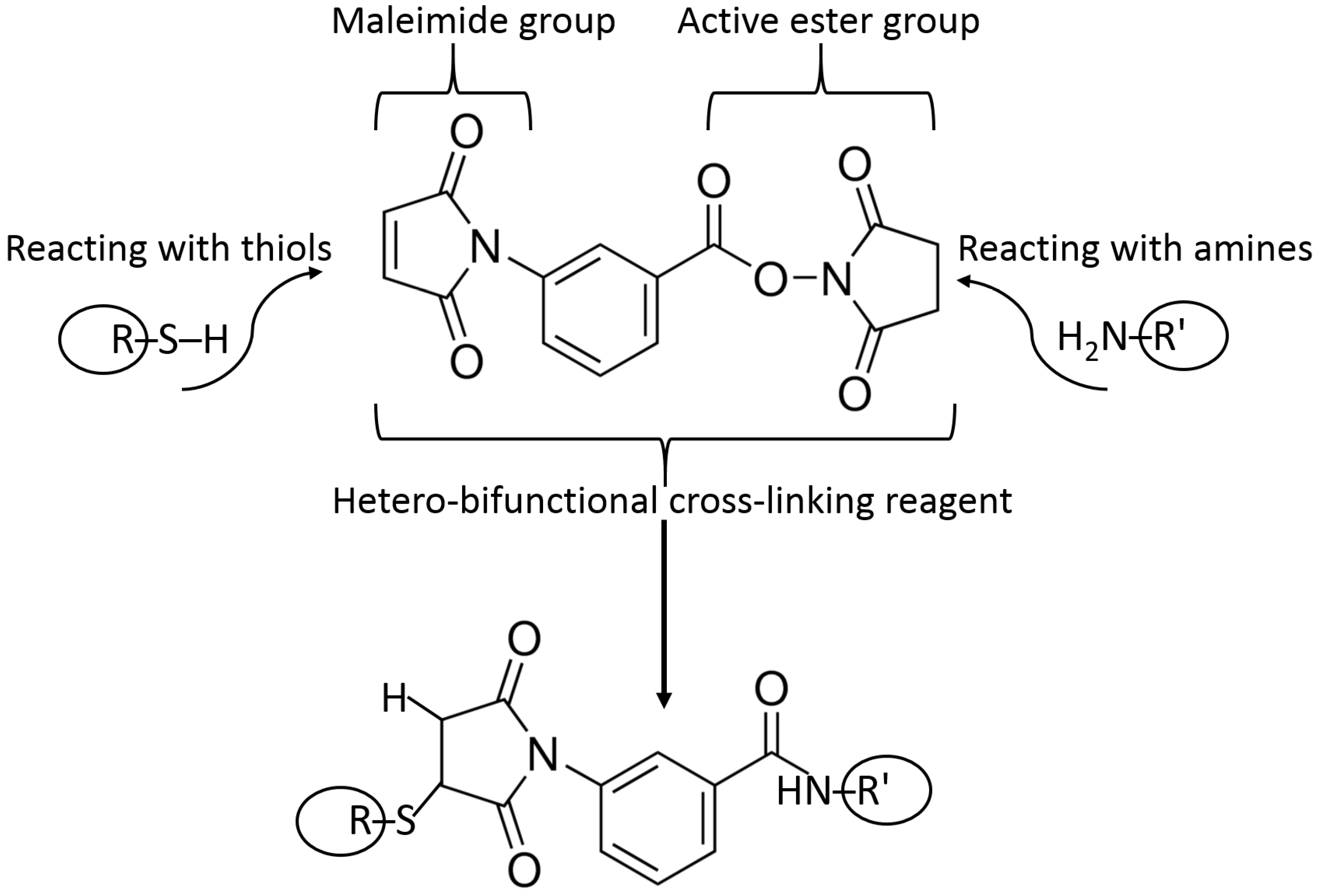
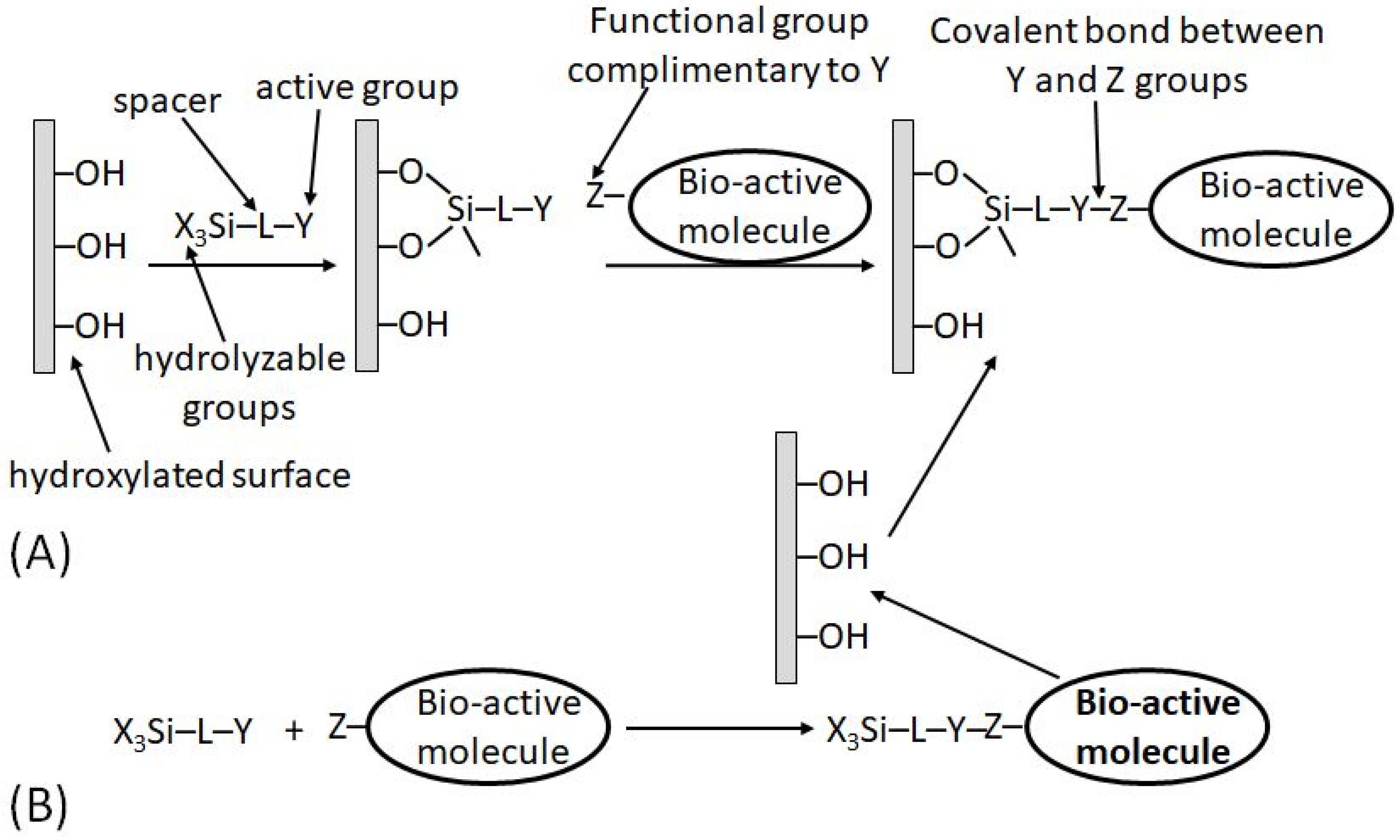
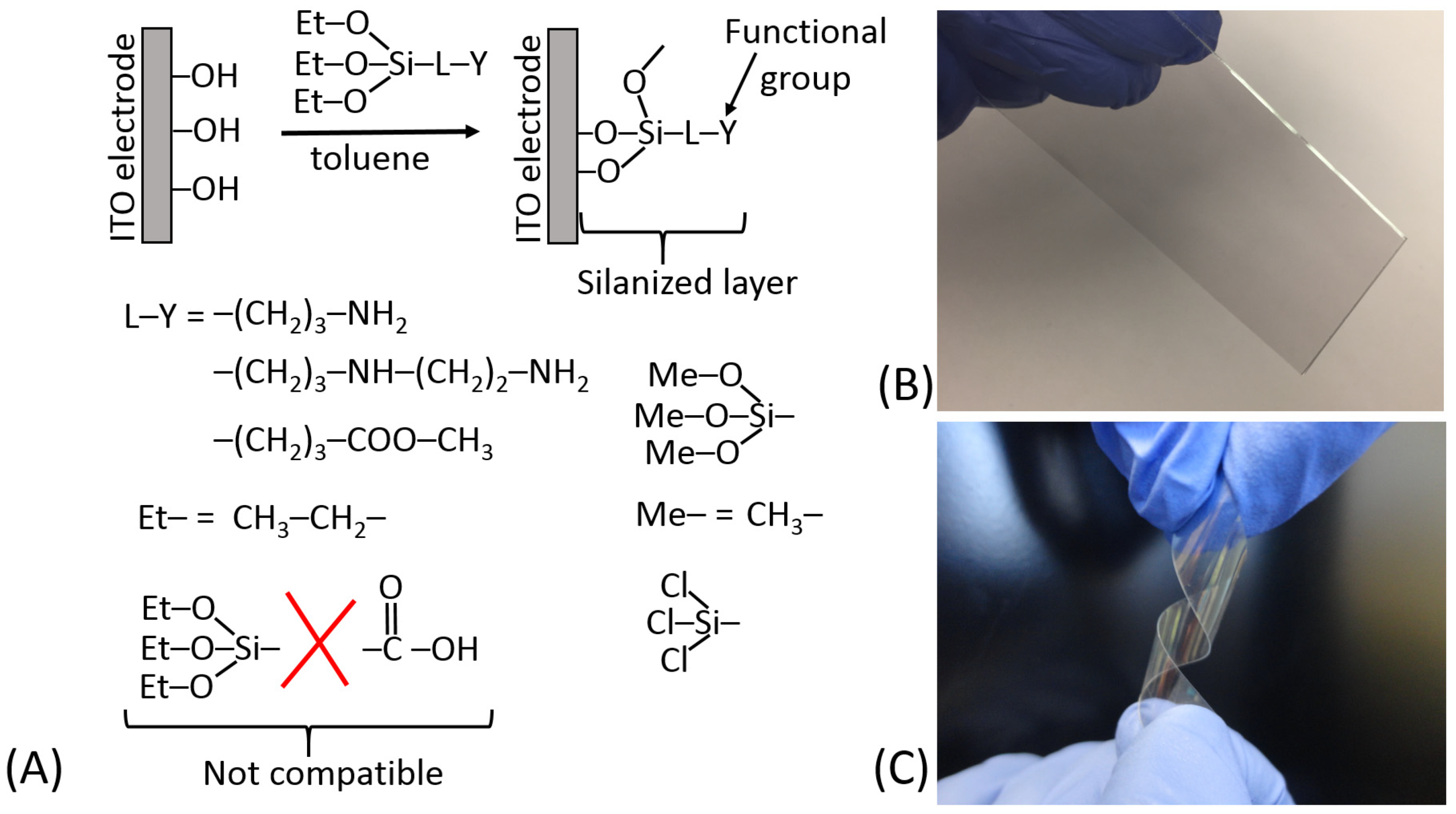
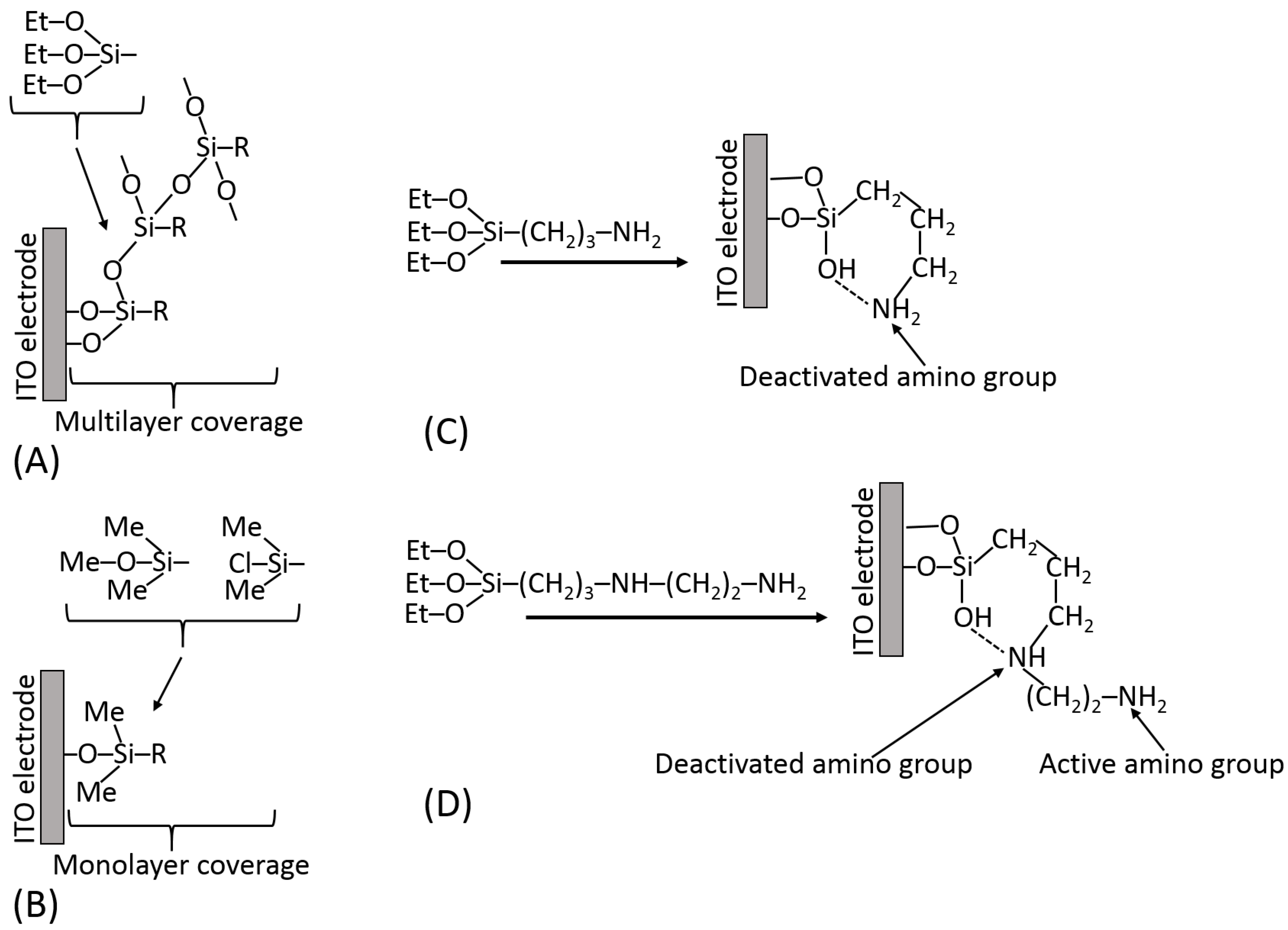
5.3. Surface Modification
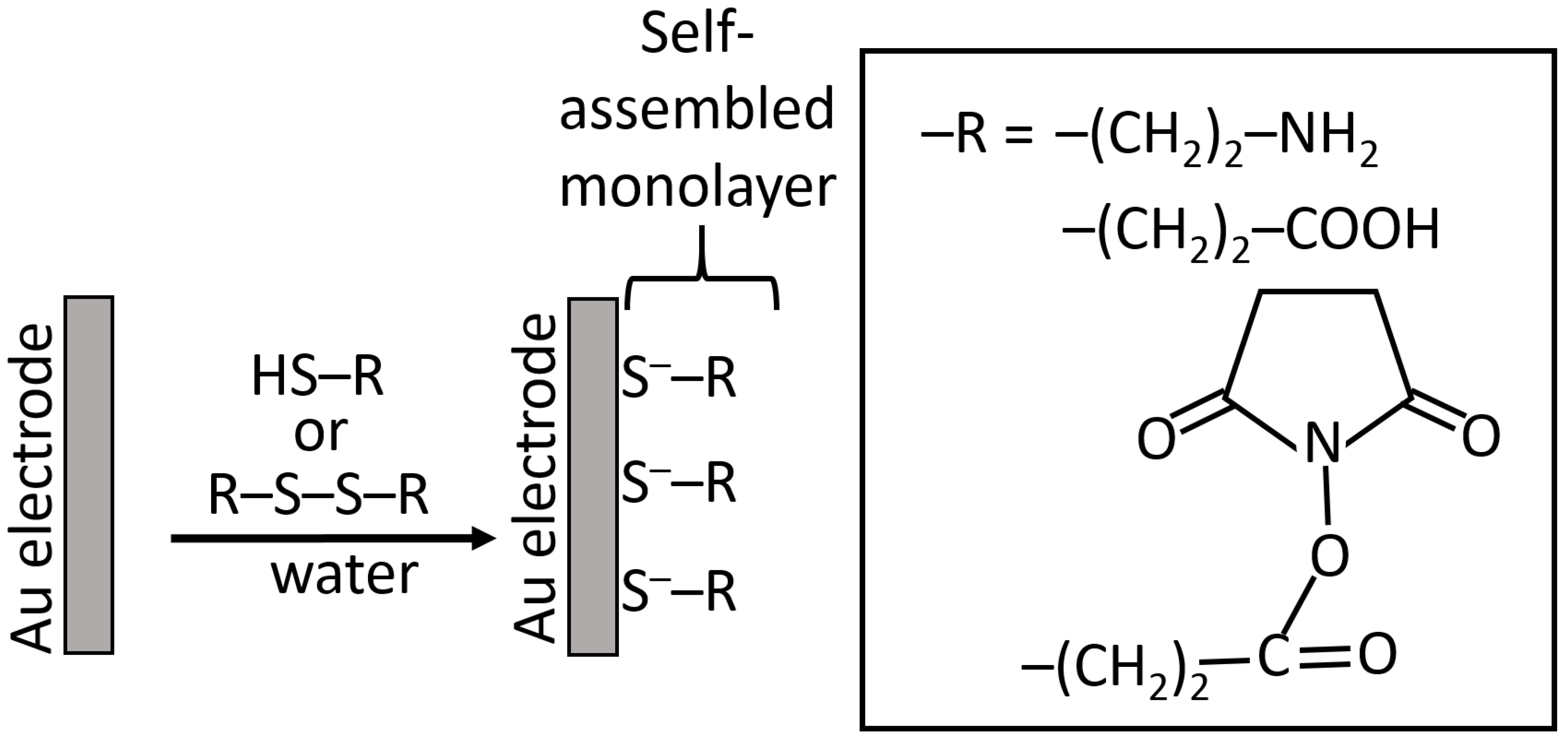
5.4. Organizing Biosensor Interfaces
6. Signal Transduction in Biosensors
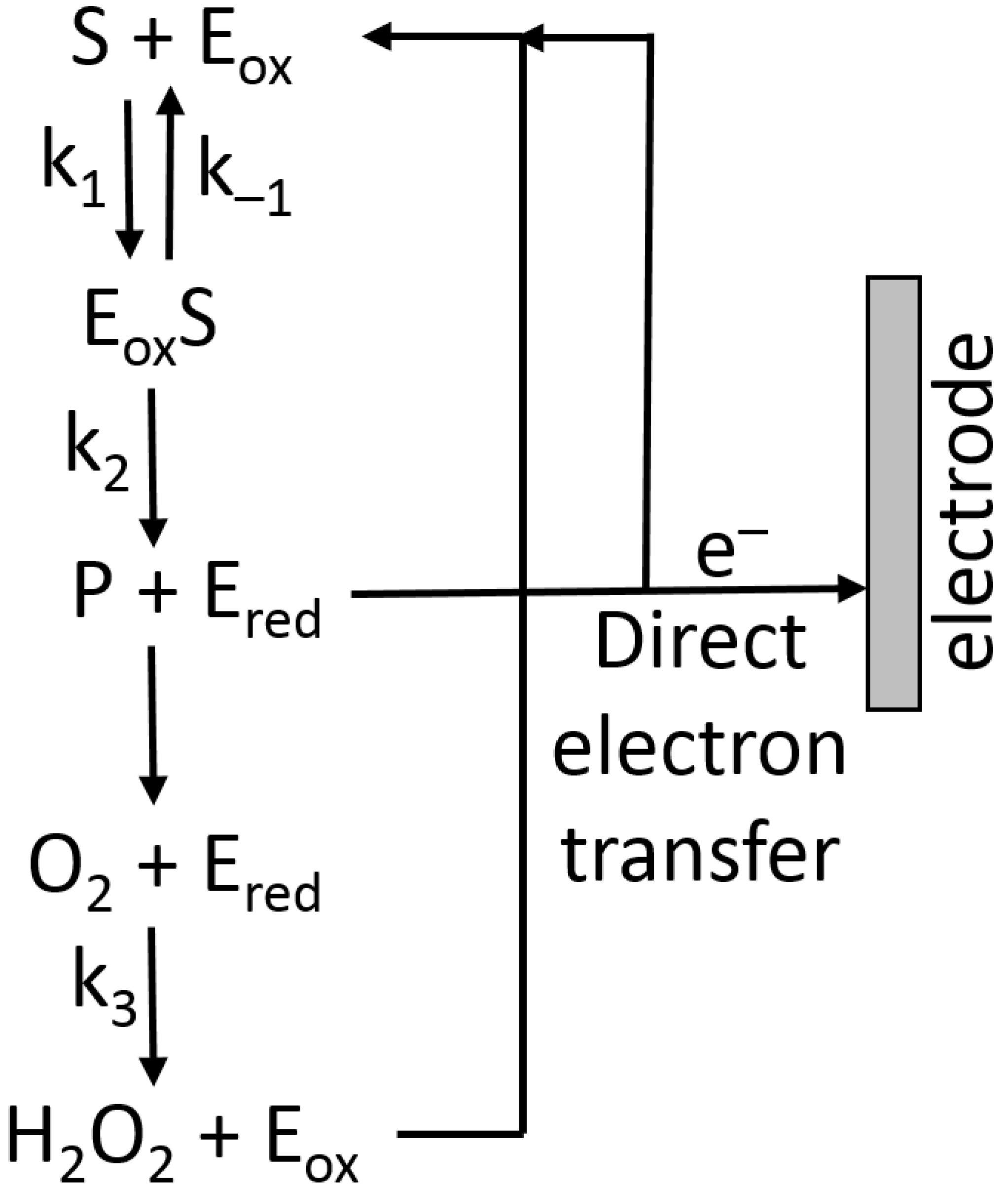
6.1. Amperometric Signal Transduction—Theoretical Consideration and Practical Applications
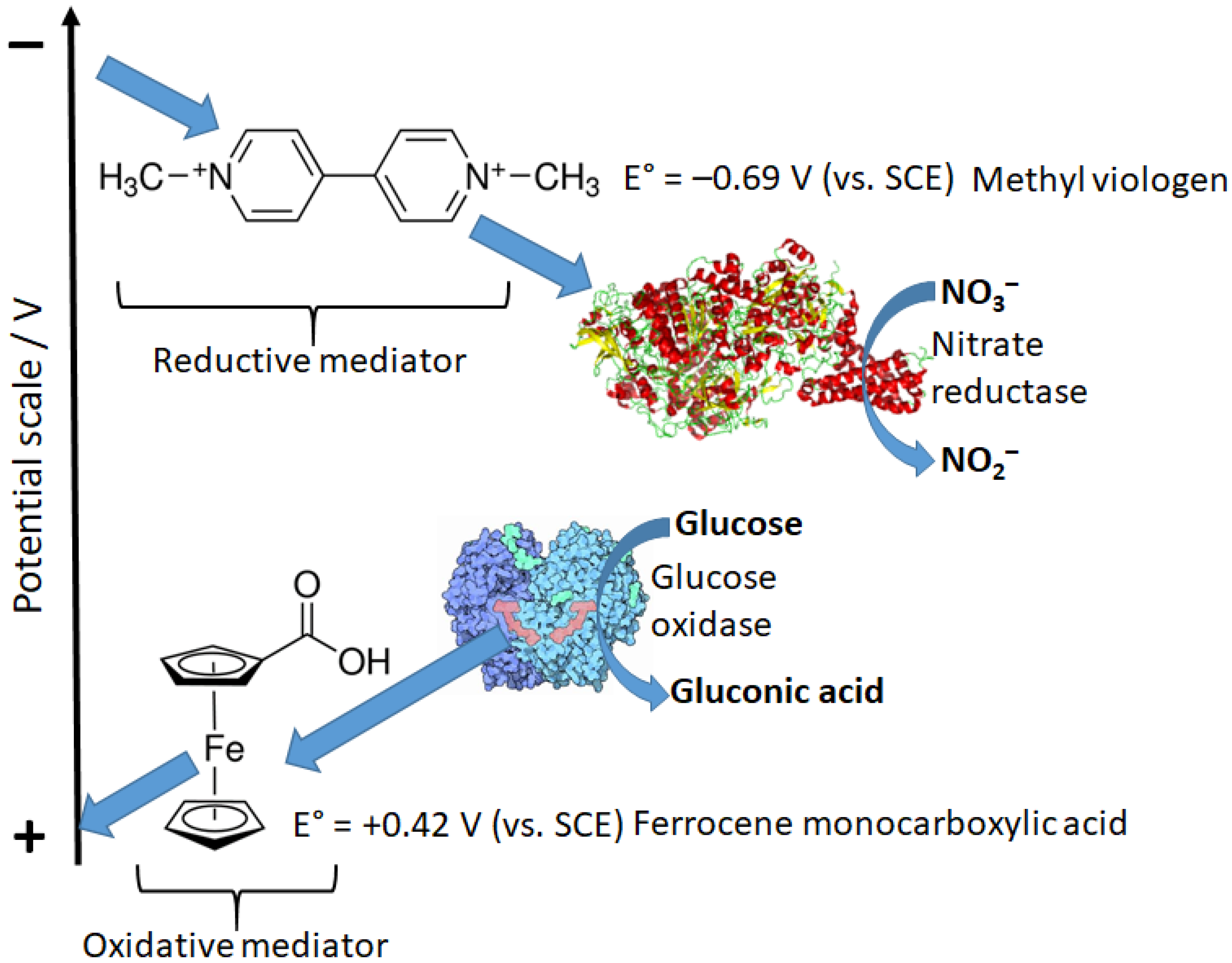
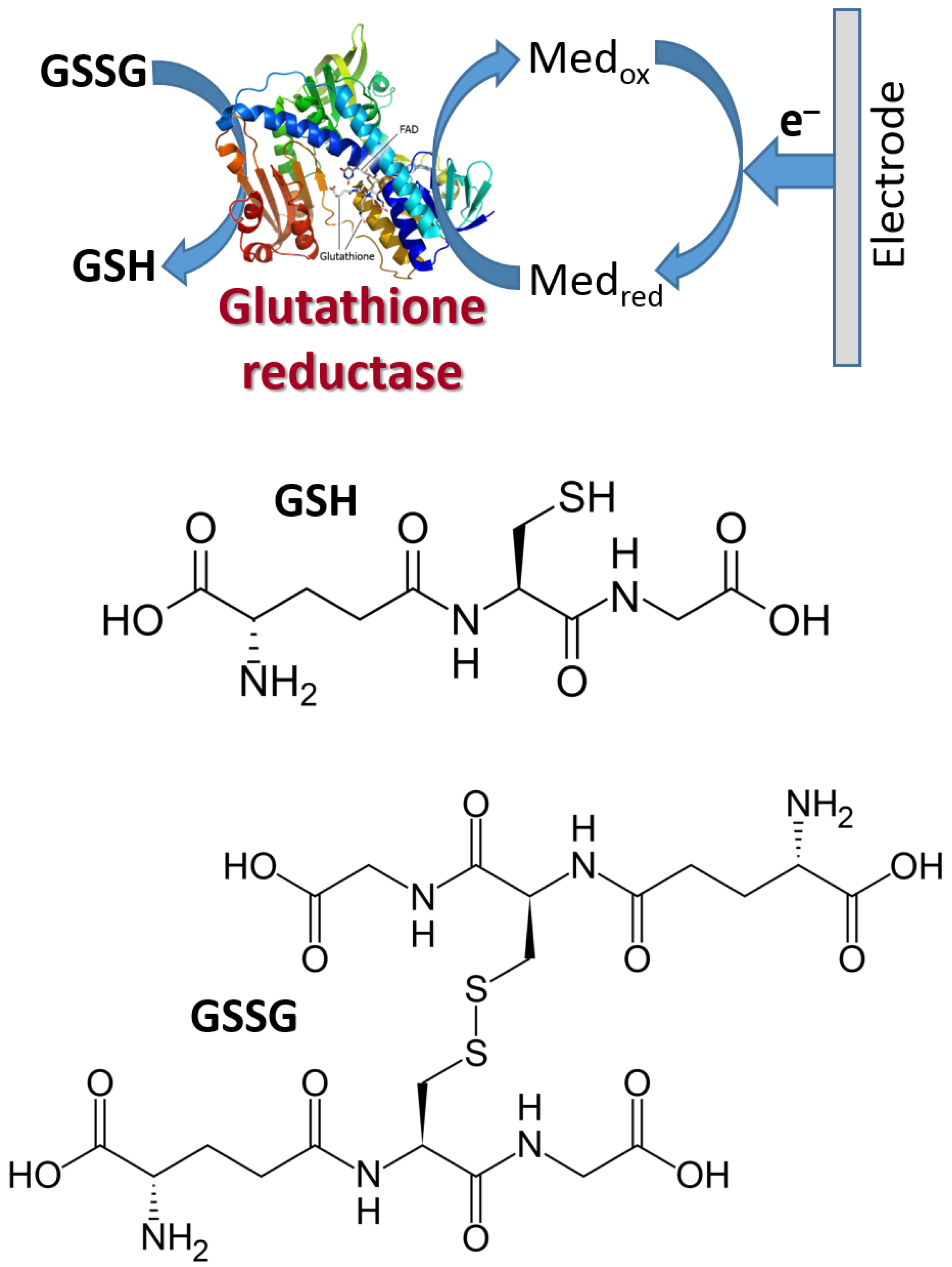
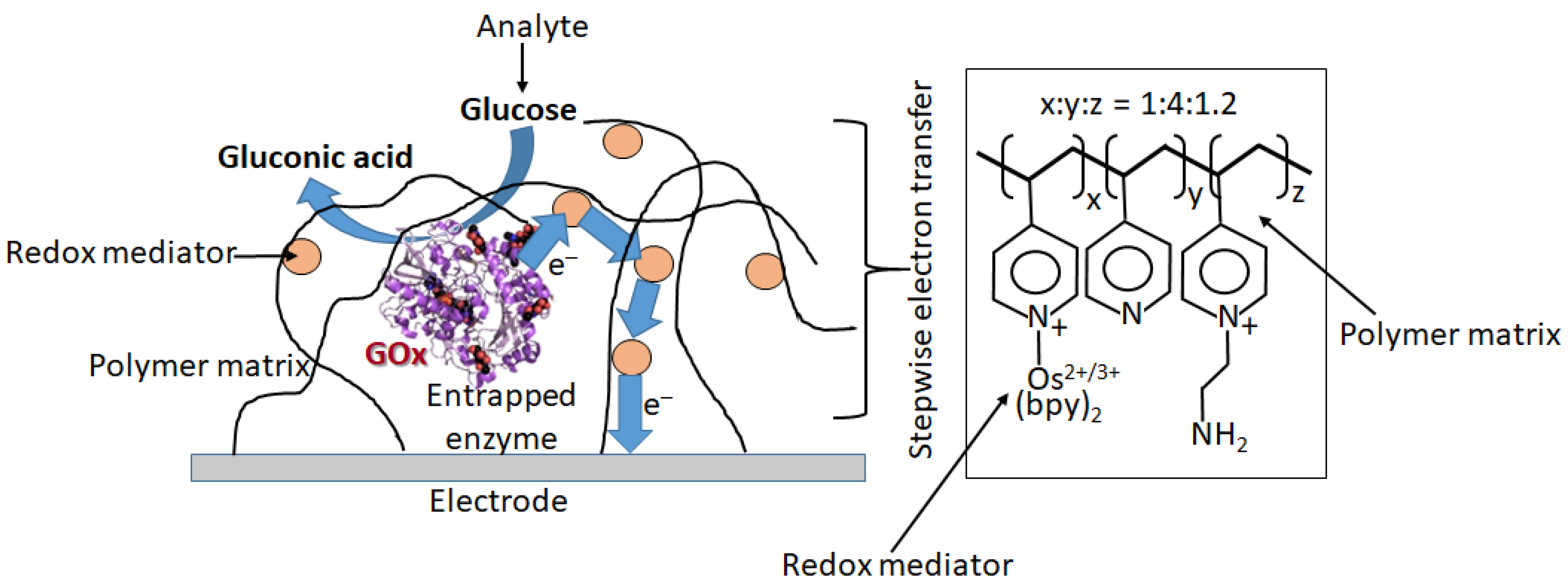
6.2. Electron-Transfer Mediators
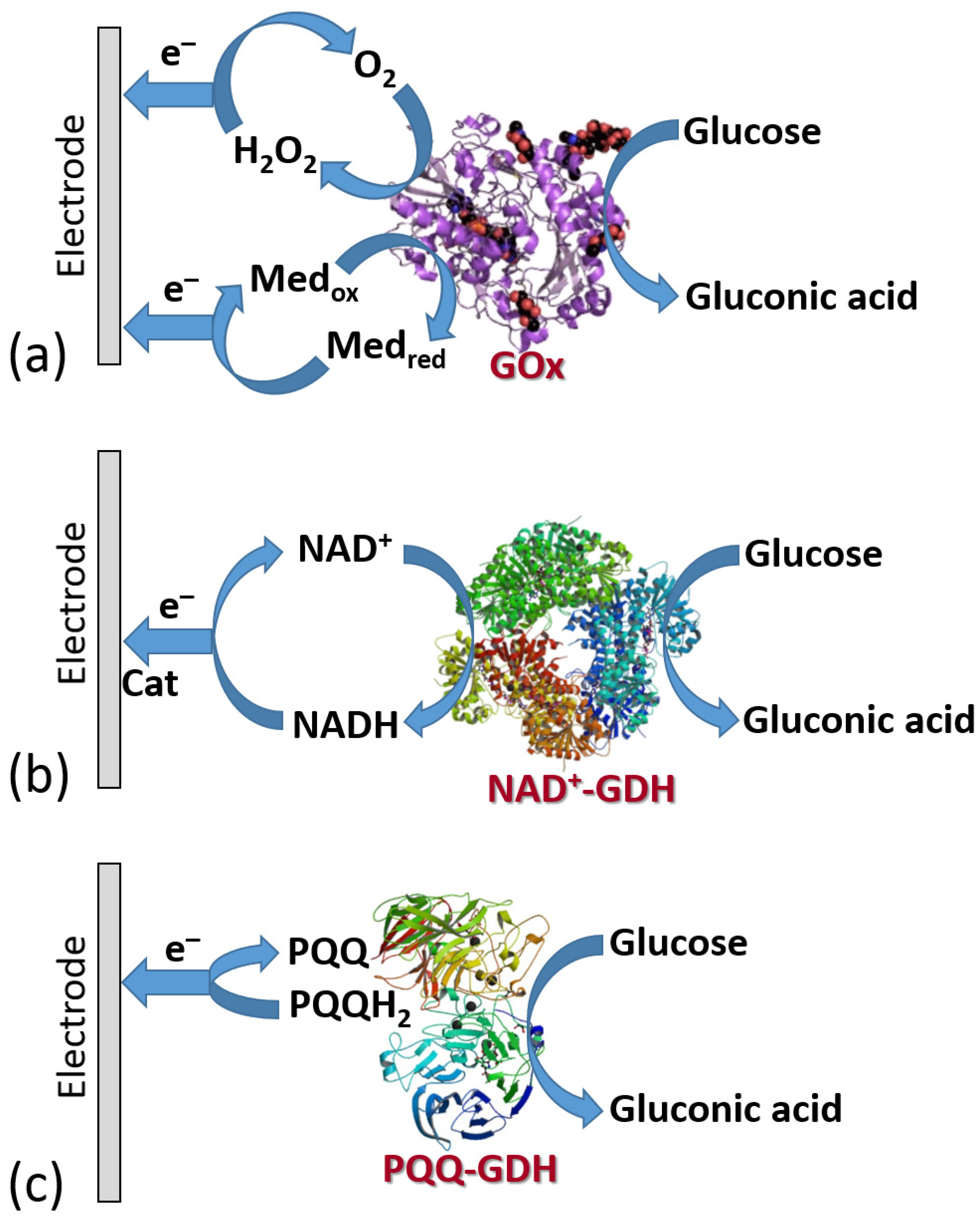
6.3. Electrodes Functionalized with Oxidase Enzymes and Some Other Redox Enzymes
6.4. Biosensors Based on Cells and Cellular Fragments
6.5. Potentiometric Biosensors
7. Brief Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engels, J.W.; Lottspeich, F. (Eds.) Bioanalytics: Analytical Methods and Concepts in Biochemistry and Molecular Biology; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Ozkan, S.A.; Uslu, B.; Mustafa Kemal Sezgintürk, M.K. (Eds.) Biosensors–Fundamentals, Emerging Technologies, and Applications; CRC Press: Boca Raton, FL, USA, 2022. [Google Scholar]
- Ligler, F.; Taitt, C. (Eds.) Optical Biosensors–Today and Tomorrow; Elsevier Science: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Singh, P. Electrochemical Biosensors–Applications in Diagnostics, Therapeutics, Environment, and Food Management; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Vu, C.-A.; Chen, W.-Y. Field-effect transistor biosensors for biomedical applications: Recent advances and future prospects. Sensors 2019, 19, 4214. [Google Scholar] [CrossRef] [Green Version]
- Shpacovitch, V.; Hergenröder, R. Surface plasmon resonance (SPR)-based biosensors as instruments with high versatility and sensitivity. Sensors 2020, 20, 3010. [Google Scholar] [CrossRef]
- Basu, A.K.; Chattopadhyay, P.; Roychoudhuri, U.; Chakraborty, R. Development of cholesterol biosensor based on immobilized cholesterol esterase and cholesterol oxidase on oxygen electrode for the determination of total cholesterol in food samples. Bioelectrochemistry 2007, 70, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Dhull, V.; Gahlaut, A.; Dilbaghi, N.; Hooda, V. Acetylcholinesterase biosensors for electrochemical detection of organophosphorus compounds: A Review. Biochem. Res. Int. 2013, 2013, 731501. [Google Scholar] [CrossRef] [Green Version]
- Wujcik, E.K.; Wei, H.; Zhang, X.; Guo, J.; Yan, X.; Sutrave, N.; Wei, S.; Guo, Z. Antibody nanosensors: A detailed review. RSC Adv. 2014, 4, 43725–43745. [Google Scholar] [CrossRef]
- Hosseini, S.; Vázquez-Villegas, P.; Rito-Palomares, M.; Martinez-Chapa, S.O. Enzyme-Linked Immunosorbent Assay (ELISA)–From A to Z; Springer: Singapore, 2018. [Google Scholar]
- Akama, K.; Iwanaga, N.; Yamawaki, K.; Okuda, M.; Jain, K.; Ueno, H.; Soga, N.; Minagawa, Y.; Noji, H. Wash- and amplification-free digital immunoassay based on single-particle motion analysis. ACS Nano 2019, 13, 13116–13126. [Google Scholar] [CrossRef]
- Teles, F.R.R.; Fonseca, L.P. Trends in DNA biosensors. Talanta 2008, 77, 606–623. [Google Scholar] [CrossRef]
- Han, J.; Wu, J.; Du, J. Fluorescent DNA biosensor for single-base mismatch detection assisted by cationic comb-type copolymer. Molecules 2019, 24, 575. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Renugopalakrishnan, V.; Liepmann, D.; Paulmurugan, R.; Malhotra, B.D. Cell-based biosensors: Recent trends, challenges and future perspectives. Biosens. Bioelectron. 2019, 141, 111435. [Google Scholar] [CrossRef]
