Systematic Assessment of Human CCR7 Signalling Using NanoBRET Biosensors Points towards the Importance of the Cellular Context

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Plasmids and Reagents
2.2. G Protein Activation Assay
2.3. β-Arrestin Recruitment Assay
2.4. Data Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stone, M.J.; Hayward, J.A.; Huang, C.; Huma, Z.E.; Sanchez, J. Mechanisms of Regulation of the Chemokine-Receptor Network. Int. J. Mol. Sci. 2017, 18, 342. [Google Scholar] [CrossRef]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M. Dancing to the Tune of Chemokines. Nat. Immunol. 2001, 2, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, R.J.B.; Graham, G.J. Immune Regulation by Atypical Chemokine Receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Eiger, D.S.; Boldizsar, N.; Honeycutt, C.C.; Gardner, J.; Rajagopal, S. Biased Agonism at Chemokine Receptors. Cell. Signal. 2020, 78, 109862. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching Old Receptors New Tricks: Biasing Seven-Transmembrane Receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar] [CrossRef]
- Kenakin, T.; Christopoulos, A. Signalling Bias in New Drug Discovery: Detection, Quantification and Therapeutic Impact. Nat. Rev. Drug Discov. 2012, 12, 205–216. [Google Scholar] [CrossRef]
- Rajagopal, S.; Bassoni, D.L.; Campbell, J.J.; Gerard, N.P.; Gerard, C.; Wehrman, T.S. Biased Agonism as a Mechanism for Differential Signaling by Chemokine Receptors. J. Biol. Chem. 2013, 288, 35039–35048. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased Signalling: From Simple Switches to Allosteric Microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Kolb, P.; Kenakin, T.; Alexander, S.P.H.; Bermudez, M.; Bohn, L.M.; Breinholt, C.S.; Bouvier, M.; Hill, S.J.; Kostenis, E.; Martemyanov, K.A.; et al. Community Guidelines for GPCR Ligand Bias: IUPHAR Review 32. Br. J. Pharmacol. 2022, 179, 3651–3674. [Google Scholar] [CrossRef]
