An Extracellular Matrix Overlay Model for Bioluminescence Microscopy to Measure Single-Cell Heterogeneous Responses to Antiandrogens in Prostate Cancer Cells

 ,
,
Abstract
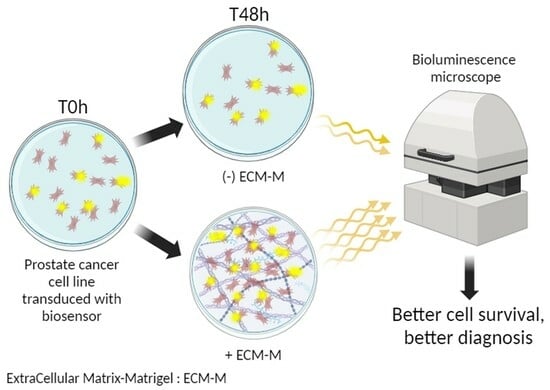
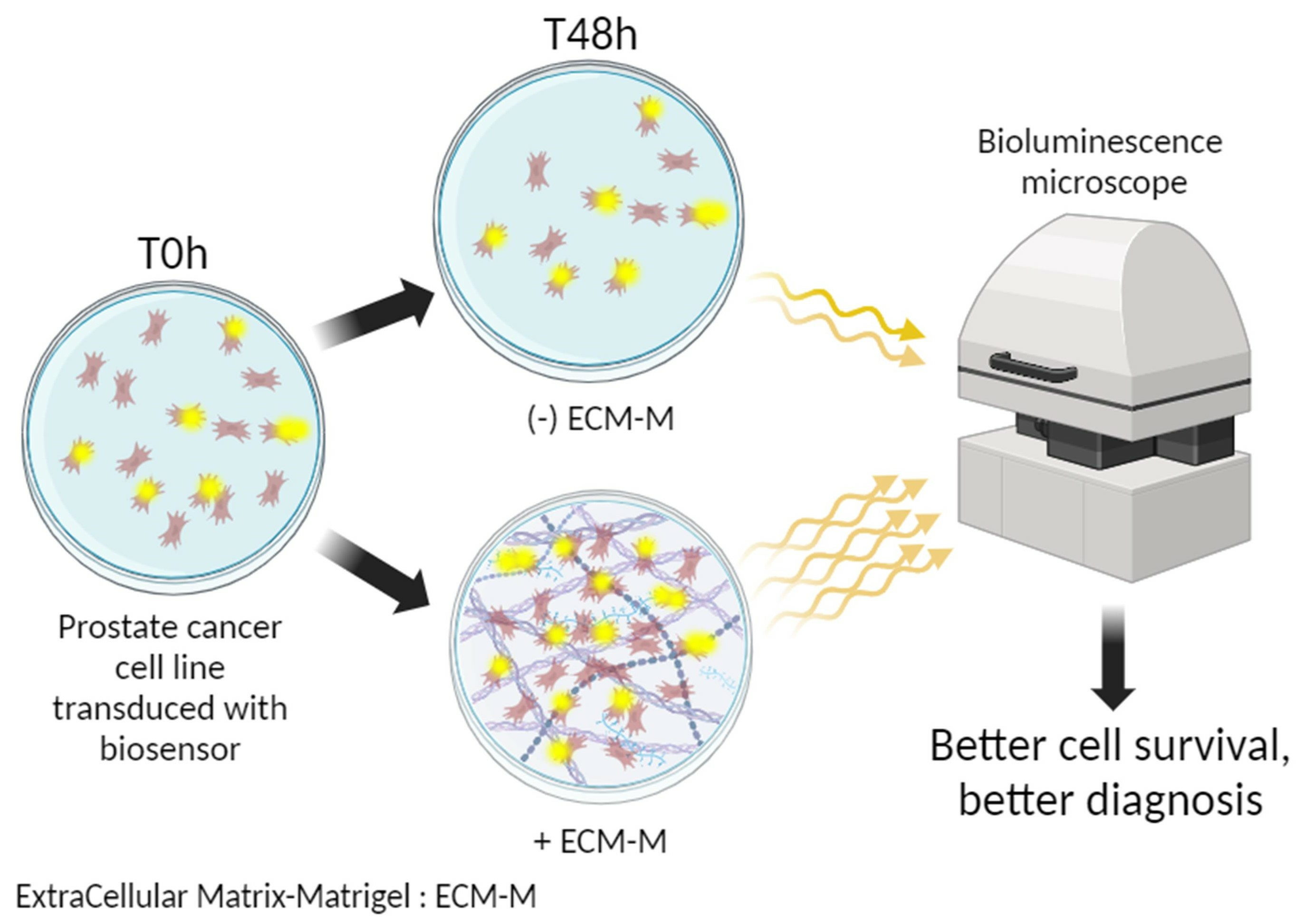
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Adenoviral Production
2.2. Cell Line Culture
2.3. Cell Line Adenoviral Infection and Treatment for Dynamic Bioluminescence Imaging
2.4. ECM-M Culture Conditions and Cell Tracking
2.5. Viability Assay
2.6. Bioluminescence Microscopy
2.7. Statistical Analysis
3. Results
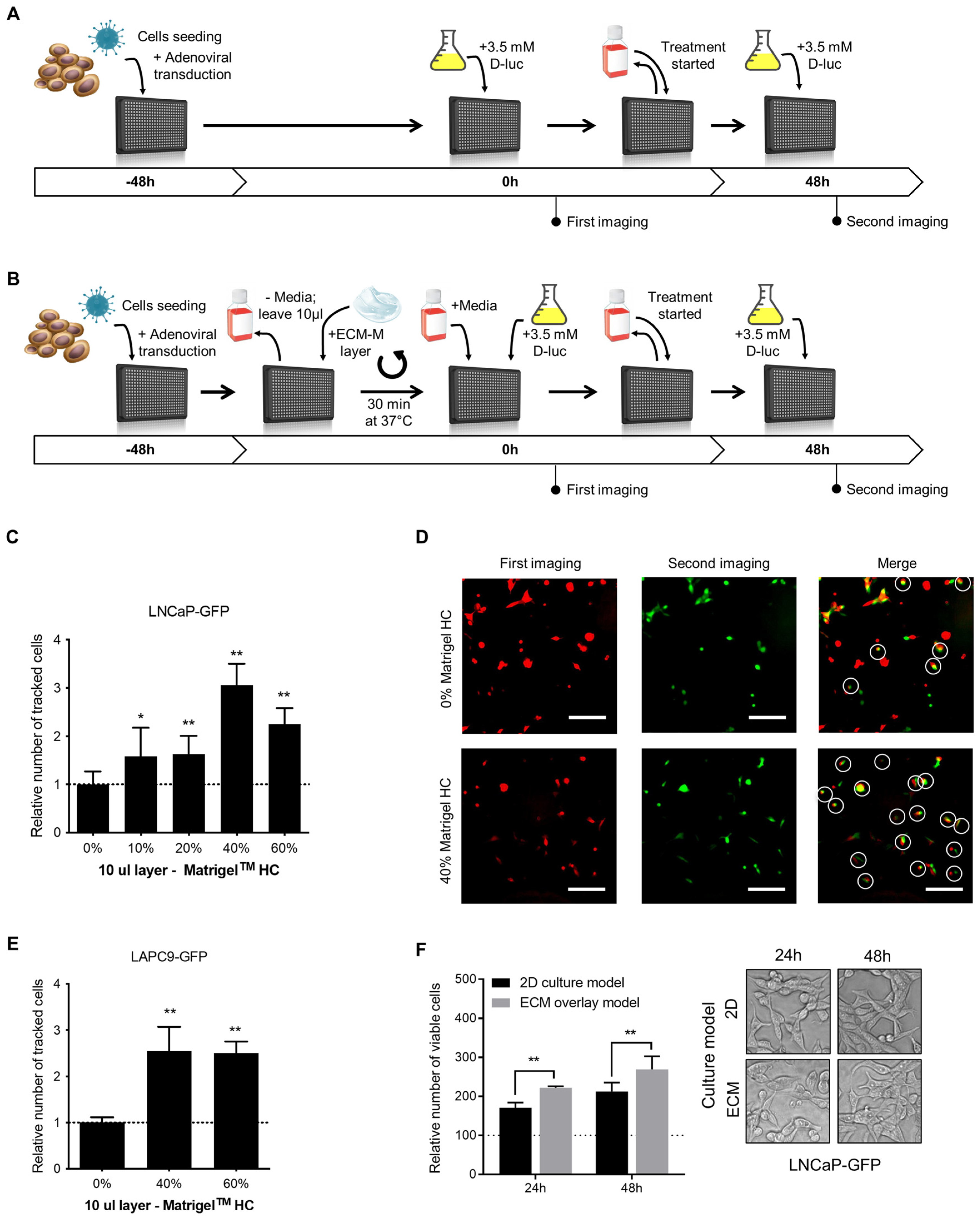
3.1. ECM-M-Based Culture Model Facilitates Cellular Tracking during Dynamic Bioluminescence Imaging and Improves Cell Viability
3.2. ECM-M-Based Culture Model Is Compatible with Bioluminescence Imaging of PCa Cells
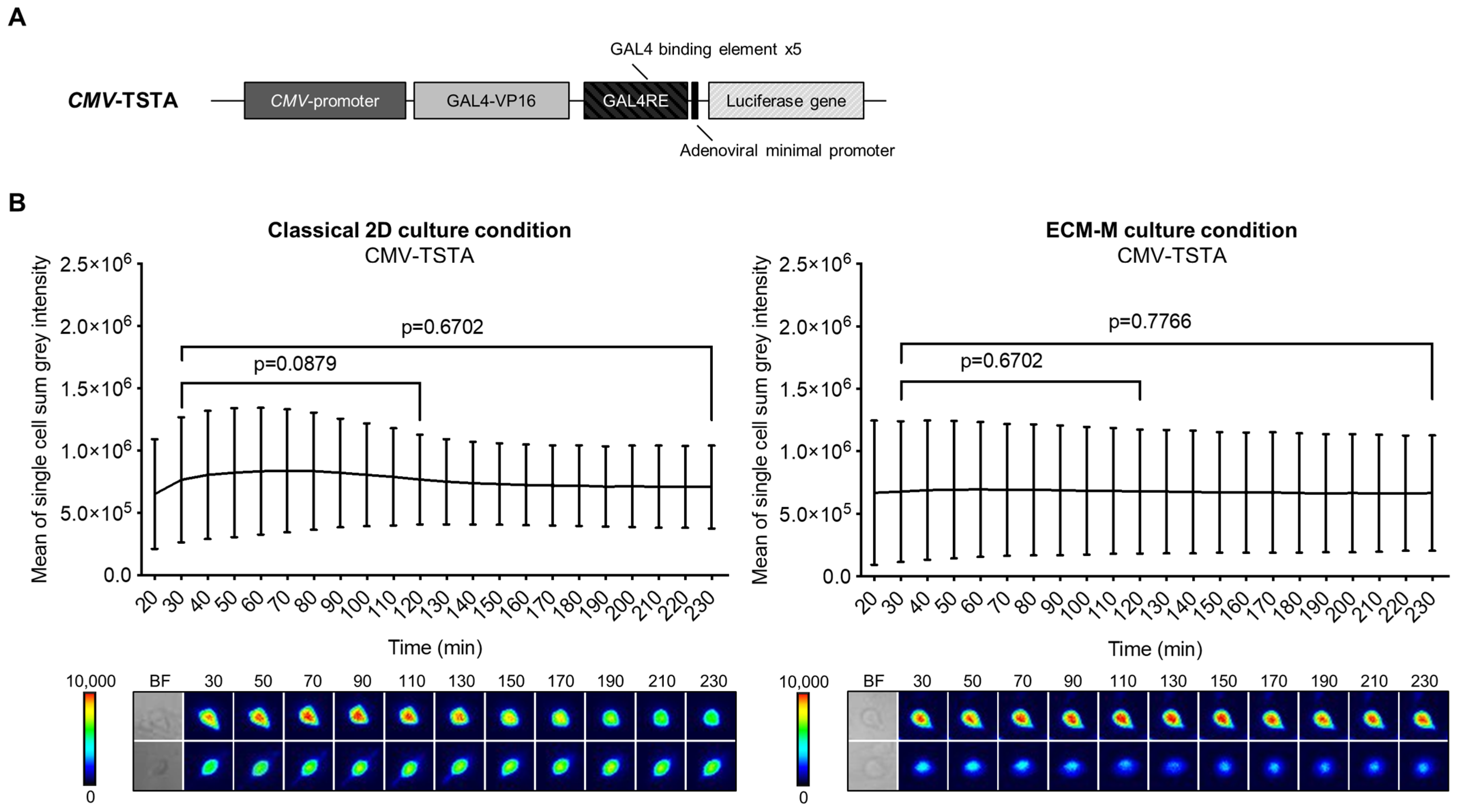
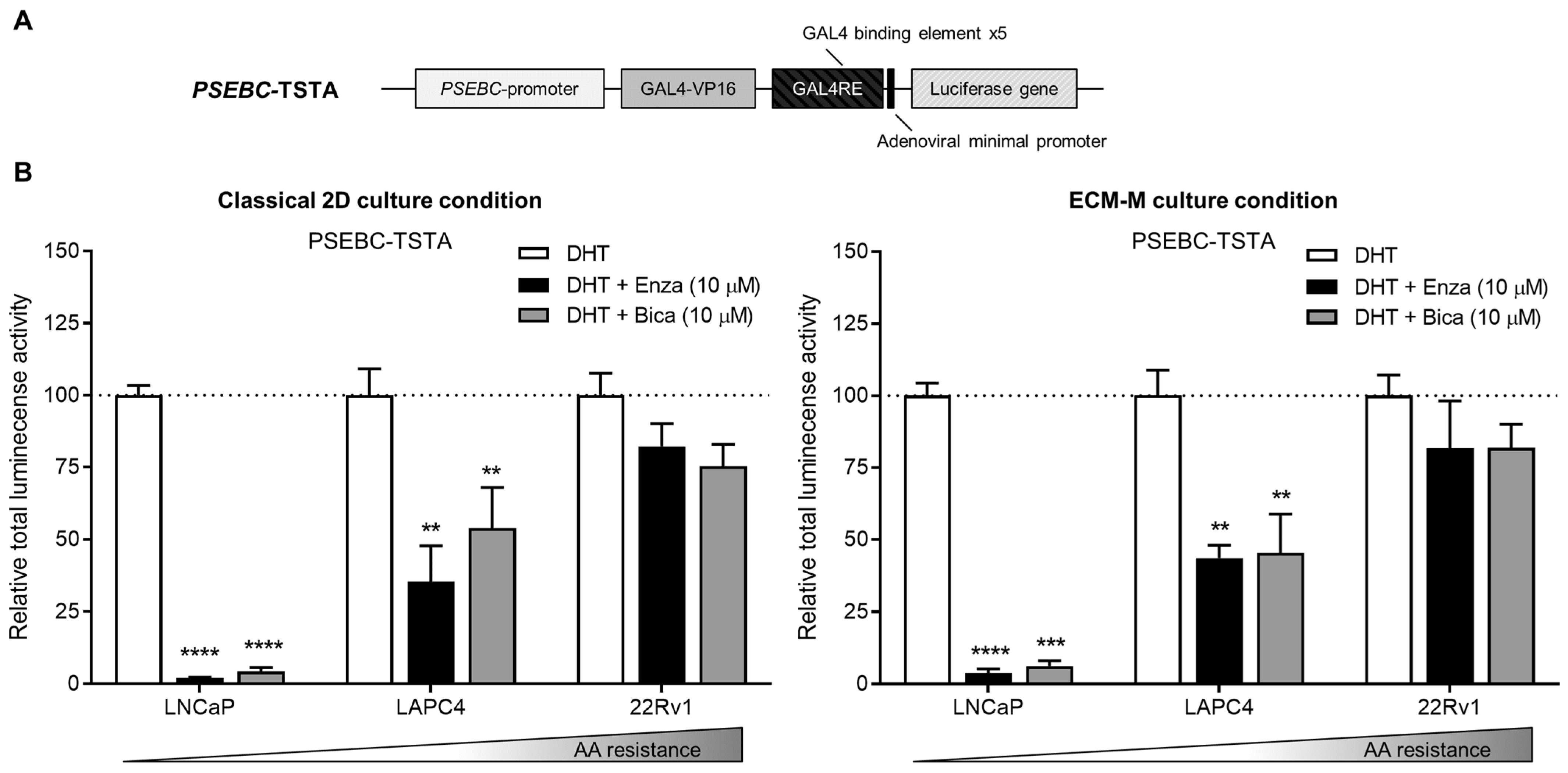
3.3. ECM-M Culture Model Supports Bioluminescence Microscopy Accuracy to Measure Androgen Receptor Activity Modulation by Antiandrogens
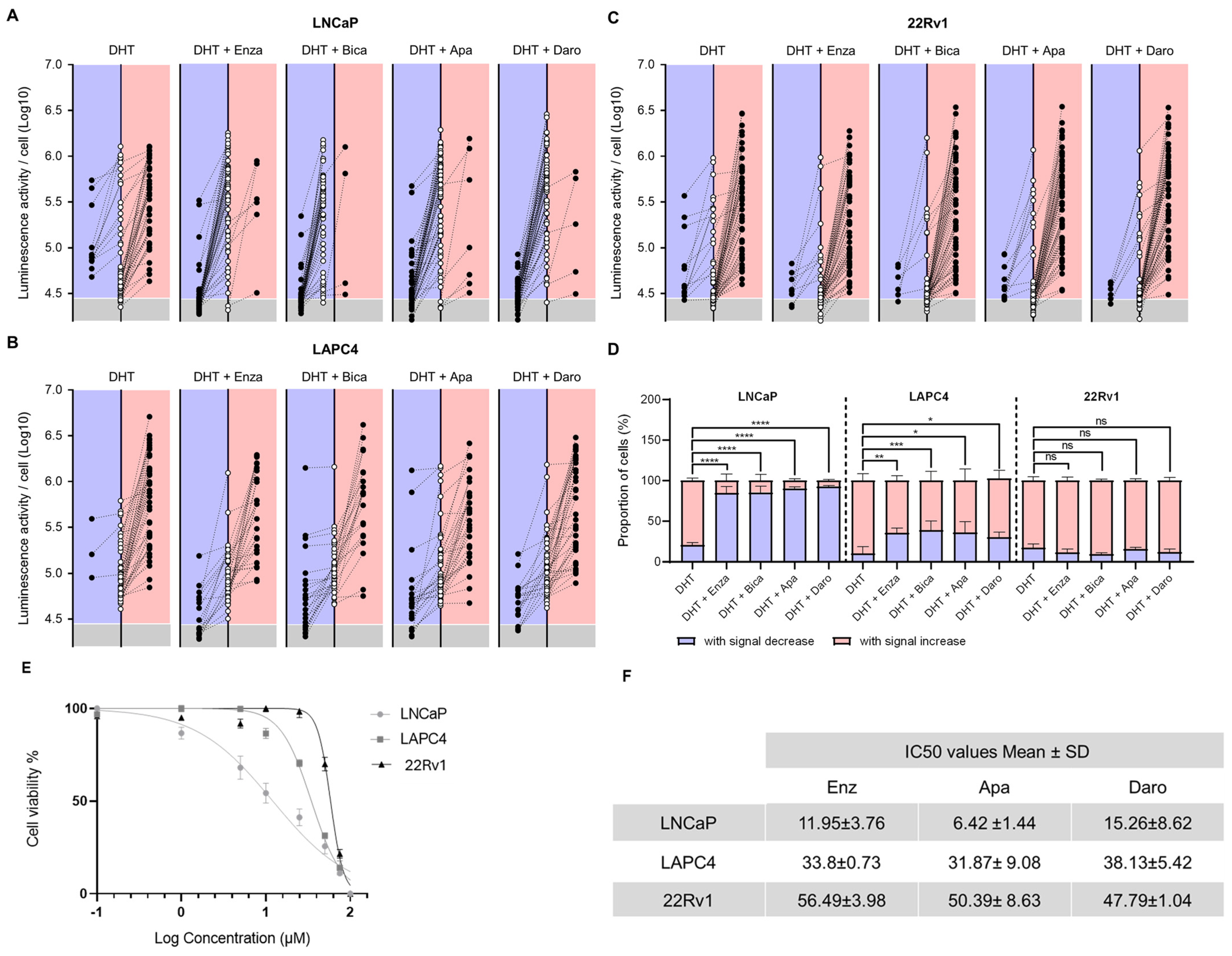
3.4. ECM-M-Based Culture Model Allows for Accurate Tracking of Each Single Cell to Determine its Sensitivity to ARi by Bioluminescence Microscopy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Cyll, K.; Ersvær, E.; Vlatkovic, L.; Pradhan, M.; Kildal, W.; Kjær, M.A.; Kleppe, A.; Hveem, T.S.; Carlsen, B.; Gill, S.; et al. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br. J. Cancer 2017, 117, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.M.; Maire, C.L.; Chou, N.; Murakami, M.A.; Knoff, D.S.; Kikuchi, Y.; Kimmerling, R.J.; Liu, H.; Haidar, S.; Calistri, N.L.; et al. Drug sensitivity of single cancer cells is predicted by changes in mass accumulation rate. Nat. Biotechnol. 2016, 34, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Calistri, N.L.; Kimmerling, R.J.; Malinowski, S.W.; Touat, M.; Stevens, M.M.; Olcum, S.; Ligon, K.L.; Manalis, S.R. Microfluidic active loading of single cells enables analysis of complex clinical specimens. Nat. Commun. 2018, 9, 4784. [Google Scholar] [CrossRef] [PubMed]
- Cetin, A.E.; Stevens, M.M.; Calistri, N.L.; Fulciniti, M.; Olcum, S.; Kimmerling, R.J.; Munshi, N.C.; Manalis, S.R. Determining therapeutic susceptibility in multiple myeloma by single-cell mass accumulation. Nat. Commun. 2017, 8, 1613. [Google Scholar] [CrossRef]
- Weitsman, G.; Mitchell, N.J.; Evans, R.; Cheung, A.; Kalber, T.L.; Bofinger, R.; Fruhwirth, G.; Keppler, M.; Wright, Z.V.F.; Barber, P.R.; et al. intratumoral heterogeneity of EGFR activity by liposome-based in vivo transfection of a fluorescent biosensor. Oncogene 2017, 36, 3618–3628. [Google Scholar] [CrossRef] [PubMed]
- Xulu, K.R.; Nweke, E.E.; Augustine, T.N. Delineating intra-tumoral heterogeneity and tumor evolution in breast cancer using precision-based approaches. Front. Genet. 2023, 14, 1087432. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Chung, D.Y.; Ha, J.S.; Cho, K.S. Novel Treatment Strategy Using Second-Generation Androgen Receptor Inhibitors for Non-Metastatic Castration-Resistant Prostate Cancer. Biomedicines 2021, 9, 661. [Google Scholar] [CrossRef]