- Wei, Y.; Wei, Y.Y.; Jiao, Y.; An, D.; Li, D.; Li, W.; Wei, Q. Review of dissolved oxygen detection technology: From laboratory analysis to online intelligent detection. Sensors 2019, 19, 3995. [Google Scholar] [CrossRef]
- Wang, X.; Guo, W.; Hu, Y.; Wu, J.; Wei, H. Nanozymes: Next Wave of Artificial Enzymes; Springer: Berlin, Germany, 2016. [Google Scholar]
- Hong, C.; Meng, X.; He, J.; Fan, K.; Yan, X. Nanozyme: A promising tool from clinical diagnosis and environmental monitoring to wastewater treatment. Particuology 2022, 71, 90–107. [Google Scholar] [CrossRef]
- Willner, I.; Katz, E. Integration of layered redox-proteins and conductive supports for bioelectronic applications. Angew. Chem. Int. Ed. 2000, 39, 1180–1218. [Google Scholar] [CrossRef]
- Trilling, A.K.; Beekwilder, J.; Zuilhof, H. Antibody orientation on biosensor surfaces: A minireview. Analyst 2013, 138, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Wasserberg, D.; Cabanas-Danés, J.; Prangsma, J.; O’Mahony, S.; Cazade, P.-A.; Tromp, E.; Blum, C.; Thompson, D.; Huskens, J.; Subramaniam, V.; et al. Controlling protein surface orientation by strategic placement of oligo-histidine tags. ACS Nano 2017, 11, 9068–9083. [Google Scholar] [CrossRef] [Green Version]
- Sassolas, A.; Hayat, A.; Marty, J.-L. Enzyme immobilization by entrapment within a gel network. Methods Mol. Biol. 2013, 1051, 229–239. [Google Scholar]
- Trevan, M.D. Enzyme immobilization by covalent bonding. In New Protein Techniques. Methods in Molecular Biology; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 1988; Volume 3. [Google Scholar]
- Saleemuddin, M. Bioaffinity based immobilization of enzymes. Adv. Biochem. Eng. Biotechnol. 1999, 64, 203–226. [Google Scholar]
- Tverdokhlebova, A.; Sterin, I.; Darie, C.C.; Katz, E.; Smutok, O. Stimulation/inhibition of protein release from alginate hydrogel using electrochemically generated local pH changes. ACS Appl. Mater. Interfaces 2022. [Google Scholar] [CrossRef]
- Hall, E. Biosensors. In Analytical Chemistry: A Modern Approach to Analytical Science; Kellner, R., Mermet, J.-M., Otto Valcárcel, M., Widmer, H.M., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp. 1079–1109. [Google Scholar]
- Al-Ghouti, M.A.; Dana, A.; Da’ana, D. Guidelines for the use and interpretation of adsorption isotherm models: A review. J. Hazard. Mater. 2020, 393, 122383. [Google Scholar] [CrossRef]
- Smith, J.R. (Ed.) Theory of Chemisorption; Springer: Berlin/Heidelberg, Germany, 1980. [Google Scholar]
- Kreuzer, H.J.; Gortel, Z.W.J.; Toennies, P. (Eds.) Physisorption Kinetics; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Bartlett, P.N.; Lipkowski, J.; Alkire, R.C. (Eds.) Electrochemistry of Carbon Electrodes; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Zittel, H.E.; Miller, F.J. A glassy-carbon electrode for voltammetry. Anal. Chem. 1965, 37, 200–203. [Google Scholar] [CrossRef]
- Miller, F.J.; Zittel, H.E. Fabrication and use of pyrolytic graphite electrode for voltammetry in aqueous solutions. Anal. Chem. 1963, 35, 1866–1869. [Google Scholar] [CrossRef]
- Chatterjee, J.; Cardenal, J.; Shellikeri, A. Engineered carbon nanotube buckypaper: A platform for electrochemical biosensors. J. Biomed. Nanotechnol. 2015, 11, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Uchiyama, S. Polymers for biosensors construction. In State of the Art in Biosensors; Rinken, T., Ed.; IntechOpen: Rijeka, Croatia, 2012. [Google Scholar]