- Pineyro, G.; Nagi, K. Signaling Diversity of Mu- and Delta-Opioid Receptor Ligands: Re-Evaluating the Benefits of β-Arrestin/G Protein Signaling Bias. Cell. Signal. 2021, 80, 109906. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N.; et al. Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 2019, 177, 1933–1947.e25. [Google Scholar] [CrossRef] [PubMed]
- Avet, C.; Mancini, A.; Breton, B.; Le Gouill, C.; Hauser, A.S.; Normand, C.; Kobayashi, H.; Gross, F.; Hogue, M.; Lukasheva, V.; et al. Effector Membrane Translocation Biosensors Reveal G Protein and Βarrestin Coupling Profiles of 100 Therapeutically Relevant GPCRs. eLife 2022, 11, e74101. [Google Scholar] [CrossRef] [PubMed]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and Its Ligands: Balancing Immunity and Tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Campbell, J.J.; Butcher, E.C. Chemokines in Tissue-Specific and Microenvironment-Specific Lymphocyte Homing. Curr. Opin. Immunol. 2000, 12, 336–341. [Google Scholar] [CrossRef]
- Brandum, E.P.; Jørgensen, A.S.; Rosenkilde, M.M.; Hjortø, G.M. Dendritic Cells and CCR7 Expression: An Important Factor for Autoimmune Diseases, Chronic Inflammation, and Cancer. Int. J. Mol. Sci. 2021, 22, 8340. [Google Scholar] [CrossRef]
- Moussouras, N.A.; Hjortø, G.M.; Peterson, F.C.; Szpakowska, M.; Chevigné, A.; Rosenkilde, M.M.; Volkman, B.F.; Dwinell, M.B. Structural Features of an Extended C-Terminal Tail Modulate the Function of the Chemokine CCL21. Biochemistry 2020, 59, 1338–1350. [Google Scholar] [CrossRef]
- Kiermaier, E.; Moussion, C.; Veldkamp, C.T.; Gerardy-Schahn, R.; de Vries, I.; Williams, L.G.; Chaffee, G.R.; Phillips, A.J.; Freiberger, F.; Imre, R.; et al. Polysialylation Controls Dendritic Cell Trafficking by Regulating Chemokine Recognition. Science 2016, 351, 186–190. [Google Scholar] [CrossRef]
- Zidar, D.A.; Violin, J.D.; Whalen, E.J.; Lefkowitz, R.J. Selective Engagement of G Protein Coupled Receptor Kinases (GRKs) Encodes Distinct Functions of Biased Ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 9649–9654. [Google Scholar] [CrossRef]
