Extract2Chip—Bypassing Protein Purification in Drug Discovery Using Surface Plasmon Resonance

 , , , , , , , , , ,
, , , , , , , , , , 
Abstract
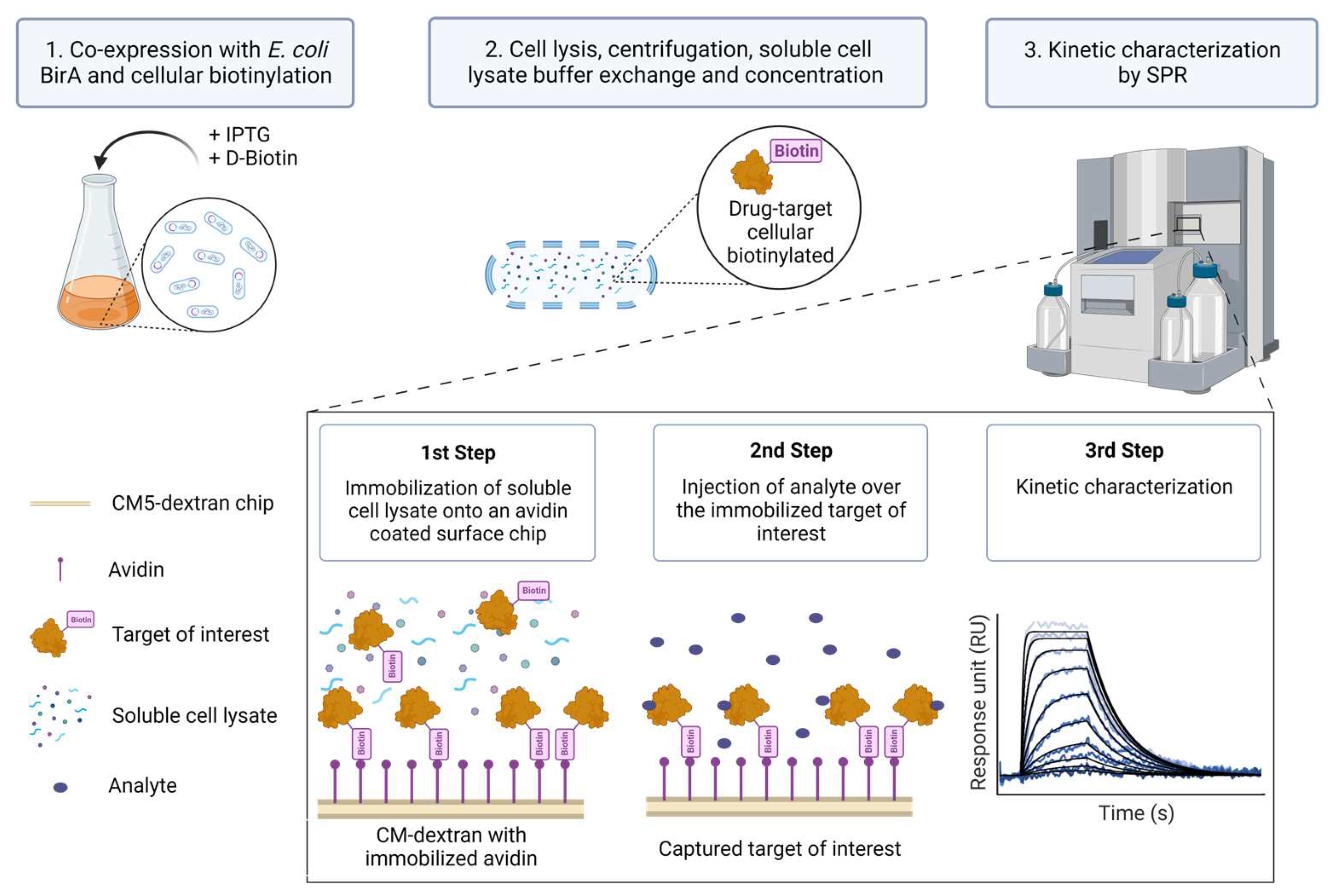
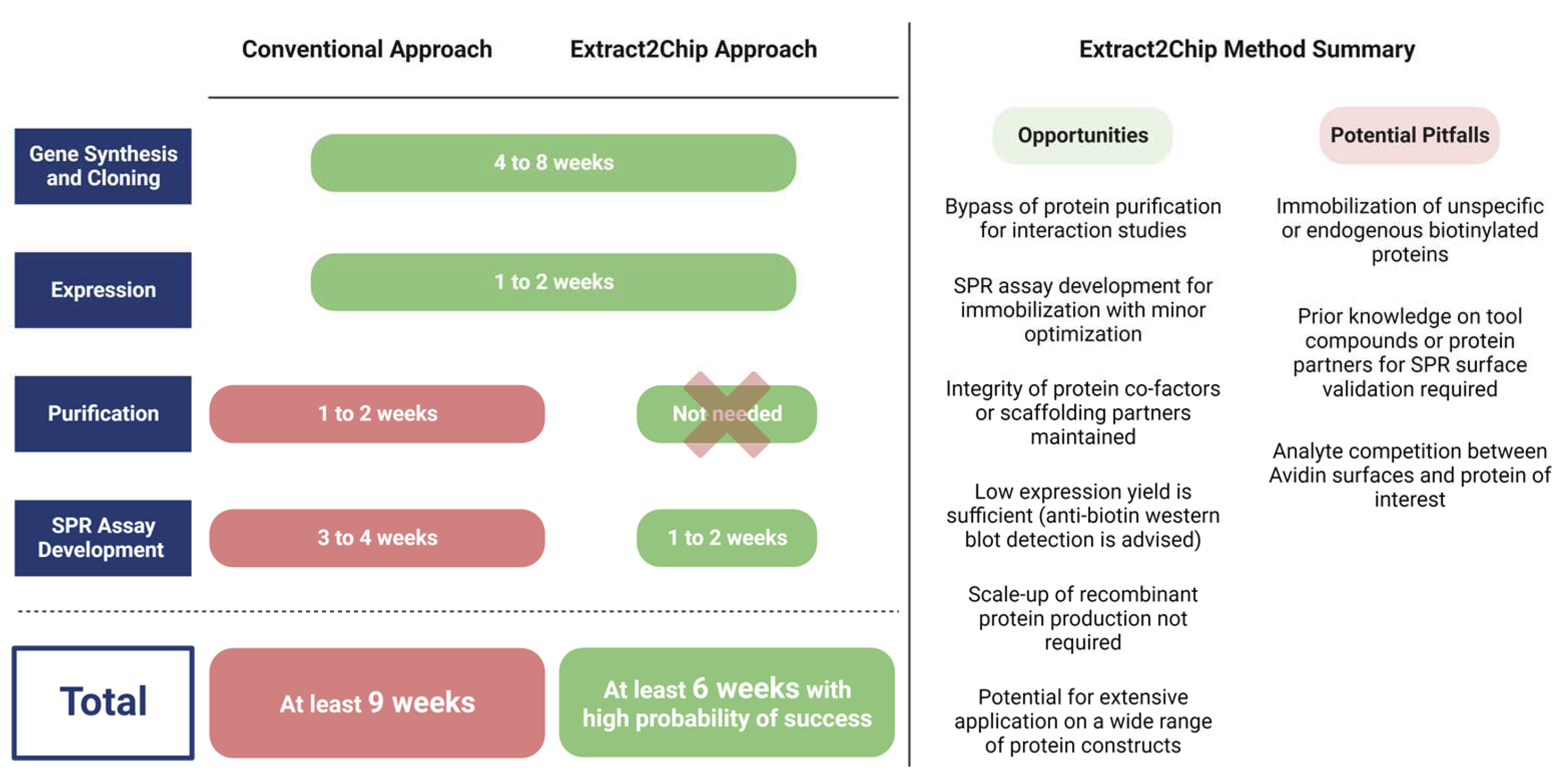
:1. Introduction
2. Materials and Methods
2.1. Extract2Chip Method Validation—Proof of Concept
2.1.1. DNA Constructs
2.1.2. Expression and Purification of CypD and BirA
2.1.3. In Vitro Biotinylation of CypD Protein
2.1.4. Cellular Biotinylation of CypD
2.2. Extract2Chip Applied on a Recalcitrant Protein
2.2.1. DNA Construct
2.2.2. Expression and Purification of TEAD1
2.2.3. Cellular Biotinylation of YAP
2.3. Extract2Chip Applied on a Drug Target Surrogate Validation
2.3.1. DNA Construct
2.3.2. Expression and Purification of MARK3
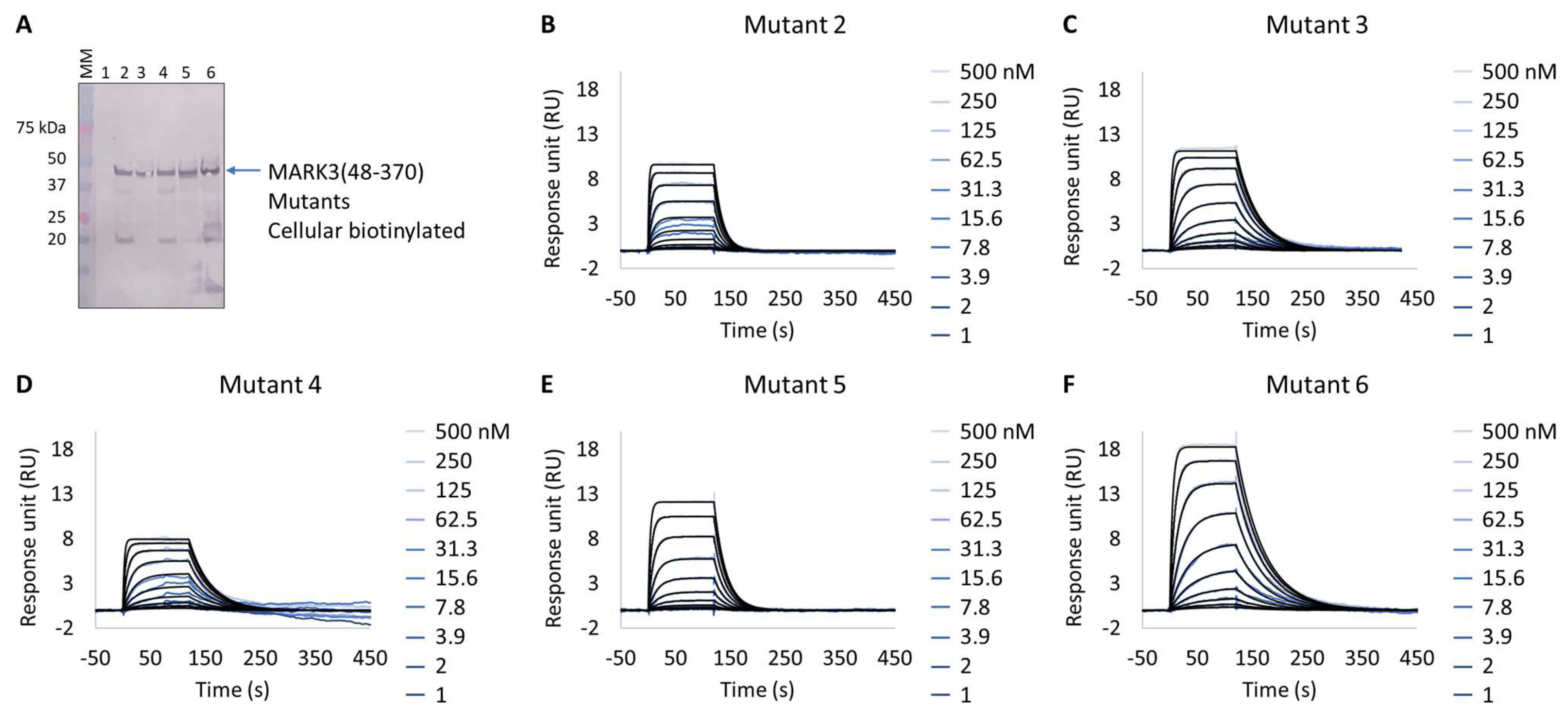
2.3.3. Cellular Biotinylation of MARK3 Kinase Domain Mutants
2.4. Extract2Chip Applied on Validation of Ternary Protein Complexes
2.4.1. DNA Construct
2.4.2. Expression and Purification of RuvBL1/RuvBL2
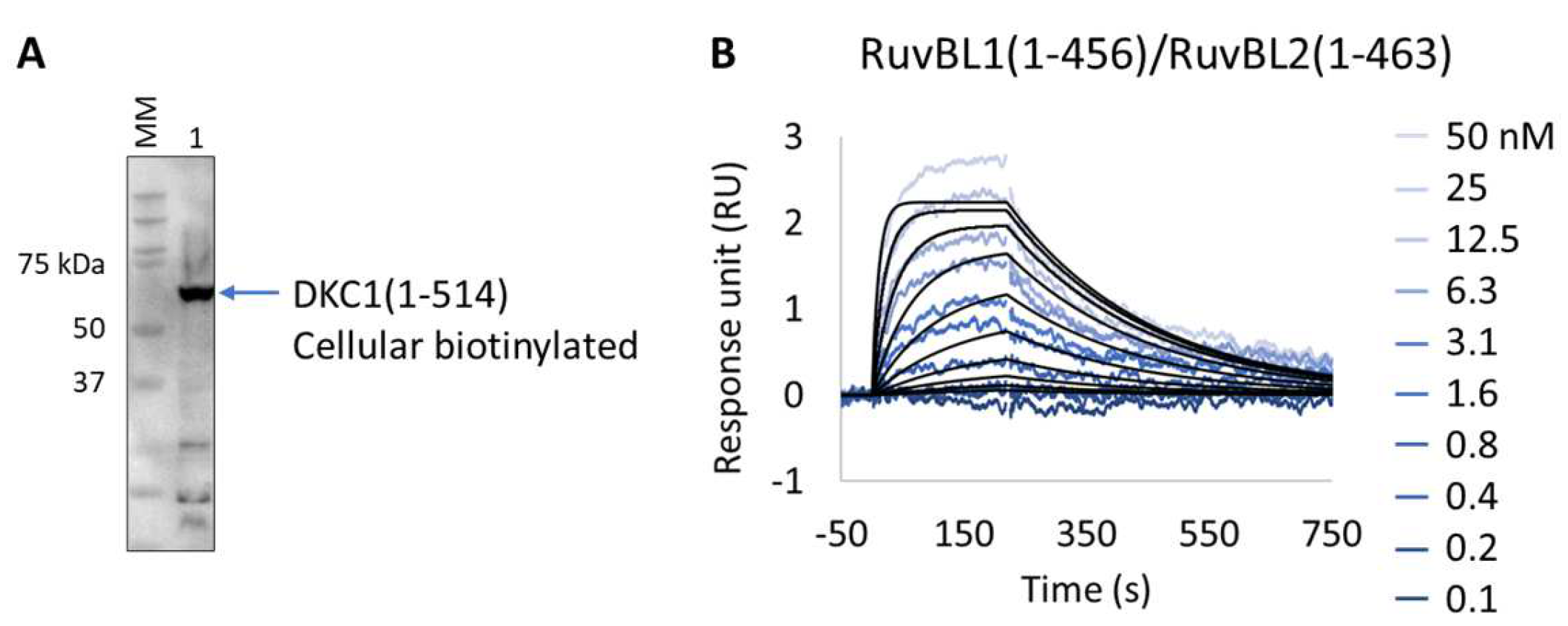
2.4.3. Cellular Biotinylation of DKC1
2.5. SPR Assays
2.5.1. Characterization of CypD Interaction with Ligand CYPD-27
2.5.2. Characterization of YAP:TEAD1 Interaction in Presence of Small Molecules
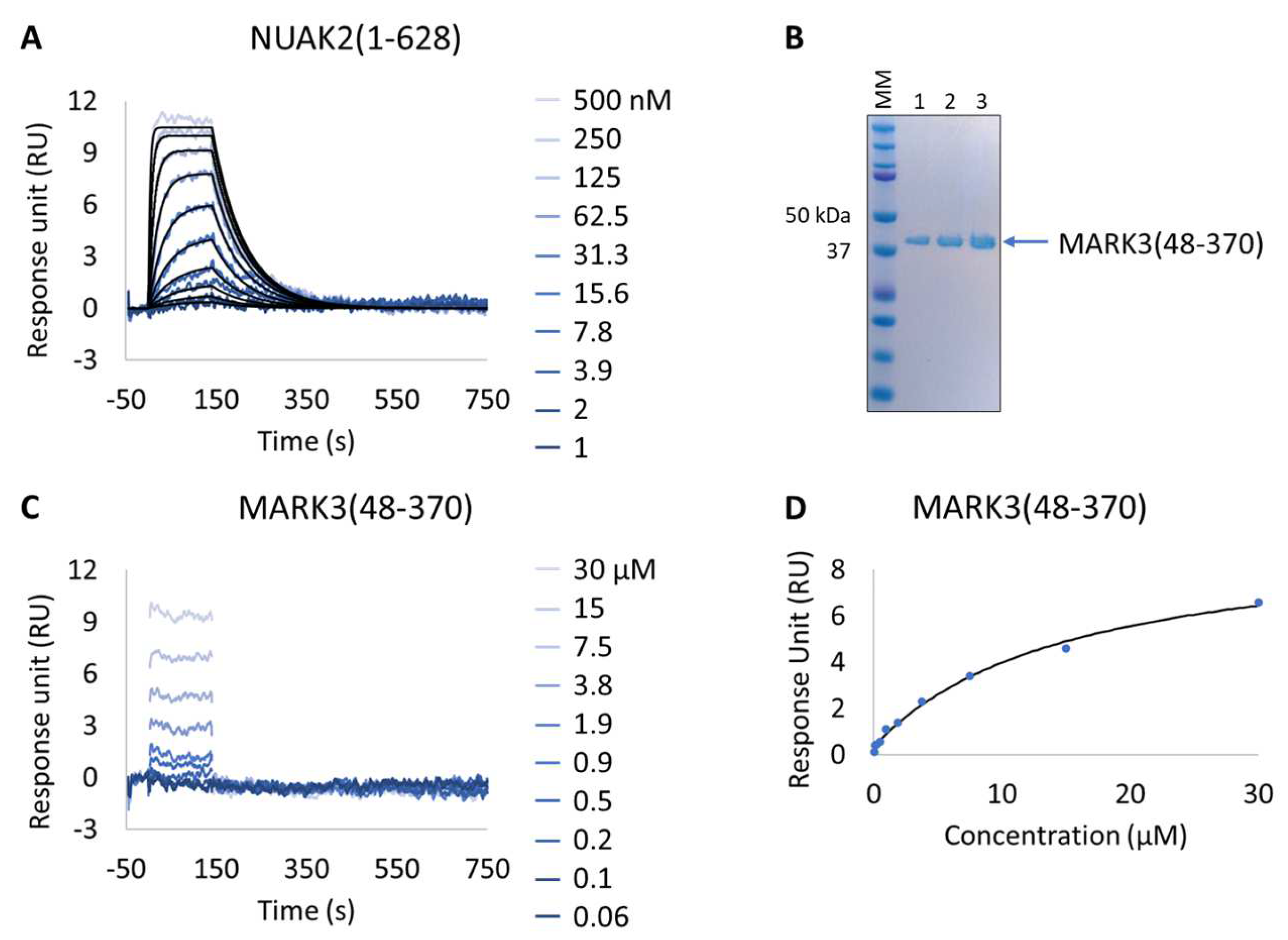
2.5.3. Characterization of NUAK2 Surrogate (MARK3 WT and Mutants) with GSK461364A
2.5.4. Characterization of DCK1:RuvBL1/RuvBL2 Interaction
3. Results and Discussion
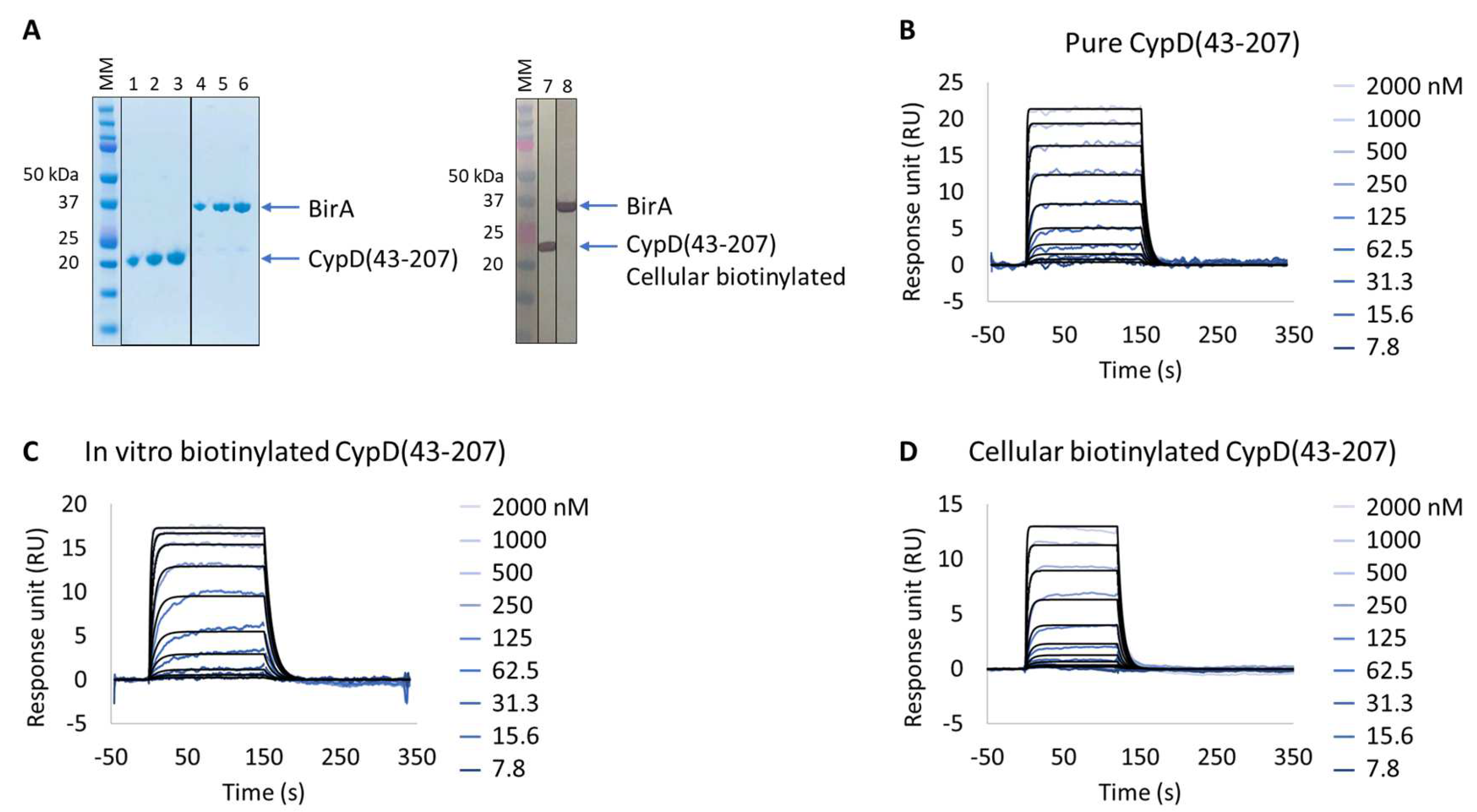
3.1. Extract2Chip Method Validation
The Study of CypD:Inhibitor Interaction Kinetics
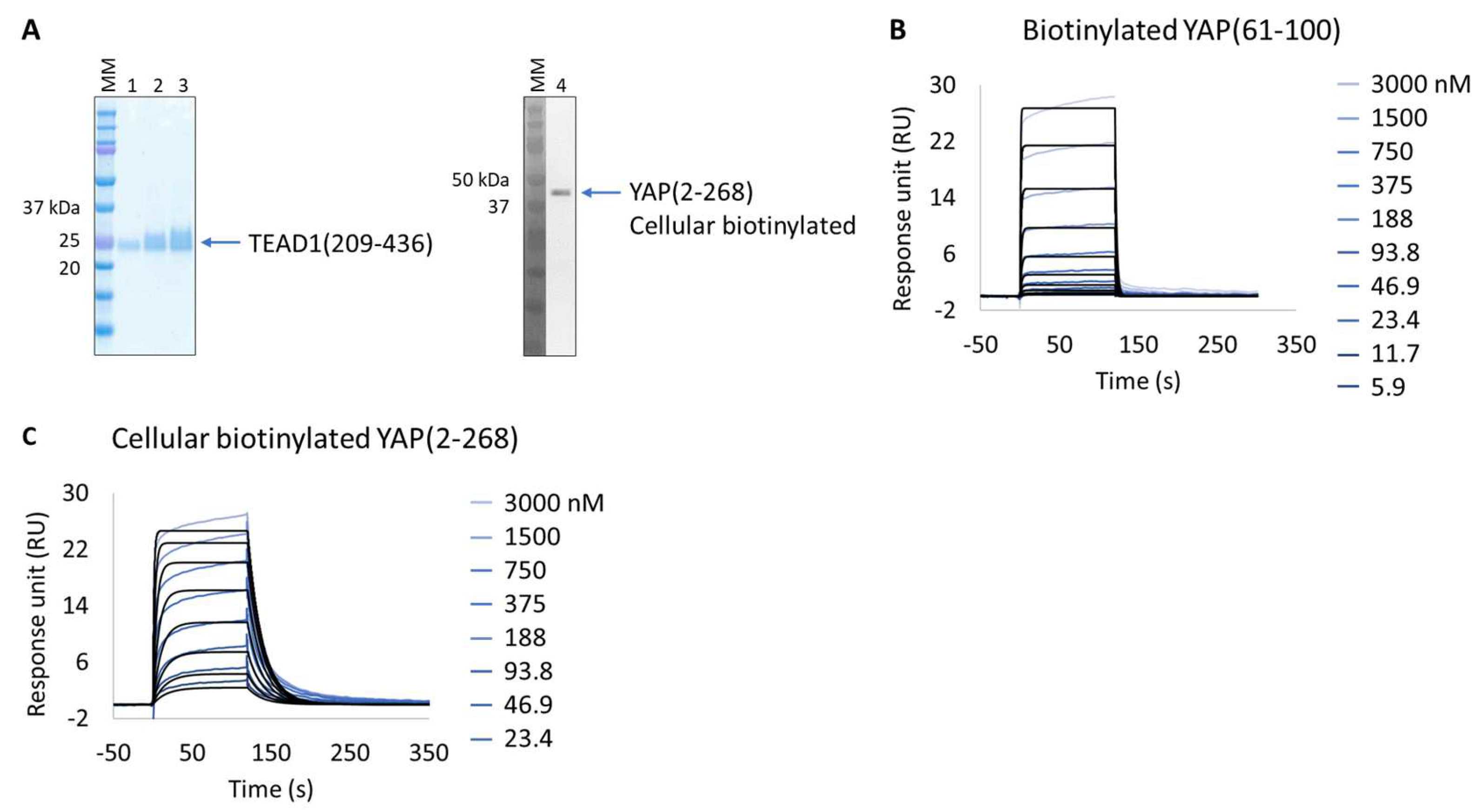
3.2. Extract2Chip Applied on a Recalcitrant Drug-Target
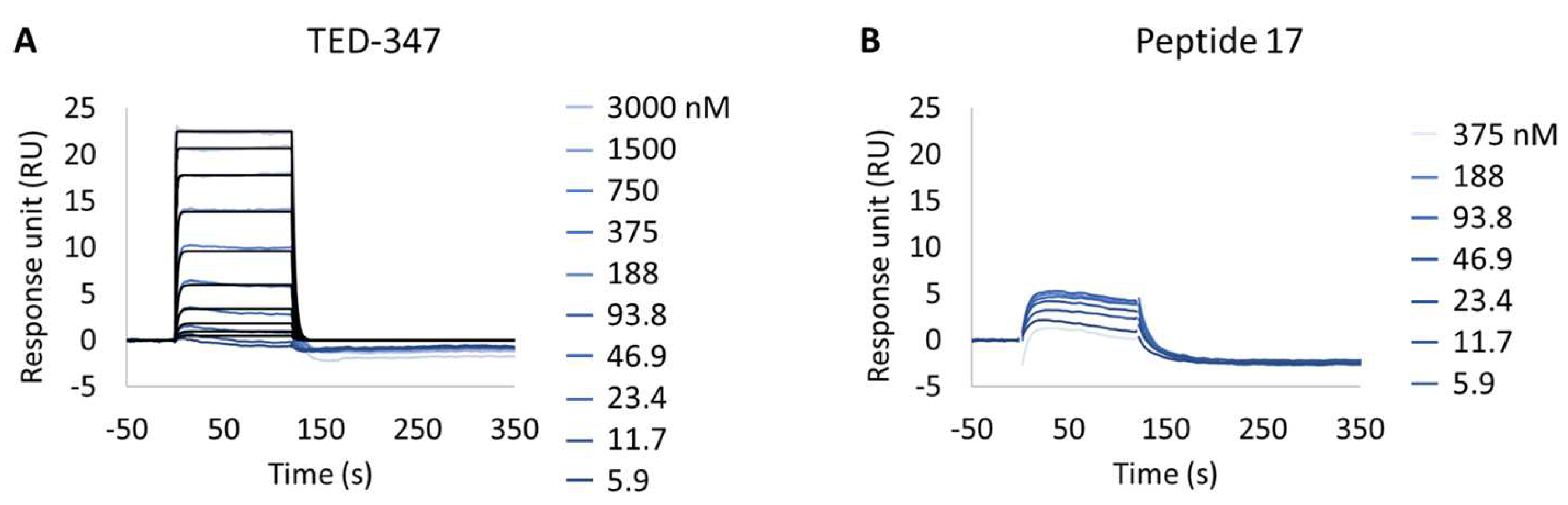
Evaluation of YAP:TEAD1 Interaction Inhibition with Small Molecules
3.3. Extract2chip for Drug Target Surrogate Validation
Unraveling NUAK2 Optimal Surrogates Using MARK3
3.4. Extract2Chip for Validation of Ternary Protein Complexes
Exploring the DKC1:RuvBL1/RuvBL2 Cellular Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, C.L.; Britton, Z.T.; Robinson, A.S. Recombinant protein expression and purification: A comprehensive review of affinity tags and microbial applications. Biotechnol. J. 2012, 7, 620–634. [Google Scholar] [CrossRef]