- Kulkarni, T.; Slaughter, G. Application of semipermeable membranes in glucose biosensing. Membranes 2016, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Meyer, L.-E.; Kara, S. Enzyme immobilization in hydrogels: A perfect liaison forefficient and sustainable biocatalysis. Eng. Life Sci. 2022, 22, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Angar, N.-E.; Aliouche, D. An enhanced immobilization of BSA biomolecule on anionic hydrogels: Swelling and adsorption modeling. Chem. Pap. 2017, 71, 1389–1397. [Google Scholar] [CrossRef]
- Gombotz, W.R.; Hoffman, A.S. Immobilization of biomolecules and cells on and within synthetic polymeric hydrogels. In Hydrogels in Medicine and Pharmacy; Peppas, N.A., Ed.; CRC Press: Boca Raton, FL, USA, 1986. [Google Scholar]
- Ismail, H.; Irani, M.; Ahmad, Z. Starch-based hydrogels: Present status and applications. Int. J. Polym. Mater. Polym. Biomater. 2013, 62, 411–420. [Google Scholar] [CrossRef]
- Xu, X.; Jha, A.K.; Harrington, D.A.; Farach-Carson, M.C.; Jia, X. Hyaluronic acid-based hydrogels: From a natural polysaccharide to complex networks. Soft Matter. 2012, 8, 3280–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Yanga, F.; Guo, Z. The chitosan hydrogels: From structure to function. New J. Chem. 2018, 42, 17162–17180. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [Green Version]
- Fijul Kabir, S.M.; Sikdar, P.P.; Haque, B.; Rahman Bhuiyan, M.A.; Ali, A.; Islam, M.N. Cellulose-based hydrogel materials: Chemistry, properties and their prospective applications. Prog. Biomater. 2018, 7, 153–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaipan, P.; Nguyen, A.; Narayan, R.J. Gelatin-based hydrogels for biomedical applications. MRS Commun. 2017, 7, 416–426. [Google Scholar] [CrossRef]
- Antoine, E.E.; Vlachos, P.P.; Rylander, M.N. Review of collagen I hydrogels for bioengineered tissue microenvironments: Characterization of mechanics, structure, and transport. Tissue Eng. B Rev. 2014, 20, 683–696. [Google Scholar] [CrossRef] [Green Version]
- Cross-Linking Reagents (Technical Handbook) Pierce. Available online: https://www.korambiotech.com/upload/bbs/2/Cross-LinkingTechHB.pdf (accessed on 27 December 2022).
- Farris, S.; Song, J.; Huang, Q. Alternative reaction mechanism for the cross-linking of gelatin with glutaraldehyde. J. Agric. Food Chem. 2010, 58, 998–1003. [Google Scholar] [CrossRef]
- Wong, S.S.; Jameson, D.M. Homobifunctional cross-linking reagents. In Chemistry of Protein and Nucleic Acid Cross-Linking and Conjugation; Wong, S.S., Jameson, D.M., Eds.; Chapter 5; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Wong, S.S.; Jameson, D.M. Heterobifunctional cross-linkers. In Chemistry of Protein and Nucleic Acid Cross-Linking and Conjugation; Wong, S.S., Jameson, D.M., Eds.; Chapter 6; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Gholamian, S.; Nourani, M.; Bakhshi, N. Formation and characterization of calcium alginate hydrogel beads filled with cumin seeds essential oil. Food Chem. 2021, 338, 128143. [Google Scholar] [CrossRef]
- Massana Roquero, D.; Othman, A.; Melman, A.; Katz, E. Iron(III)-cross-linked alginate hydrogels: A critical review. Mater. Adv. RSC 2022, 3, 1849–1873. [Google Scholar] [CrossRef]