- Soifer, H.S.; Souleimanian, N.; Wu, S.; Voskresenskiy, A.M.; Collak, F.K.; Cinar, B.; Stein, C.A. Direct Regulation of Androgen Receptor Activity by Potent CYP17 Inhibitors in Prostate Cancer Cells. J. Biol. Chem. 2012, 287, 3777–3787. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.; Lim, A.C.; Hay, C.W.; Taylor, A.E.; Wingate, A.; Nowakowska, K.; Pezaro, C.; Carreira, S.; Goodall, J.; Arlt, W.; et al. Interactions of Abiraterone, Eplerenone, and Prednisolone with Wild-type and Mutant Androgen Receptor: A Rationale for Increasing Abiraterone Exposure or Combining with MDV3100. Cancer Res. 2012, 72, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef] [PubMed]
- Messner, E.A.; Steele, T.M.; Tsamouri, M.M.; Hejazi, N.; Gao, A.C.; Mudryj, M.; Ghosh, P.M. The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines 2020, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Sawyers, C.L. Biology of Progressive, Castration-Resistant Prostate Cancer: Directed Therapies Targeting the Androgen-Receptor Signaling Axis. J. Clin. Oncol. 2005, 23, 8253–8261. [Google Scholar] [CrossRef] [PubMed]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Esther, J.; Maughan, B.L.; Anderson, N.; Agarwal, N.; Hahn, A.W. Management of Nonmetastatic Castration-Resistant Prostate Cancer: Recent Advances and Future Direction. Curr. Treat. Options Oncol. 2019, 20, 14. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Couñago, F.; López-Campos, F.; Díaz-Gavela, A.A.; Almagro, E.; Fenández-Pascual, E.; Henríquez, I.; Lozano, R.; Espinós, E.L.; Gómez-Iturriaga, A.; de Velasco, G.; et al. Clinical Applications of Molecular Biomarkers in Prostate Cancer. Cancers 2020, 12, 1550. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Porzycki, P.; Ciszkowicz, E. Modern biomarkers in prostate cancer diagnosis. Cent. Eur. J. Urol. 2020, 73, 300–306. [Google Scholar]
- Gaudreau, P.O.; Stagg, J.; Soulières, D.; Saad, F. The Present and Future of Biomarkers in Prostate Cancer: Proteomics, Genomics, and Immunology Advancements. Biomark. Cancer 2016, 8 (Suppl. S2), 15–33. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lei, Z.; Gong, Z.; Sun, Z.; Xu, D.; Piao, M. Clinical implication of prognostic and predictive biomarkers for castration-resistant prostate cancer: A systematic review. Cancer Cell Int. 2020, 20, 409. [Google Scholar] [CrossRef] [PubMed]
- Løvf, M.; Zhao, S.; Axcrona, U.; Johannessen, B.; Bakken, A.C.; Carm, K.T.; Hoff, A.M.; Myklebost, O.; Meza-Zepeda, L.A.; Lie, A.K.; et al. Multifocal Primary Prostate Cancer Exhibits High Degree of Genomic Heterogeneity. Eur. Urol. 2019, 75, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Taavitsainen, S.; Engedal, N.; Cao, S.; Handle, F.; Erickson, A.; Prekovic, S.; Wetterskog, D.; Tolonen, T.; Vuorinen, E.M.; Kiviaho, A.; et al. Single-cell ATAC and RNA sequencing reveal pre-existing and persistent cells associated with prostate cancer relapse. Nat. Commun. 2021, 12, 5307. [Google Scholar] [CrossRef]