- Mashalidis, E.H.; Sledz, P.; Lang, S.; Abell, C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat. Protoc. 2013, 8, 2309–2324. [Google Scholar] [CrossRef]
- Dias, D.M.; Ciulli, A. NMR approaches in structure-based lead discovery: Recent developments and new frontiers for targeting multi-protein complexes. Prog. Biophys. Mol. Biol. 2014, 116, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Yasgar, A.; Peryea, T.; Braisted, J.C.; Jadhav, A.; Simeonov, A.; Coussens, N.P. Fluorescence polarization assays in high-throughput screening and drug discovery: A review. Methods Appl. Fluoresc. 2016, 4, 022001. [Google Scholar] [CrossRef]
- Muretta, J.M.; Rajasekaran, D.; Blat, Y.; Little, S.; Myers, M.; Nair, C.; Burdekin, B.; Yuen, S.L.; Jimenez, N.; Guhathakurta, P.; et al. HTS driven by fluorescence lifetime detection of FRET identifies activators and inhibitors of cardiac myosin. SLAS Discov. 2023, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Surface plasmon resonance spectroscopy for characterisation of membrane protein-ligand interactions and its potential for drug discovery. Biochim. Biophys. Acta 2014, 1838 Pt A, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Pina, A.S.; Lowe, C.R.; Roque, A.C. Challenges and opportunities in the purification of recombinant tagged proteins. Biotechnol. Adv. 2014, 32, 366–381. [Google Scholar] [CrossRef]
- Coleman, N.; Rodon, J. Taking Aim at the Undruggable. Am. Soc. Clin. Oncol. Educ. Book. 2021, 41, e145–e152. [Google Scholar] [CrossRef]
- Romier, C.; Ben Jelloul, M.; Albeck, S.; Buchwald, G.; Busso, D.; Celie, P.H.; Christodoulou, E.; De Marco, V.; van Gerwen, S.; Knipscheer, P.; et al. Co-expression of protein complexes in prokaryotic and eukaryotic hosts: Experimental procedures, database tracking and case studies. Acta Crystallogr. D Biol. Crystallogr. 2006, 62 Pt 10, 1232–1242. [Google Scholar] [CrossRef]
- Bayer, E.A.; Wilchek, M. Application of avidin-biotin technology to affinity-based separations. J. Chromatogr. 1990, 510, 3–11. [Google Scholar] [CrossRef]