- Massana Roquero, D.; Bollella, P.; Katz, E.; Melman, A. Controlling porosity of calcium alginate hydrogel by interpenetrating polyvinyl alcohol-diboronate polymer network. ACS Appl. Polym. Mater. 2021, 3, 1499–1507. [Google Scholar] [CrossRef]
- Arif, U.; Haider, S.; Haider, A.; Khan, N.; Alghyamah, A.A.; Jamila, N.; Khan, M.I.; Almasry, W.A.; Kang, I.-K. Biocompatible polymers and their potential biomedical applications: A review. Curr. Pharm. Des. 2019, 25, 3608–3619. [Google Scholar] [CrossRef]
- Imoto, I. The mechanism of film formation of emulsions. Prog. Org. Coat. 1974, 2, 193–205. [Google Scholar] [CrossRef]
- Jin, Z.; Güven, G.; Bocharova, V.; Halámek, J.; Tokarev, I.; Minko, S.; Melman, A.; Mandler, D.; Katz, E. Electrochemically controlled drug-mimicking protein release from iron-alginate thin-films associated with an electrode. ACS Appl. Mater. Interfaces 2012, 4, 466–475. [Google Scholar] [CrossRef]
- Jin, Z.; Harvey, A.M.; Mailloux, S.; Halámek, J.; Bocharova, V.; Twiss, M.R.; Katz, E. Electrochemically stimulated release of lysozyme from alginate matrix cross-linked with iron cations. J. Mater. Chem. 2012, 22, 19523–19528. [Google Scholar] [CrossRef]
- Monton, M.R.N.; Forsberg, E.M.; Brennan, J.D. Tailoring sol–gel-derived silica materials for optical biosensing. Chem. Mater. 2012, 24, 796–811. [Google Scholar] [CrossRef]
- Motta, N.; Guadalupe, A.R. Activated carbon paste electrodes for biosensors. Anal. Chem. 1994, 66, 566–571. [Google Scholar] [CrossRef]
- Lakard, B.; Herlem, G.; Lakard, S.; Antoniou, A.; Fahys, B. Urea potentiometric biosensor based on modified electrodes with urease immobilized on polyethylenimine films. Biosens. Bioelectron. 2004, 19, 1641–1647. [Google Scholar] [CrossRef]
- Pompe, T.; Zschoche, S.; Herold, N.; Salchert, K.; Gouzy, M.-F.; Sperling, C.; Werner, C. Maleic anhydride copolymers–a versatile platform for molecular biosurface engineering. Biomacromolecules 2003, 4, 1072–1079. [Google Scholar] [CrossRef]
- Hervás Pérez, J.P.; López-Cabarcos, E.; López-Ruiz, B. The application of methacrylate-based polymers to enzyme biosensors. Biomol. Eng. 2006, 23, 233–245. [Google Scholar] [CrossRef]
- Katz, E.; Shkuropatov, A.Y.; Vagabova, O.I.; Shuvalov, V.A. Chemical modification of the PtO electrode by naphthoquinone using aminosilane. J. Electroanal. Chem. 1989, 260, 53–62. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1170. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Benitez, G.; Carro, P.; Salvarezz, R.C. Self-assembled monolayers of thiols and dithiols on gold: New challenges for a well-known system. Chem. Soc. Rev. 2010, 39, 1805–1834. [Google Scholar] [CrossRef]
- Ishida, T.; Yamamoto, S.; Mizutani, W.; Motomatsu, M.; Tokumoto, H.; Hokari, H.; Azehara, H.; Fujihira, M. Evidence for cleavage of disulfides in the self-assembled monolayer on Au(111). Langmuir 1997, 13, 3261–3265. [Google Scholar] [CrossRef]
- Katz, E.; Borovkov, V.V.; Evstigneeva, R.P. Application of quinone thio derivatives as a basis for assembling complex molecular systems at an electrode surface. J. Electroanal. Chem. 1992, 326, 197–212. [Google Scholar] [CrossRef]
- Katz, E.; Solov’ev, A.A. Chemical modification of platinum and gold electrodes by naphthoquinones using amines containing sulfhydryl or disulphide groups. J. Electroanal. Chem. 1990, 291, 171–186. [Google Scholar] [CrossRef]
- Badenhorst, R.; Krishna Kadambar, V.; Bellare, M.; Melman, A.; Katz, E.; Smutok, O. Electrochemically produced local pH changes stimulating (bio)molecule release from pH-switchable electrode-immobilized avidin-biotin systems. Phys. Chem. Chem. Phys. 2022, 24, 6410–6414. [Google Scholar] [CrossRef]