- Kohout, T.A.; Nicholas, S.L.; Perry, S.J.; Reinhart, G.; Junger, S.; Struthers, R.S. Differential Desensitization, Receptor Phosphorylation, Beta-Arrestin Recruitment, and ERK1/2 Activation by the Two Endogenous Ligands for the CC Chemokine Receptor 7. J. Biol. Chem. 2004, 279, 23214–23222. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.D.; Lane, J.R.; Canals, M.; Stone, M.J. Systematic Assessment of Chemokine Signaling at Chemokine Receptors CCR4, CCR7 and CCR10. Int. J. Mol. Sci. 2021, 22, 4232. [Google Scholar] [CrossRef] [PubMed]
- Vanalken, N.; Boon, K.; Doijen, J.; Schols, D.; Van Loy, T. Cellular Electrical Impedance as a Method to Decipher CCR7 Signalling and Biased Agonism. Int. J. Mol. Sci. 2022, 23, 8903. [Google Scholar] [CrossRef] [PubMed]
- Hjortø, G.M.; Larsen, O.; Steen, A.; Daugvilaite, V.; Berg, C.; Fares, S.; Hansen, M.; Ali, S.; Rosenkilde, M.M. Differential CCR7 Targeting in Dendritic Cells by Three Naturally Occurring CC-Chemokines. Front. Immunol. 2016, 7, 568. [Google Scholar] [CrossRef]
- Jørgensen, A.S.; Larsen, O.; Uetz-von Allmen, E.; Lückmann, M.; Legler, D.F.; Frimurer, T.M.; Veldkamp, C.T.; Hjortø, G.M.; Rosenkilde, M.M. Biased Signaling of CCL21 and CCL19 Does Not Rely on N-Terminal Differences, but Markedly on the Chemokine Core Domains and Extracellular Loop 2 of CCR7. Front. Immunol. 2019, 10, 2156. [Google Scholar] [CrossRef]
- Jørgensen, A.S.; Brandum, E.P.; Mikkelsen, J.M.; Orfin, K.A.; Boilesen, D.R.; Egerod, K.L.; Moussouras, N.A.; Vilhardt, F.; Kalinski, P.; Basse, P.; et al. The C-Terminal Peptide of CCL21 Drastically Augments CCL21 Activity through the Dendritic Cell Lymph Node Homing Receptor CCR7 by Interaction with the Receptor N-Terminus. Cell Mol. Life Sci. 2021, 78, 6963–6978. [Google Scholar] [CrossRef]
- Corbisier, J.; Galès, C.; Huszagh, A.; Parmentier, M.; Springael, J.-Y. Biased Signaling at Chemokine Receptors. J. Biol. Chem. 2015, 290, 9542–9554. [Google Scholar] [CrossRef]
- Artinger, M.; Gerken, O.J.; Legler, D.F. Heparin Specifically Interacts with Basic BBXB Motifs of the Chemokine CCL21 to Define CCR7 Signaling. Int. J. Mol. Sci. 2023, 24, 1670. [Google Scholar] [CrossRef]