- Jain, P.; Neveu, B.; Velot, L.; Wu, L.; Fradet, Y.; Pouliot, F. Bioluminescence Microscopy as a Method to Measure Single Cell Androgen Receptor Activity Heterogeneous Responses to Antiandrogens. Sci. Rep. 2016, 6, 33968. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, F.; Johnson, M.; Wu, L. Non-invasive molecular imaging of prostate cancer lymph node metastasis. Trends Mol. Med. 2009, 15, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, F.; Sato, M.; Jiang, Z.K.; Huyn, S.; Karanikolas, B.D.; Wu, L. A molecular imaging system based on both transcriptional and genomic amplification to detect prostate cancer cells in vivo. Mol. Ther. 2013, 21, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, F.; Karanikolas, B.D.; Johnson, M.; Sato, M.; Priceman, S.J.; Stout, D.; Sohn, J.; Satyamurthy, N.; Dekernion, J.B.; Wu, L. In vivo imaging of intraprostatic-specific gene transcription by PET. J. Nucl. Med. 2011, 52, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Champagne, A.; Jain, P.; Vélot, L.; Riopel, J.; Lefebvre, V.; Neveu, B.; Pouliot, F. A transcriptional biosensor to monitor single cancer cell therapeutic responses by bioluminescence microscopy. Theranostics 2022, 12, 474–492. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Renugopalakrishnan, V.; Liepmann, D.; Paulmurugan, R.; Malhotra, B.D. Cell-based biosensors: Recent trends, challenges and future perspectives. Biosens. Bioelectron. 2019, 141, 111435. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yan, C.; Zheng, Z. Functional polymer surfaces for controlling cell behaviors. Mater. Today 2018, 21, 38–59. [Google Scholar] [CrossRef]