- Gao, Z.F.; Gao, J.B.; Zhou, L.Y.; Zhang, Y.; Si, J.C.; Luo, H.Q.; Li, N.B. Rapid assembly of ssDNA on gold electrode surfaces at low pH and high salt concentration conditions. RSC Adv. 2013, 3, 12334–12340. [Google Scholar] [CrossRef]
- Singh, S.; Wang, J.; Cinti, S. Review—An overview on recent progress in screen-printed electroanalytical (bio)sensors. ECS Sens. Plus 2022, 1, 023401. [Google Scholar] [CrossRef]
- Sui, Y.; Zorman, C.A. Review—Inkjet printing of metal structures for electrochemical sensor applications. J. Electrochem. Soc. 2020, 167, 037571. [Google Scholar] [CrossRef]
- Luo, C.; Xu, C.; Lv, L.; Li, H.; Huang, X.; Liu, W. Review of recent advances in inorganic photoresists. RSC Adv. 2020, 10, 8385–8395. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L. Enzyme based amperometric biosensors. Curr. Opin. Electrochem. 2018, 10, 157–173. [Google Scholar] [CrossRef]
- Limoges, B.; Marchal, D.; Mavré, F.; Savéant, J.-M.; Schöllhorn, B. Theory and practice of enzyme bioaffinity electrodes. Direct electrochemical product detection. J. Am. Chem. Soc. 2008, 130, 7259–7275. [Google Scholar]
- Bourdillon, C.; Demaille, C.; Moiroux, J.; Saveant, J.-M. Catalysis and mass transport in spatially ordered enzyme assemblies on electrodes. J. Am. Chem. Soc. 1995, 117, 11499–11506. [Google Scholar] [CrossRef]
- Freire, R.S.; Pessoa, C.A.; Mello, L.D.; Kubota, L.T. Direct electron transfer: An approach for electrochemical biosensors with higher selectivity and sensitivity. J. Braz. Chem. Soc. 2003, 14, 230–243. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.S. Native glucose oxidase does not undergo direct electron transfer. Biosens. Bioelectron. 2016, 82, vii–viii. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Al-Lolage, F.A. There is no evidence to support literature claims of direct electron transfer (DET) for native glucose oxidase (GOx) at carbon nanotubes or graphene. J. Electroanal. Chem. 2018, 819, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, T.P. Marcus theory: Thermodynamics CAN control the kinetics of electron transfer reactions. J. Chem. Educ. 2012, 89, 1159–1167. [Google Scholar] [CrossRef]
- Marcus, R.A. On the theory of electron-transfer reaction VI. Unified treatment of homogeneous and electrode reactions. J. Chem. Phys. 1965, 43, 679–701. [Google Scholar]
- Katz, E.; Shipway, A.N.; Willner, I. Mediated electron-transfer between redox-enzymes and electrode supports. In Encyclopedia of Electrochemistry, Vol. 9: Bioelectrochemistry; Wilson, G.S., Bard, A.J., Stratmann, M., Eds.; Chapter 17; Wiley-VCH: Weinheim, Germany, 2002; pp. 559–626. [Google Scholar]
- Martens, N.; Hindle, A.; Hall, E.A.H. An assessment of mediators as oxidants for glucose oxidase in the presence of oxygen. Biosens. Bioelectron. 1995, 10, 393–403. [Google Scholar] [CrossRef]
- Willner, I.; Katz, E.; Riklin, A.; Kasher, R. Mediated electron transfer in gluthathione reductase organized in self-assembled monolayers on gold electrodes. J. Am. Chem. Soc. 1992, 114, 10965–10966. [Google Scholar] [CrossRef]
- Willner, I.; Katz, E.; Lapidot, N.; Baüerle, P. Bioelectrocatalysed reduction of nitrate utilizing polythiophene bipyridinium enzyme electrodes. Bioelectrochem. Bioenerg. 1992, 29, 29–45. [Google Scholar] [CrossRef]