- Boon, K.; Vanalken, N.; Meyen, E.; Schols, D.; Van Loy, T. REGA-SIGN: Development of a Novel Set of NanoBRET-Based G Protein Biosensors. Biosensors 2023, 13, 767. [Google Scholar] [CrossRef]
- Luís, R.; D’Uonnolo, G.; Palmer, C.B.; Meyrath, M.; Uchański, T.; Wantz, M.; Rogister, B.; Janji, B.; Chevigné, A.; Szpakowska, M. Nanoluciferase-Based Methods to Monitor Activation, Modulation and Trafficking of Atypical Chemokine Receptors. In Methods in Cell Biology; Shukla, A.K., Ed.; Biomolecular Interactions Part B; Academic Press: Cambridge, MA, USA, 2022; Volume 169, Chapter 13; pp. 279–294. [Google Scholar]
- Mansour, A.; Nagi, K.; Dallaire, P.; Lukasheva, V.; Le Gouill, C.; Bouvier, M.; Pineyro, G. Comprehensive Signaling Profiles Reveal Unsuspected Functional Selectivity of δ-Opioid Receptor Agonists and Allow the Identification of Ligands with the Greatest Potential for Inducing Cyclase Superactivation. ACS Pharmacol. Transl. Sci. 2021, 4, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Winpenny, D.; Clark, M.; Cawkill, D. Biased Ligand Quantification in Drug Discovery: From Theory to High Throughput Screening to Identify New Biased μ Opioid Receptor Agonists. Br. J. Pharmacol. 2016, 173, 1393–1403. [Google Scholar] [CrossRef] [PubMed]





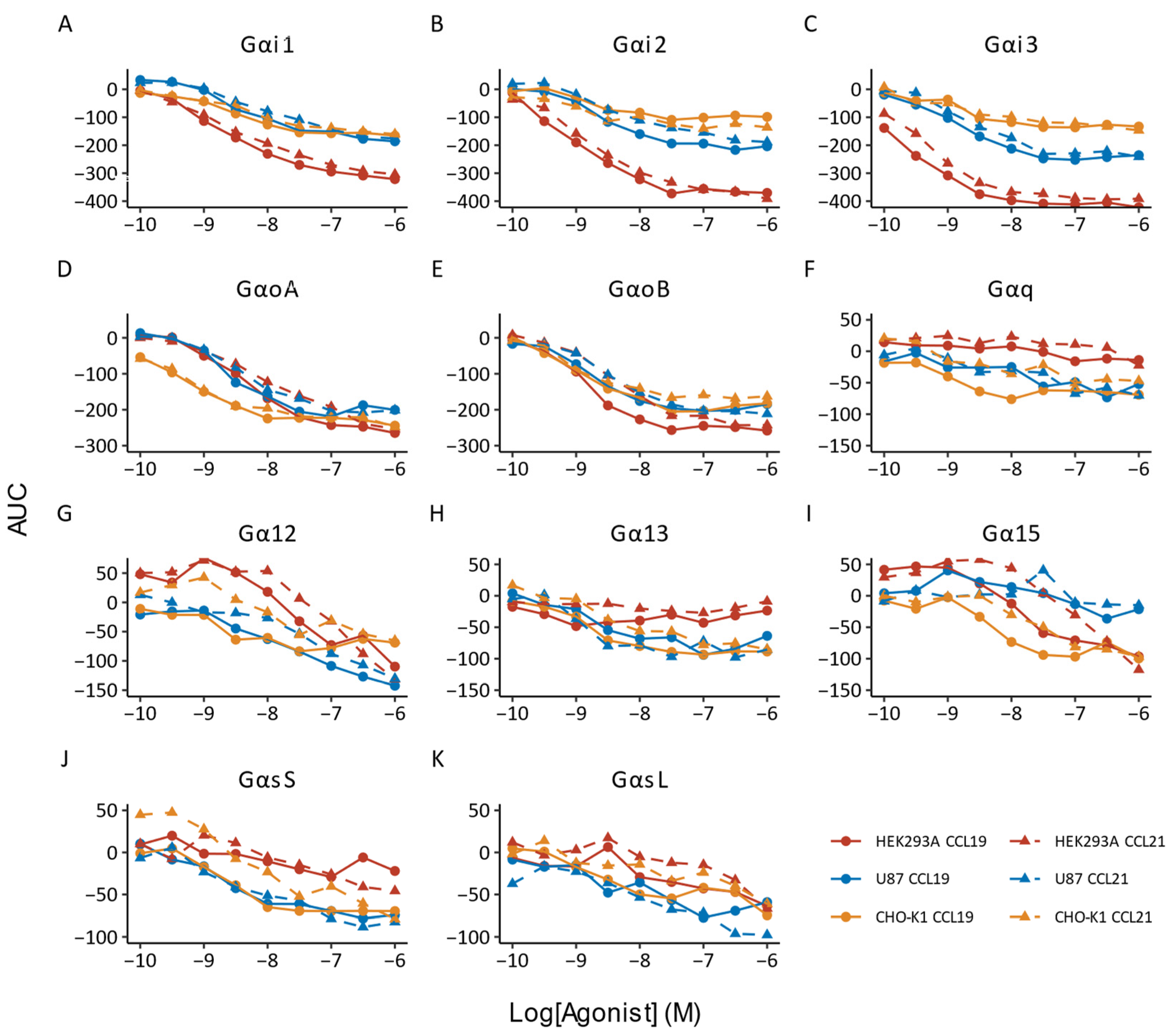{kind=link}
{kind=link}
{kind=link}
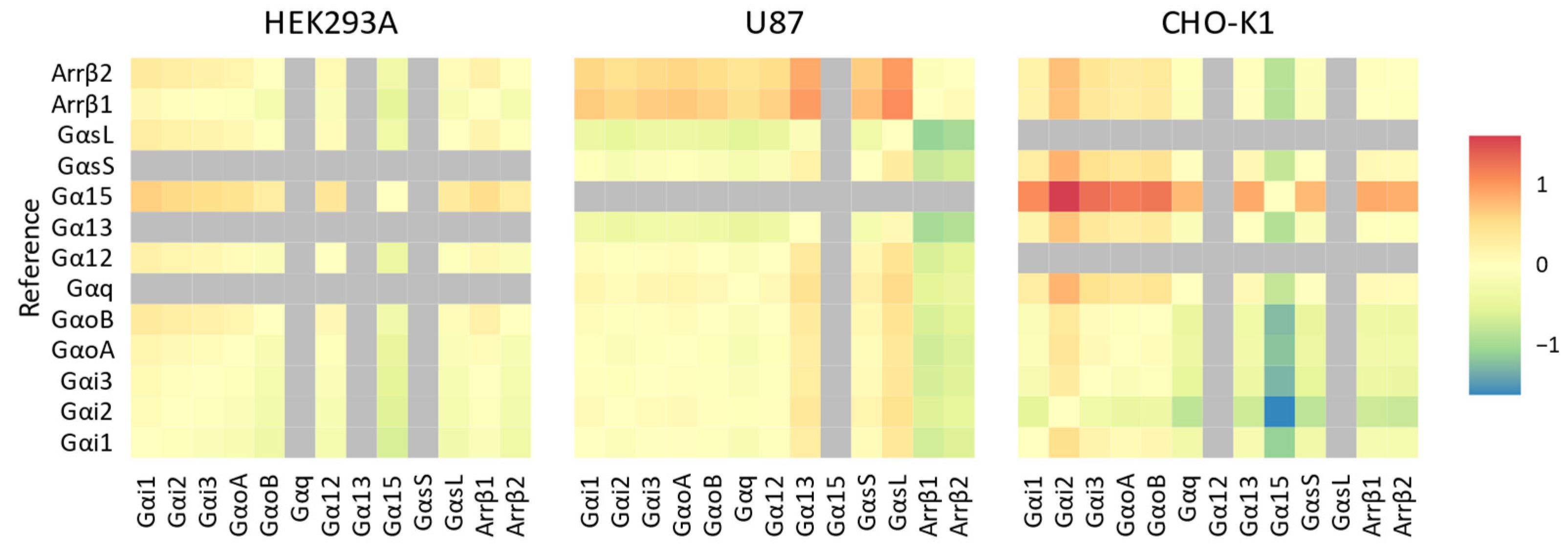{kind=link}
| Transducer | Ligand | HEK293A | U87 | CHO-K1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 | Emax | Log(Emax/EC50) | pEC50 | Emax | Log(Emax/EC50) | pEC50 | Emax | Log(Emax/EC50) | ||
| Gαi1 | CCL19 | 8.63 ± 0.06 | 94.69 ± 1.39 | 8.61 ± 0.06 | 8.28 ± 0.34 | 95.77 ± 1.30 | 8.26 ± 0.34 | 8.54 ± 0.09 | 96.06 ± 2.50 * | 8.82 ± 0.09 |
| CCL21 | 8.47 ± 0.38 | 89.00 ± 7.21 | 8.41 ± 0.41 | 8.02 ± 0.54 | 93.08 ± 4.36 | 7.98 ± 0.55 | 8.37 ± 0.34 | 89.50 ± 14.03 | 8.32 ± 0.35 | |
| Gαi2 | CCL19 | 9.03 ± 0.15 | 94.99 ± 4.25 | 9.01 ± 0.15 | 8.56 ± 0.09 | 94.22 ± 1.06 | 8.53 ± 0.08 | 8.69 ± 0.39 | 83.61 ± 6.65 | 8.61 ± 0.38 |
| CCL21 | 8.76 ± 0.24 | 95.68 ± 2.29 | 8.74 ± 0.24 | 8.27 ± 0.20 | 80.01 ± 16.70 | 8.16 ± 0.29 | 8.91 ± 0.40 | 103.51 ± 14.10 | 8.93 ± 0.41 | |