- Hutsell, S.Q.; Kimple, R.J.; Siderovski, D.P.; Willard, F.S.; Kimple, A.J. High-affinity immobilization of proteins using biotin- and GST-based coupling strategies. Methods Mol. Biol. 2010, 627, 75–90. [Google Scholar] [CrossRef]
- Bayer, E.A.; Wilchek, M. The use of the avidin-biotin complex as a tool in molecular biology. In Methods of Biochemichal Analysis; Wiley: Hoboken, NJ, USA, 1980; Volume 26, pp. 1–45. [Google Scholar] [CrossRef]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef]
- Cull, M.G.; Schatz, P.J. Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzymol. 2000, 326, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Fairhead, M.; Howarth, M. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 2015, 1266, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, B.; Zauner, D.; Lehtonen, S.I.; Koskinen, M.; Thomson, C.; Kahkonen, N.; Kukkurainen, S.; Maatta, J.A.; Ihalainen, T.O.; Kulomaa, M.S.; et al. Switchavidin: Reversible biotin-avidin-biotin bridges with high affinity and specificity. Bioconjugate Chem. 2014, 25, 2233–2243. [Google Scholar] [CrossRef]
- Gunnarsson, A.; Stubbs, C.J.; Rawlins, P.B.; Taylor-Newman, E.; Lee, W.C.; Geschwindner, S.; Hytonen, V.; Holdgate, G.; Jha, R.; Dahl, G. Regenerable Biosensors for Small-Molecule Kinetic Characterization Using SPR. SLAS Discov. 2021, 26, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Belkacem, A.; Colliandre, L.; Ahnou, N.; Nevers, Q.; Gelin, M.; Bessin, Y.; Brillet, R.; Cala, O.; Douguet, D.; Bourguet, W.; et al. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat. Commun. 2016, 7, 12777. [Google Scholar] [CrossRef]
- Shore, E.R.; Awais, M.; Kershaw, N.M.; Gibson, R.R.; Pandalaneni, S.; Latawiec, D.; Wen, L.; Javed, M.A.; Criddle, D.N.; Berry, N.; et al. Small Molecule Inhibitors of Cyclophilin D To Protect Mitochondrial Function as a Potential Treatment for Acute Pancreatitis. J. Med. Chem. 2016, 59, 2596–2611. [Google Scholar] [CrossRef]