- Katz, E.; Lötzbeyer, T.; Schlereth, D.D.; Schuhmann, W.; Schmidt, H.-L. Electrocatalytic oxidation of reduced nicotinamide coenzymes at gold and platinum electrode surfaces modified with a monolayer of pyrroloquinoline quinone. Effect of Ca2+ cations. J. Electroanal. Chem. 1994, 373, 189–200. [Google Scholar] [CrossRef]
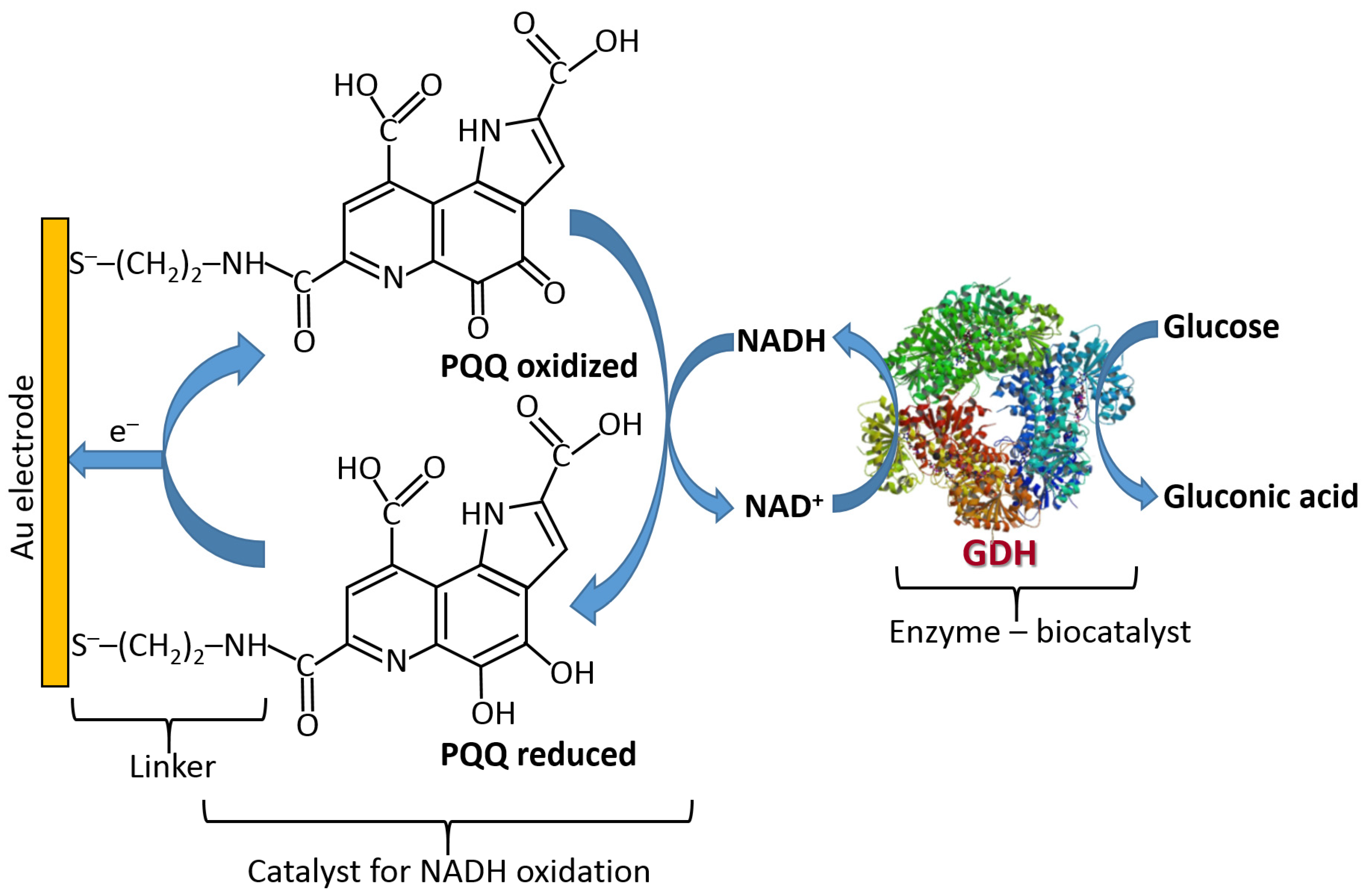
- Gorton, L.; Domínguez, E. Electrocatalytic oxidation of NAD(P) H at mediator-modified electrodes. J. Biotechnol. 2002, 82, 371–392. [Google Scholar] [CrossRef]
- Göbel, G.; Schubart, I.W.; Scherbahn, V.; Lisdat, F. Direct electron transfer of PQQ-glucose dehydrogenase at modified carbon nanotubes electrodes. Electrochem. Commun. 2011, 13, 1240–1243. [Google Scholar] [CrossRef]
- Heller, A.; Feldman, B. Electrochemical glucose sensors and their applications in diabetes management. Chem. Rev. 2008, 108, 2482–2505. [Google Scholar] [CrossRef] [Green Version]
- Clark, L.C., Jr.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef]
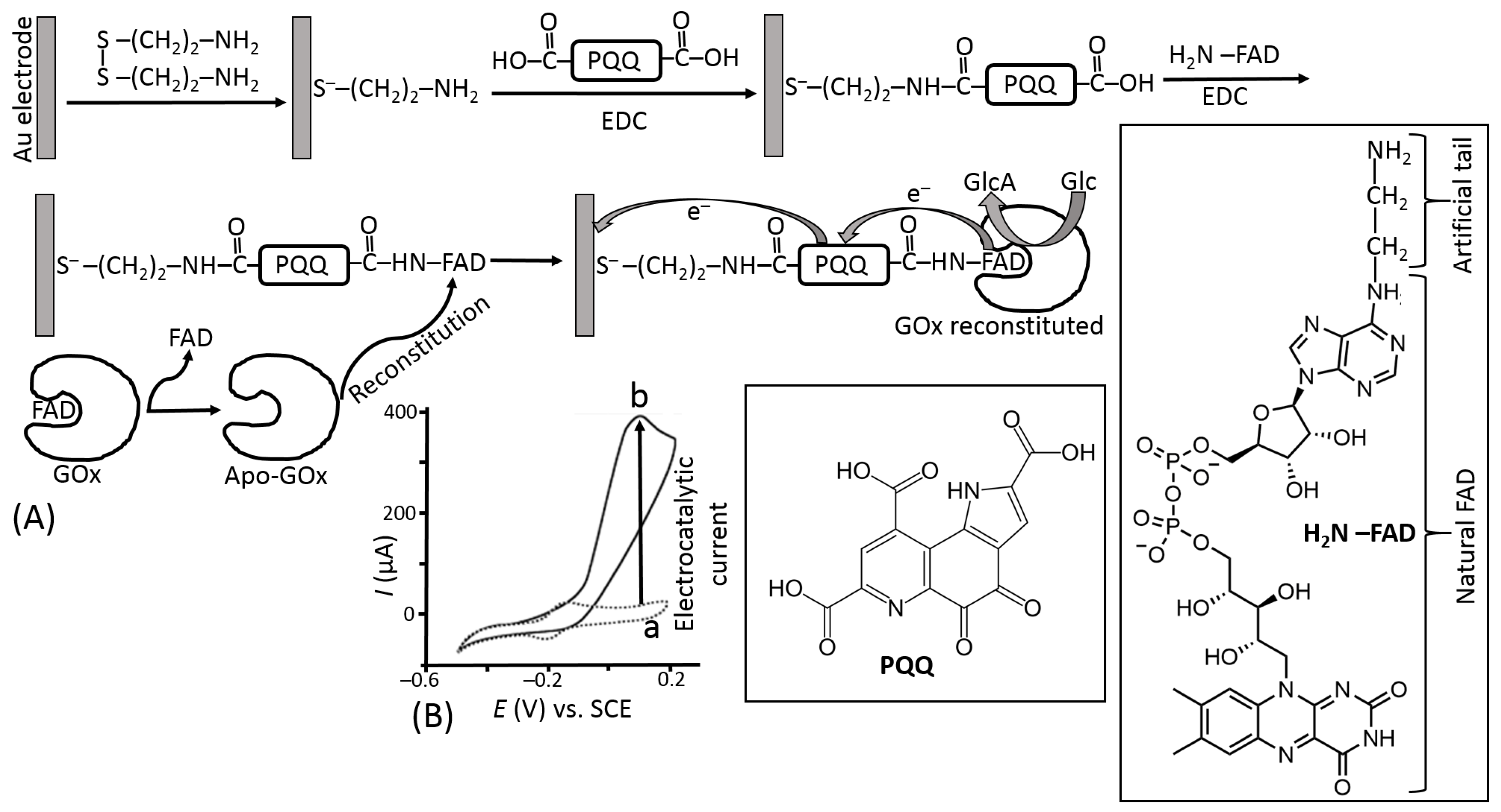
- Willner, I.; Heleg-Shabtai, V.; Blonder, R.; Katz, E.; Tao, G.; Bückmann, A.F.; Heller, A. Electrical wiring of glucose oxidase by reconstitution of FAD-modified monolayers assembled onto Au-electrodes. J. Am. Chem. Soc. 1996, 118, 10321–10322. [Google Scholar] [CrossRef]
- Zayats, M.; Katz, E.; Willner, I. Electrical contacting of glucose oxidase by surface reconstitution of the apo-protein on a relay-boronic acid-FAD cofactor monolayer. J. Am. Chem. Soc. 2002, 124, 2120–2121. [Google Scholar] [CrossRef]
- Eshkenazi, I.; Maltz, E.; Zion, B.; Rishpon, J. A three-cascaded-enzymes biosensor to determine lactose concentration in raw milk. J. Dairy Sci. 2000, 83, 1939–1945. [Google Scholar] [CrossRef]
- Ibadullaeva, S.Z.; Appazov, N.O.; Tarahovsky, Y.S.; Zamyatina, E.A.; Fomkina, M.G.; Kim, Y.A. Amperometric multi-enzyme biosensors: Development and application, a short review. Biophysics 2019, 64, 696–707. [Google Scholar] [CrossRef]
- Kucherenko, S.; Soldatkin, O.O.; Dzyadevych, S.V.; Soldatkin, A.P. Electrochemical biosensors based on multienzyme systems: Main groups, advantages and limitations–A review. Anal. Chim. Acta 2020, 1111, 114–131. [Google Scholar] [CrossRef]
- Willner, I.; Riklin, A. Electrical communication between electrodes and NAD(P)+-dependent enzymes using pyrroloquinolinequinone-enzyme electrodes in a self-assembled monolayer configuration: Design of a new class of amperometric biosensors. Anal. Chem. 1994, 66, 1535–1539. [Google Scholar] [CrossRef]
- Lim, J.W.; Ha, D.; Lee, J.; Lee, S.K.; Kim, T. Review of micro/nanotechnologies for microbial biosensors. Front. Bioeng. Biotechnol. 2015, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- Moraskie, M.; Roshid, M.H.O.; O’Connor, G.; Dikici, E.; Zingg, J.-M.; Deo, S.; Daunert, S. Microbial whole-cell biosensors: Current applications, challenges, and future perspectives. Biosens. Bioelectron. 2021, 191, 113359. [Google Scholar] [CrossRef]
- Ravikumar, S.; Baylon, M.G.; Park, S.J.; Choi, J. Engineered microbial biosensors based on bacterial two-component systems as synthetic biotechnology platforms in bioremediation and biorefinery. Microb. Cell Factories 2017, 16, 62. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Chen, W.; Mulchandani, A. Microbial biosensors. Anal. Chim. Acta 2006, 568, 200–210. [Google Scholar] [CrossRef]
- Chiappini, S.A.; Kormes, D.J.; Bonetto, M.C.; Sacco, N.; Cortón, E. A new microbial biosensor for organic water pollution based on measurement of carbon dioxide production. Sens. Actuat. B 2010, 148, 103–109. [Google Scholar] [CrossRef]