| Gαi3 | CCL19 | 9.61 ± 0.16 | 96.94 ± 1.21 | 9.60 ± 0.16 | 8.87 ± 0.21 | 94.05 ± 5.35 | 8.85 ± 0.20 | 8.84 ± 0.19 | 88.66 ± 2.67 | 8.78 ± 0.20 |
| CCL21 | 9.33 ± 0.05 | 91.97 ± 3.19 | 9.30 ± 0.04 | 8.60 ± 0.26 | 90.76 ± 2.47 | 8.55 ± 0.26 | 8.84 ± 0.24 | 86.61 ± 20.68 | 8.77 ± 0.34 | |
| GαoA | CCL19 | 8.30 ± 0.12 * | 96.18 ± 3.38 | 8.28 ± 0.13 * | 8.59 ± 0.21 | 94.63 ± 5.54 | 8.57 ± 0.21 | 9.37 ± 0.38 | 93.04 ± 4.17 | 9.34 ± 0.37 |
| CCL21 | 7.97 ± 0.08 | 89.12 ± 1.59 | 7.92 ± 0.08 | 8.33 ± 0.37 | 96.17 ± 23.05 | 8.30 ± 0.84 | 9.27 ± 0.26 | 90.49 ± 9.19 | 9.22 ± 0.28 | |
| GαoB | CCL19 | 8.85 ± 0.21 | 98.46 ± 0.84 | 8.85 ± 0.21 | 8.80 ± 0.06 | 97.07 ± 2.33 | 8.78 ± 0.05 | 8.96 ± 0.22 | 93.86 ± 3.45 | 8.93 ± 0.23 |
| CCL21 | 8.34 ± 0.27 | 92.10 ± 9.87 | 8.30 ± 0.32 | 8.45 ± 0.19 | 101.87 ± 0.83 | 8.45 ± 0.19 | 8.97 ± 0.46 | 81.95 ± 17.33 | 8.87 ± 0.46 | |
| Gαq | CCL19 | n.d. | 8.41 ± 0.87 | 72.23 ± 26.73 | 8.24 ± 0.68 | 8.77 ± 0.63 | 78.38 ± 15.20 | 8.65 ± 0.65 | ||
| CCL21 | n.d. | 8.10 ± 0.65 | 78.50 ± 59.03 | 7.80 ± 0.47 | 8.45 ± 0.66 | 53.05 ± 22.37 | 8.15 ± 0.77 | |||
| Gα12 | CCL19 | 7.44 ± 0.26 * | 91.44 ± 8.48 * | 7.40 ± 0.25 | 7.81 ± 0.21 | 91.27 ± 7.16 | 7.77 ± 0.18 | n.d. | ||
| CCL21 | 6.79 ± 0.20 | 149.83 ± 22.74 | 6.97 ± 0.13 | 7.50 ± 0.46 | 88.78 ± 21.52 | 7.44 ± 0.40 | 7.94 ± 0.11 | 70.30 ± 29.18 | 7.77 ± 0.07 | |
| Gα13 | CCL19 | n.d. | 8.77 ± 0.23 | 84.19 ± 17.93 | 8.69 ± 0.33 | 8.87 ± 0.67 | 87.66 ± 6.56 | 8.81 ± 0.68 | ||
| CCL21 | n.d. | 8.70 ± 0.6 | 104.69 ± 31.37 | 8.71 ± 0.67 | 8.53 ± 0.73 | 77.97 ± 22.39 | 8.40 ± 0.68 | |||
| Gα15 | CCL19 | 7.80 ± 0.26 ** | 97.07 ± 5.41 | 7.78 ± 0.23 * | n.d. | 8.91 ± 1.02 | 78.62 ± 10.63 | 8.80 ± 0.96 | ||
| CCL21 | 6.74 ± 0.22 | 164.09 ± 57.96 | 6.94 ± 0.11 | n.d. | 7.56 ± 0.31 | 87.78 ± 15.35 | 7.50 ± 0.29 | |||
| GαsS | CCL19 | n.d. | 8.65 ± 0.41 | 89.30 ± 4.17 | 8.60 ± 0.39 | 8.58 ± 0.29 * | 83.38 ± 8.74 | 8.50 ± 0.28 * | ||
| CCL21 | 231.28 ± 174.49 | 7.13 ± 0.83 | 7.42 ± 0.53 | 8.39 ± 0.49 | 104.80 ± 29.32 | 8.40 ± 0.50 | 8.10 ± 0.15 | 76.48 ± 16.16 | 7.98 ± 0.19 | |