- Benton, G.; Arnaoutova, I.; George, J.; Kleinman, H.K.; Koblinski, J. Matrigel: From discovery and ECM mimicry to assays and models for cancer research. Adv. Drug Deliv. Rev. 2014, 79–80, 3–18. [Google Scholar] [CrossRef]
- Frégeau-Proulx, L.; Lacouture, A.; Berthiaume, L.; Weidmann, C.; Harvey, M.; Gonthier, K.; Pelletier, J.-F.; Neveu, B.; Jobin, C.; Bastien, D.; et al. Multiple metabolic pathways fuel the truncated tricarboxylic acid cycle of the prostate to sustain constant citrate production and secretion. Mol. Metab. 2022, 62, 101516. [Google Scholar] [CrossRef]
- Evans, R.K.; Nawrocki, D.K.; Isopi, L.A.; Williams, D.M.; Casimiro, D.R.; Chin, S.; Chen, M.; Zhu, D.-M.; Shiver, J.W.; Volkin, D.B. Development of stable liquid formulations for adenovirus-based vaccines. J. Pharm. Sci. 2004, 93, 2458–2475. [Google Scholar] [CrossRef]
- Lee, Y.; Schwarz, E.; Davies, M.; Jo, M.; Gates, J.; Wu, J.; Zhang, X.; Lieberman, J.R. Differences in the cytokine profiles associated with prostate cancer cell induced osteoblastic and osteolytic lesions in bone. J. Orthop. Res. 2003, 21, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2, e17. [Google Scholar] [CrossRef] [PubMed]