- Endo, H.; Kamata, A.; Hoshi, M.; Hayashi, T.; Watanabe, E. Microbial biosensor system for rapid determination of vitamin B6. Food Sci. 1995, 60, 554–557. [Google Scholar] [CrossRef]
- Riedel, K. Microbial biosensors based on oxygen electrodes. In Enzyme and Microbial Biosensors; Mulchandani, A., Rogers, K.R., Eds.; Humana Press: Totowa, NJ, USA, 1998; pp. 199–223. [Google Scholar]
- Hassan, R.Y.A.; Febbraio, F.; Andreescu, S. Microbial electrochemical systems: Principles, construction and biosensing applications. Sensors 2021, 21, 1279. [Google Scholar] [CrossRef]
- Park, M.; Tsai, S.-L.; Chen, W. Microbial biosensors: Engineered microorganisms as the sensing machinery. Sensors 2013, 13, 5777–5795. [Google Scholar] [CrossRef] [Green Version]
- Andriukonis, E.; Celiesiute-Germaniene, R.; Ramanavicius, S.; Viter, R.; Ramanavicius, A. From microorganism-based amperometric biosensors towards microbial fuel cells. Sensors 2021, 21, 2442. [Google Scholar] [CrossRef]
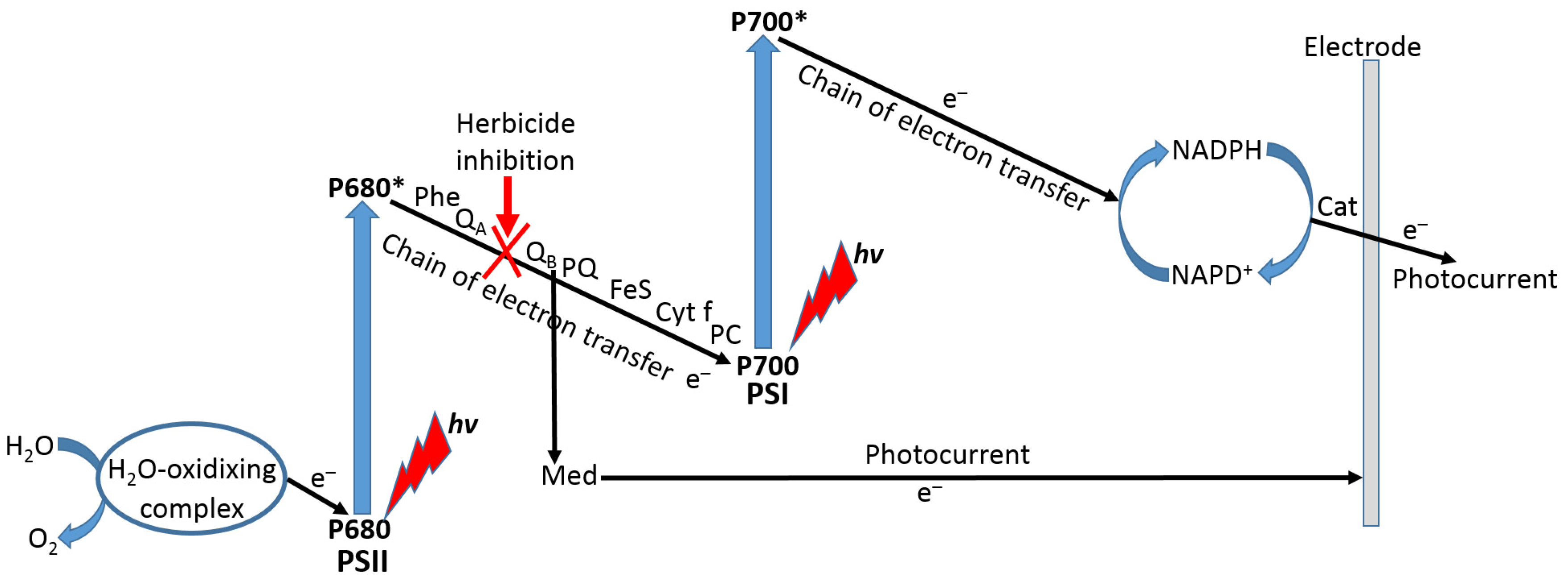
- Shao, C.Y.; Howe, C.J.; Porter, A.J.R.; Glover, L.A. Novel cyanobacterial biosensor for detection of herbicides. Appl. Environ. Microbiol. 2002, 68, 5026–5033. [Google Scholar] [CrossRef] [Green Version]
- Shing, W.L.; Heng, L.Y.; Surif, S. Performance of a cyanobacteria whole cell-based fluorescence biosensor for heavy metal and pesticide detection. Sensors 2013, 13, 6394–6404. [Google Scholar]
- Li, J.; Wei, X.; Peng, T. Fabrication of herbicide biosensors based on the inhibition of enzyme activity that catalyzes the scavenging of hydrogen peroxide in a thylakoid membrane. Anal. Sci. 2005, 21, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Bettazzi, F.; Laschi, S.; Mascini, M. One-shot screen-printed thylakoid membrane-based biosensor for the detection of photosynthetic inhibitors in discrete samples. Anal. Chim. Acta 2007, 589, 14–21. [Google Scholar] [CrossRef]
- Mikhelson, K.N. Ion-Selective Electrodes; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Jackson, D.T.; Nelson, P.N. Preparation and properties of some ion selective membranes: A review. J. Molec. Struct. 2019, 1182, 241–259. [Google Scholar] [CrossRef]
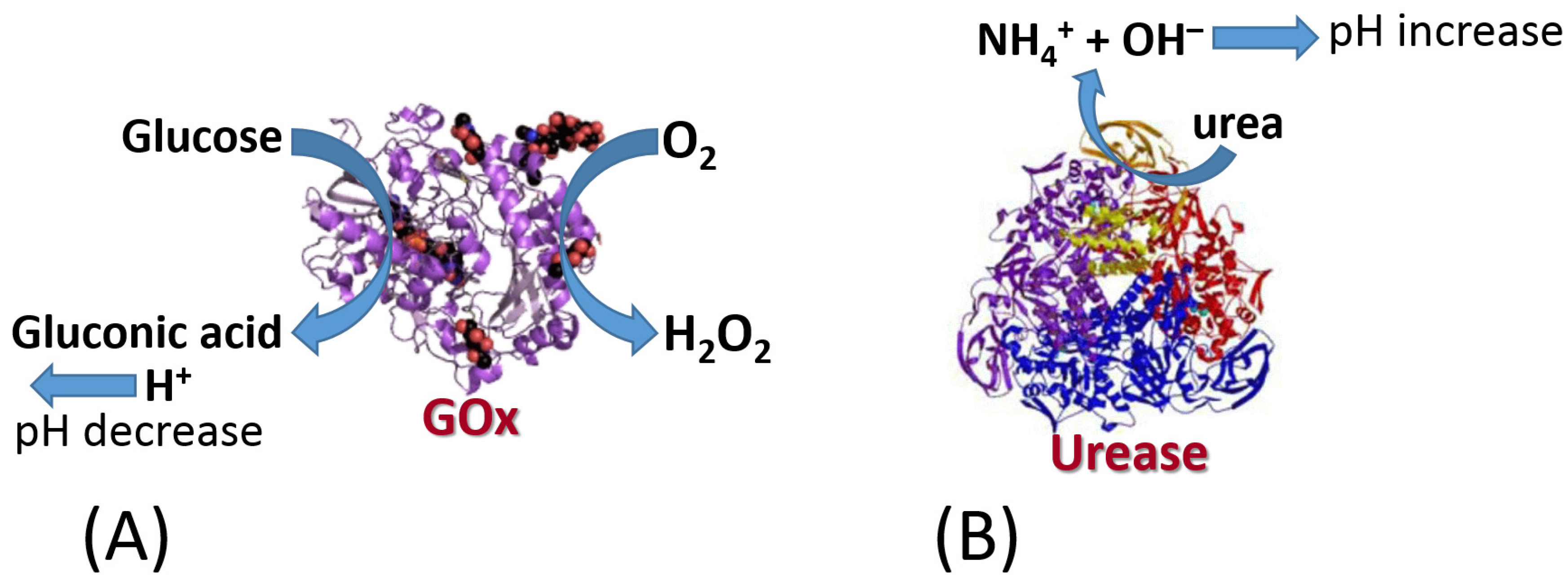
- Bailey, J.E.; Chow, M.T.C. Immobilized enzyme catalysis with reaction-generated pH change. Biotechnol. Bioeng. 1974, 16, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Wells, P.K.; Smutok, O.; Melman, A.; Katz, E. Switchable biocatalytic reactions controlled by interfacial pH changes produced by orthogonal biocatalytic processes. ACS Appl. Mater. Interfaces 2021, 13, 33830–33839. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Suni, I.I.; Bever, C.S.; Hammock, B.D. Impedance biosensors: Applications to sustainability and remaining technical challenges. ACS Sustain. Chem. Eng. 2014, 2, 1649–1655. [Google Scholar] [CrossRef]
- Lisdat, F.; Schäfer, D. The use of electrochemical impedance spectroscopy for biosensing. Anal. Bioanal. Chem. 2008, 391, 1555–1567. [Google Scholar] [CrossRef]
- Sadana, A.; Sadana, N. Handbook of Biosensors and Biosensor Kinetics; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Karunakaran, C.; Bhargava, K.; Benjamin, R. (Eds.) Biosensors and Bioelectronics; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Naresh, V.; Lee, N. A review on biosensors and recent development of nanostructured materials-enabled biosensors. Sensors 2021, 21, 1109. [Google Scholar] [CrossRef]