| GαsL | CCL19 | 7.49 ± 0.54 | 88.20 ± 20.66 | 7.43 ± 0.45 | 8.95 ± 1.33 | 82.58 ± 11.20 | 8.86 ± 1.27 | 8.97 ± 0.74 | 72.43 ± 7.06 | 8.82 ± 0.76 |
| CCL21 | 6.79 ± 0.73 | 170.51 ± 117.5 | 6.94 ± 0.44 | 8.99 ± 1.32 | 101.11 ± 16.89 | 8.99 ± 1.31 | n.d. | |||
| Arrβ1 | CCL19 | 7.46 ± 0.14 | 108.79 ± 3.73 | 7.49 ± 0.13 | 7.86 ± 0.24 ** | 104.49 ± 3.69 | 7.88 ± 0.23 ** | 7.92 ± 0.25 | 105.06 ± 2.93 | 7.94 ± 0.24 |
| CCL21 | 7.15 ± 0.17 | 107.66 ± 3.12 | 7.19 ± 0.17 | 6.90 ± 0.15 | 106.43 ± 5.12 | 6.92 ± 0.13 | 7.52 ± 0.25 | 102.60 ± 4.96 | 7.53 ± 0.23 | |
| Arrβ2 | CCL19 | 7.57 ± 0.13 * | 106.42 ± 5.36 | 7.59 ± 0.11 ** | 7.86 ± 0.16 ** | 102.06 ± 4.66 | 7.86 ± 0.15 ** | 8.11 ± 0.29 | 102.10 ± 5.79 | 8.12 ± 0.27 |
| CCL21 | 7.02 ± 0.05 | 106.86 ± 1.87 | 7.05 ± 0.05 | 7.01 ± 0.15 | 97.20 ± 5.27 | 7.00 ± 0.16 | 7.69 ± 0.27 | 98.30 ± 4.19 | 7.68 ± 0.28 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanalken, N.; Boon, K.; Szpakowska, M.; Chevigné, A.; Schols, D.; Van Loy, T. Systematic Assessment of Human CCR7 Signalling Using NanoBRET Biosensors Points towards the Importance of the Cellular Context. Biosensors 2024, 14, 142. https://doi.org/10.3390/bios14030142
Vanalken N, Boon K, Szpakowska M, Chevigné A, Schols D, Van Loy T. Systematic Assessment of Human CCR7 Signalling Using NanoBRET Biosensors Points towards the Importance of the Cellular Context. Biosensors. 2024; 14(3):142. https://doi.org/10.3390/bios14030142
Chicago/Turabian StyleVanalken, Nathan, Katrijn Boon, Martyna Szpakowska, Andy Chevigné, Dominique Schols, and Tom Van Loy. 2024. "Systematic Assessment of Human CCR7 Signalling Using NanoBRET Biosensors Points towards the Importance of the Cellular Context" Biosensors 14, no. 3: 142. https://doi.org/10.3390/bios14030142
APA StyleVanalken, N., Boon, K., Szpakowska, M., Chevigné, A., Schols, D., & Van Loy, T. (2024). Systematic Assessment of Human CCR7 Signalling Using NanoBRET Biosensors Points towards the Importance of the Cellular Context. Biosensors, 14(3), 142. https://doi.org/10.3390/bios14030142