- Gradler, U.; Schwarz, D.; Blaesse, M.; Leuthner, B.; Johnson, T.L.; Bernard, F.; Jiang, X.; Marx, A.; Gilardone, M.; Lemoine, H.; et al. Discovery of novel Cyclophilin D inhibitors starting from three dimensional fragments with millimolar potencies. Bioorg Med. Chem. Lett. 2019, 29, 126717. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Peterson, C.; Schneider, R.; Garg, S.; Schwarz, D.; Gunera, J.; Seshire, A.; Kotzner, L.; Schlesiger, S.; Musil, D.; et al. Optimization of TEAD P-Site Binding Fragment Hit into In Vivo Active Lead MSC-4106. J. Med. Chem. 2022, 65, 9206–9229. [Google Scholar] [CrossRef]
- Dos Santos Morais, R.; Santo, P.E.; Ley, M.; Schelcher, C.; Abel, Y.; Plassart, L.; Desligniere, E.; Chagot, M.E.; Quinternet, M.; Paiva, A.C.F.; et al. Deciphering cellular and molecular determinants of human DPCD protein in complex with RUVBL1/RUVBL2 AAA-ATPases. J. Mol. Biol. 2022, 434, 167760. [Google Scholar] [CrossRef] [PubMed]
- Bum-Erdene, K.; Zhou, D.; Gonzalez-Gutierrez, G.; Ghozayel, M.K.; Si, Y.; Xu, D.; Shannon, H.E.; Bailey, B.J.; Corson, T.W.; Pollok, K.E.; et al. Small-Molecule Covalent Modification of Conserved Cysteine Leads to Allosteric Inhibition of the TEAD⋅Yap Protein-Protein Interaction. Cell Chem. Biol. 2019, 26, 378–389 e313. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, Z.; Zhou, Z.; Shen, H.C.; Yan, S.F.; Mayweg, A.V.; Xu, Z.; Qin, N.; Wong, J.C.; Zhang, Z.; et al. Structure-Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP-TEAD Protein-Protein Interaction. ACS Med. Chem. Lett. 2014, 5, 993–998. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Richter, M.C.; Erskine, S.G.; Kruger, R.G.; Madden, L.; Hassler, D.F.; Smith, G.K.; Gontarek, R.R.; Courtney, M.P.; et al. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. 2009, 69, 6969–6977. [Google Scholar] [CrossRef]
- Abel, Y.; Paiva, A.C.F.; Bizarro, J.; Chagot, M.E.; Santo, P.E.; Robert, M.C.; Quinternet, M.; Vandermoere, F.; Sousa, P.M.F.; Fort, P.; et al. NOPCHAP1 is a PAQosome cofactor that helps loading NOP58 on RUVBL1/2 during box C/D snoRNP biogenesis. Nucleic Acids Res. 2021, 49, 1094–1113. [Google Scholar] [CrossRef]
- Fayaz, S.M.; Raj, Y.V.; Krishnamurthy, R.G. CypD: The Key to the Death Door. CNS Neurol. Disord. Drug Targets 2015, 14, 654–663. [Google Scholar] [CrossRef]