- Poincloux, R.; Collin, O.; Lizárraga, F.; Romao, M.; Debray, M.; Piel, M.; Chavrier, P. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc. Natl. Acad. Sci. USA 2011, 108, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.L.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.M.; Khoo, U.S.; Ng, I.O.; et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Estrada, M.F.; Vidic, S.; Gjerde, K.; Rudisch, A.; Santo, V.E.; Barbier, M.; Blom, S.; Arundkar, S.C.; Selvam, I.; et al. Capturing tumor complexity in vitro: Comparative analysis of 2D and 3D tumor models for drug discovery. Sci. Rep. 2016, 6, 28951. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Qin, L.; Wei, W.; Geng, F.; Fan, R.; Shin, Y.S.; Guo, D.; Hood, L.; Mischel, P.S.; Heath, J.R. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc. Natl. Acad. Sci. USA 2012, 109, 419–424. [Google Scholar] [CrossRef]
- Kim, K.-T.; Lee, H.W.; Lee, H.-O.; Kim, S.C.; Seo, Y.J.; Chung, W.; Eum, H.H.; Nam, D.-H.; Kim, J.; Joo, K.M.; et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015, 16, 127. [Google Scholar] [CrossRef]
- Heath, J.R.; Ribas, A.; Mischel, P.S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 2016, 15, 204–216. [Google Scholar] [CrossRef] [PubMed]
- McLeod, C.M.; Mauck, R.L. On the origin and impact of mesenchymal stem cell heterogeneity: New insights and emerging tools for single cell analysis. Eur. Cells Mater. 2017, 34, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Tombal, B.; Saad, F.; Fizazi, K.; Sternberg, C.N.; Crawford, E.D.; Shore, N.; Kopyltsov, E.; Kalebasty, A.R.; Bögemann, M.; et al. Darolutamide Plus Androgen-Deprivation Therapy and Docetaxel in Metastatic Hormone-Sensitive Prostate Cancer by Disease Volume and Risk Subgroups in the Phase III ARASENS Trial. J. Clin. Oncol. 