- Mehrotra, P. Biosensors and their applications–A review. J. Oral Biol. Craniofacial Res. 2016, 6, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Bard, A.J.; Mirkin, M.V. (Eds.) Scanning Electrochemical Microscopy, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2022. [Google Scholar]
- Salazar, G.A.E.; Helú, M.A.B.; Walcarius, A.; Liu, L. Scanning gel electrochemical microscopy: Combination with quartz crystal microbalance for studying the electrolyte residue. Electrochim. Acta 2023, 437, 141455. [Google Scholar] [CrossRef]
- Kubota, L.T.; Da Silva, J.A.F.; Sena, M.M.; Alves, W.A. (Eds.) Tools and Trends in Bioanalytical Chemistry; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Dutta, G.; Biswas, A. (Eds.) Next Generation Smart Nano-Bio-Devices. In Smart Innovation, Systems and Technologies; Springer: Singapore, 2023; Volume 322. [Google Scholar]
- Amreen, K.; Guha, K.; Goel, S. An overview of integrated miniaturized/microfluidic electrochemical biosensor platforms for health care applications. In Next Generation Smart Nano-Bio-Devices. Smart Innovation, Systems and Technologies; Dutta, G., Biswas, A., Eds.; Springer: Singapore, 2023; Volume 322. [Google Scholar]
- Titus, R.; Mandal, M.; Dutta, G. Electrochemical biosensor designs used for detecting SARS-CoV-2 virus: A review. In Next Generation Smart Nano-Bio-Devices. Smart Innovation, Systems and Technologies; Dutta, G., Biswas, A., Eds.; Springer: Singapore, 2023; Volume 322. [Google Scholar]
- Singh, L.; Mahapatra, D.; Kumar, S. (Eds.) Multifaceted Bio-Sensing Technology; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Schofield, Z.; Meloni, G.N.; Tran, P.; Zerfass, C.; Sena, G.; Hayashi, Y.; Grant, M.; Contera, S.A.; Minteer, S.D.; Kim, M.; et al. Bioelectrical understanding and engineering of cell biology. J. Royal Soc. Interface 2020, 17, 166. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Hall, W.C.; LaMantia, A.-S.; Mooney, R.D.; Platt, M.L.; White, L.E. (Eds.) Neuroscience, 6th ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Leopold, A.V.; Shcherbakova, D.M.; Verkhusha, V.V. Fluorescent biosensors for neurotransmission and neuromodulation: Engineering and applications. Front. Cell Neurosci. 2019, 13, 474. [Google Scholar] [CrossRef] [Green Version]
- Lakard, S.; Pavel, I.-A.; Lakard, B. Electrochemical biosensing of dopamine neurotransmitter: A review. Biosensors 2021, 11, 179. [Google Scholar] [CrossRef]
- Wang, Y.; Mishra, D.; Bergman, J.; Keighron, J.D.; Skibicka, K.P.; Cans, A.-S. Ultrafast glutamate biosensor recordings in brain slices reveal complex single exocytosis transients. ACS Chem. Neurosci. 2019, 10, 1744–1752. [Google Scholar] [CrossRef]

























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smutok, O.; Katz, E. Biosensors: Electrochemical Devices—General Concepts and Performance. Biosensors 2023, 13, 44. https://doi.org/10.3390/bios13010044
Smutok O, Katz E. Biosensors: Electrochemical Devices—General Concepts and Performance. Biosensors. 2023; 13(1):44. https://doi.org/10.3390/bios13010044
Chicago/Turabian StyleSmutok, Oleh, and Evgeny Katz. 2023. "Biosensors: Electrochemical Devices—General Concepts and Performance" Biosensors 13, no. 1: 44. https://doi.org/10.3390/bios13010044
APA StyleSmutok, O., & Katz, E. (2023). Biosensors: Electrochemical Devices—General Concepts and Performance. Biosensors, 13(1), 44. https://doi.org/10.3390/bios13010044