- Kajitani, K.; Fujihashi, M.; Kobayashi, Y.; Shimizu, S.; Tsujimoto, Y.; Miki, K. Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 0.96-A resolution. Proteins 2008, 70, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Naoumov, N.V. Cyclophilin inhibition as potential therapy for liver diseases. J. Hepatol. 2014, 61, 1166–1174. [Google Scholar] [CrossRef]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 2010, 1797, 1113–1118. [Google Scholar] [CrossRef]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef]
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef]
- Chen, L.; Chan, S.W.; Zhang, X.; Walsh, M.; Lim, C.J.; Hong, W.; Song, H. Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes. Dev. 2010, 24, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.L.; Xu, Y. Structural insights into the YAP and TEAD complex. Genes. Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef]
- Fedir, B.; Yannick, M.; Marco, M.; Patrizia, F.; Catherine, Z.; Frederic, V.; Dirk, E.; Joerg, K.; Clemens, S.; Camilo, V.V.; et al. N-terminal beta-strand in YAP is critical for stronger binding to scalloped relative to TEAD transcription factor. Protein Sci. 2023, 32, e4545. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.C.; Pepe-Mooney, B.; Galli, G.G.; Dill, M.T.; Huang, H.T.; Hao, M.; Wang, Y.; Liang, H.; Calogero, R.A.; Camargo, F.D. NUAK2 is a critical YAP target in liver cancer. Nat. Commun. 2018, 9, 4834. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Namiki, T.; Tanemura, A.; Valencia, J.C.; Coelho, S.G.; Passeron, T.; Kawaguchi, M.; Vieira, W.D.; Ishikawa, M.; Nishijima, W.; Izumo, T.; et al. AMP kinase-related kinase NUAK2 affects tumor growth, migration, and clinical outcome of human melanoma. Proc. Natl. Acad. Sci. USA 2011, 108, 6597–6602. [Google Scholar] [CrossRef]
- Fu, W.; Zhao, M.T.; Driver, L.M.; Schirmer, A.U.; Yin, Q.; You, S.; Freedland, S.J.; DiGiovanni, J.; Drewry, D.H.; Macias, E. NUAK family kinase 2 is a novel therapeutic target for prostate cancer. Mol. Carcinog. 2022, 61, 334–345. [Google Scholar] [CrossRef]
- Banerjee, S.; Buhrlage, S.J.; Huang, H.T.; Deng, X.; Zhou, W.; Wang, J.; Traynor, R.; Prescott, A.R.; Alessi, D.R.; Gray, N.S. Characterization of WZ4003 and HTH-01-015 as selective inhibitors of the LKB1-tumour-suppressor-activated NUAK kinases. Biochem. J. 2014, 457, 215–225. [Google Scholar] [CrossRef]