2023, 41, 3595–3607. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Champagne, A.; Chebra, I.; Jain, P.; Ringuette Goulet, C.; Lauzier, A.; Guyon, A.; Neveu, B.; Pouliot, F. An Extracellular Matrix Overlay Model for Bioluminescence Microscopy to Measure Single-Cell Heterogeneous Responses to Antiandrogens in Prostate Cancer Cells. Biosensors 2024, 14, 175. https://doi.org/10.3390/bios14040175
Champagne A, Chebra I, Jain P, Ringuette Goulet C, Lauzier A, Guyon A, Neveu B, Pouliot F. An Extracellular Matrix Overlay Model for Bioluminescence Microscopy to Measure Single-Cell Heterogeneous Responses to Antiandrogens in Prostate Cancer Cells. Biosensors. 2024; 14(4):175. https://doi.org/10.3390/bios14040175
Chicago/Turabian StyleChampagne, Audrey, Imene Chebra, Pallavi Jain, Cassandra Ringuette Goulet, Annie Lauzier, Antoine Guyon, Bertrand Neveu, and Frédéric Pouliot. 2024. "An Extracellular Matrix Overlay Model for Bioluminescence Microscopy to Measure Single-Cell Heterogeneous Responses to Antiandrogens in Prostate Cancer Cells" Biosensors 14, no. 4: 175. https://doi.org/10.3390/bios14040175
APA StyleChampagne, A., Chebra, I., Jain, P., Ringuette Goulet, C., Lauzier, A., Guyon, A., Neveu, B., & Pouliot, F. (2024). An Extracellular Matrix Overlay Model for Bioluminescence Microscopy to Measure Single-Cell Heterogeneous Responses to Antiandrogens in Prostate Cancer Cells. Biosensors, 14(4), 175. https://doi.org/10.3390/bios14040175