- Drewry, D.H.; Annor-Gyamfi, J.K.; Wells, C.I.; Pickett, J.E.; Dederer, V.; Preuss, F.; Mathea, S.; Axtman, A.D. Identification of Pyrimidine-Based Lead Compounds for Understudied Kinases Implicated in Driving Neurodegeneration. J. Med. Chem. 2022, 65, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Ojha, S.; Malla, S.; Lyons, S.M. snoRNPs: Functions in Ribosome Biogenesis. Biomolecules 2020, 10, 783. [Google Scholar] [CrossRef] [PubMed]
- Kiss, T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001, 20, 3617–3622. [Google Scholar] [CrossRef]
- Hall, K.B.; McLaughlin, L.W. Properties of pseudouridine N1 imino protons located in the major groove of an A-form RNA duplex. Nucleic Acids Res. 1992, 20, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Charette, M.; Gray, M.W. Pseudouridine in RNA: What, where, how, and why. IUBMB Life 2000, 49, 341–351. [Google Scholar] [CrossRef]
- Eyler, D.E.; Franco, M.K.; Batool, Z.; Wu, M.Z.; Dubuke, M.L.; Dobosz-Bartoszek, M.; Jones, J.D.; Polikanov, Y.S.; Roy, B.; Koutmou, K.S. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. USA 2019, 116, 23068–23074. [Google Scholar] [CrossRef]
- Bellodi, C.; Krasnykh, O.; Haynes, N.; Theodoropoulou, M.; Peng, G.; Montanaro, L.; Ruggero, D. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 2010, 70, 6026–6035. [Google Scholar] [CrossRef]
- Jack, K.; Bellodi, C.; Landry, D.M.; Niederer, R.O.; Meskauskas, A.; Musalgaonkar, S.; Kopmar, N.; Krasnykh, O.; Dean, A.M.; Thompson, S.R.; et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell 2011, 44, 660–666. [Google Scholar] [CrossRef]
- Grozdanov, P.N.; Roy, S.; Kittur, N.; Meier, U.T. SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA 2009, 15, 1188–1197. [Google Scholar] [CrossRef]
- Walbott, H.; Machado-Pinilla, R.; Liger, D.; Blaud, M.; Rety, S.; Grozdanov, P.N.; Godin, K.; van Tilbeurgh, H.; Varani, G.; Meier, U.T.; et al. The H/ACA RNP assembly factor SHQ1 functions as an RNA mimic. Genes. Dev. 2011, 25, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Machado-Pinilla, R.; Liger, D.; Leulliot, N.; Meier, U.T. Mechanism of the AAA+ ATPases pontin and reptin in the biogenesis of H/ACA RNPs. RNA 2012, 18, 1833–1845. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract2Chip Applications | Description and Objectives |
|---|---|
| 1. Proof of concept |
|
| 2. Recalcitrant proteins |
|
| 3. Drug target surrogate validation |
|
| 4. Validation of ternary protein complexes |
|
| Mutant 1 | I62L, V116I, E139D, T204H, V205Q # |
| Mutant 2 | I62L, V116I, E139D, T204H, V205Q, L128I, L130I |
| Mutant 3 | I62L, V116I, E139D, T204H, V205Q, L128I, L130I, N66T, F67Y, A68G |
| Mutant 4 | I62L, V116I, E139D, T204H, V205Q, L128I, L130I, N66T, F67Y, A68G, F199L |
| Mutant 5 | I62L, V116I, E139D, T204H, V205Q, L128I, L130I, N66T, F67Y, A68G, F199L, G137R |
| Mutant 6 | I62L, V116I, E139D, T204H, V205Q, L128I, L130I, N66T, F67Y, A68G, F199L, G137R, L72K |
| KDss ± SD ¥ (M) | ka ± SD (M−1·s−1) | kd ± SD (s−1) | KD ± SD (M) | |
|---|---|---|---|---|
| Pure CypD(43-207) | 2.2 × 10−7 ± 1.3 × 10−8 | 9.5 × 10+5 ± 3.1 × 10+5 | 2.0 × 10−1 ± 6.4 × 10−2 | 2.1 × 10−7 ± 1.5 × 10−8 |
| In vitro biotinylated CypD(43-207) | 1.1 × 10−7 ± 8.6 × 10−9 | 4.8 × 10+5 ± 2.2 × 10+5 | 9.2 × 10−2 ± 8.0 × 10−3 | 1.9 × 10−7 ± 1.3 × 10−7 |
| Cellular biotinylated CypD(43-207) | 2.9 × 10−7 ± 8.3 × 10−9 | 3.9 × 10+5 ± 3.1 × 10+4 | 1.5 × 10−1 ± 4.3 × 10−3 | 3.7 × 10−7 ± 1.7 × 10−8 |
| KDss ± SD (M) | ka ± SD (M−1·s−1) | kd ± SD (s−1) | KD ± SD (M) | |
|---|---|---|---|---|
| Biotinylated YAP(61-100) | 1.2 × 10−6 ± 3.5 × 10−8 | 4.6 × 10+5 ± 1.7 × 10+4 | 4.8 × 10−1 ± 6.5 × 10−3 | 1.1 × 10−6 ± 3.7 × 10−8 |
| Cellular biotinylated YAP(2-268) | ND * | 1.8 × 10+6 ± 1.4 × 10+6 | 8.0 × 10−2 ± 2.2 × 10−2 | 4.5 × 10−8 ± 1.6 × 10−8 |
| KDss ± SD (M) | ka ± SD (M−1·s−1) | kd ± SD (s−1) | KD ± SD (M) | |
|---|---|---|---|---|
| TED-347:TEAD1 | 2.6 × 10−7 ± 1.3 × 10−8 | 1.2 × 10+6 ± 1.2 × 10+5 | 3.4 × 10−1 ± 7.4 × 10−2 | 3.0 × 10−7 ± 3.3 × 10−8 |
| Peptide 17:TEAD1 | >80% Reduction ¶ | >80% Reduction ¶ | >80% Reduction ¶ | >80% Reduction ¶ |
| KDss ± SD (M) | ka ± SD (M−1·s−1) | kd ± SD (s−1) | KD ± SD (M) | |
|---|---|---|---|---|
| NUAK2(1-628) | ND | 1.6 × 10+6 ± 1.1 × 10+6 | 3.1 × 10−2 ± 1.7 × 10−2 | 1.9 × 10−8 ± 4.3 × 10−9 |
| MARK3(48-370) | 1.5 × 10−5 ± 1.6 × 10−6 | ND | ND | ND |
| ka ± SD (M−1·s−1) | kd ± SD (s−1) | KD ± SD (M) | |
|---|---|---|---|
| Mutant 2 | 1.0 × 10+6 ± 9.9 × 10+4 | 8.1 × 10−2 ± 1.5 × 10−2 | 8.1 × 10−8 ± 2.4 × 10−8 |
| Mutant 3 | 8.6 × 10+5 ± 2.0 × 10+5 | 2.8 × 10−2 ± 3.0 × 10−3 | 3.3 × 10−8 ± 4.8 × 10−9 |
| Mutant 4 | 7.6 × 10+5 ± 1.2 × 10+4 | 2.9 × 10−2 ± 2.8 × 10−3 | 3.8 × 10−8 ± 3.4 × 10−9 |
| Mutant 5 | 3.9 × 10+5 ± 1.1 × 10+5 | 3.5 × 10−2 ± 1.2 × 10−2 | 8.9 × 10−8 ± 6.5 × 10−9 |
| Mutant 6 | 4.2 × 10+5 ± 4.7 × 10+4 | 2.1 × 10−2 ± 1.2 × 10−3 | 5.1 × 10−8 ± 1.9 × 10−9 |
| ka ± SD (M−1·s−1) | kd ± SD (s−1) | KD ± SD (M) | |
|---|---|---|---|
| RuvBL1(1-456)/RuvBL2(1-463) | 6.5 × 10+5 ± 1.3 × 10+5 | 1.5 × 10−2 ± 1.2 × 10−3 | 2.3 × 10−8 ± 2.9 × 10−9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paiva, A.C.F.; Lemos, A.R.; Busse, P.; Martins, M.T.; Silva, D.O.; Freitas, M.C.; Santos, S.P.; Freire, F.; Barrey, E.J.; Manival, X.; et al. Extract2Chip—Bypassing Protein Purification in Drug Discovery Using Surface Plasmon Resonance. Biosensors 2023, 13, 913. https://doi.org/10.3390/bios13100913
Paiva ACF, Lemos AR, Busse P, Martins MT, Silva DO, Freitas MC, Santos SP, Freire F, Barrey EJ, Manival X, et al. Extract2Chip—Bypassing Protein Purification in Drug Discovery Using Surface Plasmon Resonance. Biosensors. 2023; 13(10):913. https://doi.org/10.3390/bios13100913
Chicago/Turabian StylePaiva, Ana C. F., Ana R. Lemos, Philipp Busse, Madalena T. Martins, Diana O. Silva, Micael C. Freitas, Sandra P. Santos, Filipe Freire, Evelyne J. Barrey, Xavier Manival, and et al. 2023. "Extract2Chip—Bypassing Protein Purification in Drug Discovery Using Surface Plasmon Resonance" Biosensors 13, no. 10: 913. https://doi.org/10.3390/bios13100913
APA StylePaiva, A. C. F., Lemos, A. R., Busse, P., Martins, M. T., Silva, D. O., Freitas, M. C., Santos, S. P., Freire, F., Barrey, E. J., Manival, X., Koetzner, L., Heinrich, T., Wegener, A., Grädler, U., Bandeiras, T. M., Schwarz, D., & Sousa, P. M. F. (2023). Extract2Chip—Bypassing Protein Purification in Drug Discovery Using Surface Plasmon Resonance. Biosensors, 13(10), 913. https://doi.org/10.3390/bios13100913