1. Introduction
The total volume of data generated by human society annually is experiencing exponential growth, gradually approaching the physical limits of existing storage media [
1]. Under the current paradigm of solid-state storage, the demand for storing massive data imposes enormous pressure on material resources, personnel allocation, industrial power, and land usage [
2,
3]. DNA, with its exceptionally high data density and ultra-long storage lifespan, is considered a highly promising alternative as the next-generation storage medium [
4,
5,
6,
7,
8]. The process of utilizing DNA for data storage mainly involves four steps: data encoding, DNA synthesis, DNA sequencing, and data decoding. Among them, DNA synthesis and DNA sequencing correspond to the writing and reading of the stored data, serving as the crucial foundational technologies in the entire storage process. DNA sequencing technology, benefiting from the enormous collective efforts from both academia (e.g., the Human Genome Project) and industry (including market giant Illumina and third-generation sequencing platforms like PacBio and Oxford Nanopore), has now surpassed DNA synthesis technology significantly in almost all relevant metrics including material cost, operation throughput, and time consumption [
5,
9]. Therefore, the current bottleneck hindering development in DNA data storage lies mainly in DNA synthesis and thus further advancement in high-density, high-throughput, and low-cost DNA synthesis technology is crucial for DNA data storage systems.
In the current DNA synthesis technological landscape, the most widely used platform is the conventional column synthesizers where a standard four-step reaction relying on the solid phase phosphoramidite chemistry is adopted [
10]. Due to its low synthesis throughput (i.e., the number of unique oligonucleotides produced), high reagent consumption, and high costs for each synthesized oligonucleotide, the column synthesizers have failed to meet the increasing demands of evolving biological applications, let alone the more demanding requirement for data storage [
5,
11]. Chip-based high-throughput DNA synthesis strategies have been gradually developed over the past decades. Current high-throughput DNA synthesis methods still rely on the four-step cycle of phosphoramidite chemistry, with key modification in controlling the first deprotection step by incorporating inkjet printing [
12], electrochemistry [
13], photolithography [
14], or other techniques [
5,
10] as means of paralleled DNA synthesis control at a massive scale. For instance, the inkjet DNA synthesizer involves the selective addition of nucleotides by directly spraying nucleotide monomer solution onto specified spots through an inkjet printing head [
12]. While the inkjet DNA synthesizer is relatively straightforward, its site density is always constrained by the minimum droplet size that the printing head can generate and the positioning accuracy of the mechanical system. The light-directed DNA synthesis utilizes photolabile protecting groups in combination with photolithographic masks for selective deprotection [
15]. The issue of higher cost induced by multiple masks was addressed by deploying the digital micromirror array for directing programmable light sources onto individual reaction spots [
14]. During the synthesis, light scattering may lead to incorrect deprotection of neighboring sites around the illuminated spot, resulting in insertion errors. Therefore, light-directed synthesis faces challenges related to low synthesis quality and high synthesis costs. More recently, the light-directed platform has been applied in direct DNA data writing using >10,000 independent synthesis spots, with a specially designed error correcting algorithm to cope with the higher error rates, which is not suitable in biological applications requiring precision at the base-by-base level [
16].
Most notably, electrochemically directed DNA synthesis involves selectively applying voltage to the reaction spots through an exposed electrode, initiating the electrochemical redox of hydroquinone/benzoquinone in an organic solvent. After the application of current to the microelectrodes, oxidation of hydroquinone near the anode occurs to generate protons, which removes the dimethoxytrityl (DMT) protecting group from the end of oligonucleotides. The excess protons will diffuse and continue to remove the DMT in the vicinity, until they approach the cathode and undergo a reduction reaction with benzoquinone to revert to hydroquinone [
17]. The currently available electrochemically directed platforms report 90,000 independent synthesis spots with > 100-mer oligonucleotide arrays [
18]. To restrict proton diffusion and interference among neighboring synthesis spots, Karl Maurer et al., from CustomArray Inc., introduced 2,6-lutidine as an organic base to the system for neutralizing excessive protons [
13]. Recently, Nguyen et al. reduced the electrode diameter for electrochemically directed DNA synthesis to 650 nm, achieving a synthesis density of 25 million/cm
2, with a specific aim at DNA data writing [
11]. To confine proton diffusion in such a high-density electrode array, microwells of 200 nm depth were established around each electrode as physical shields, while simultaneously four cathodes were placed nearby each anode to reduce the diffusing protons through redox reactions.
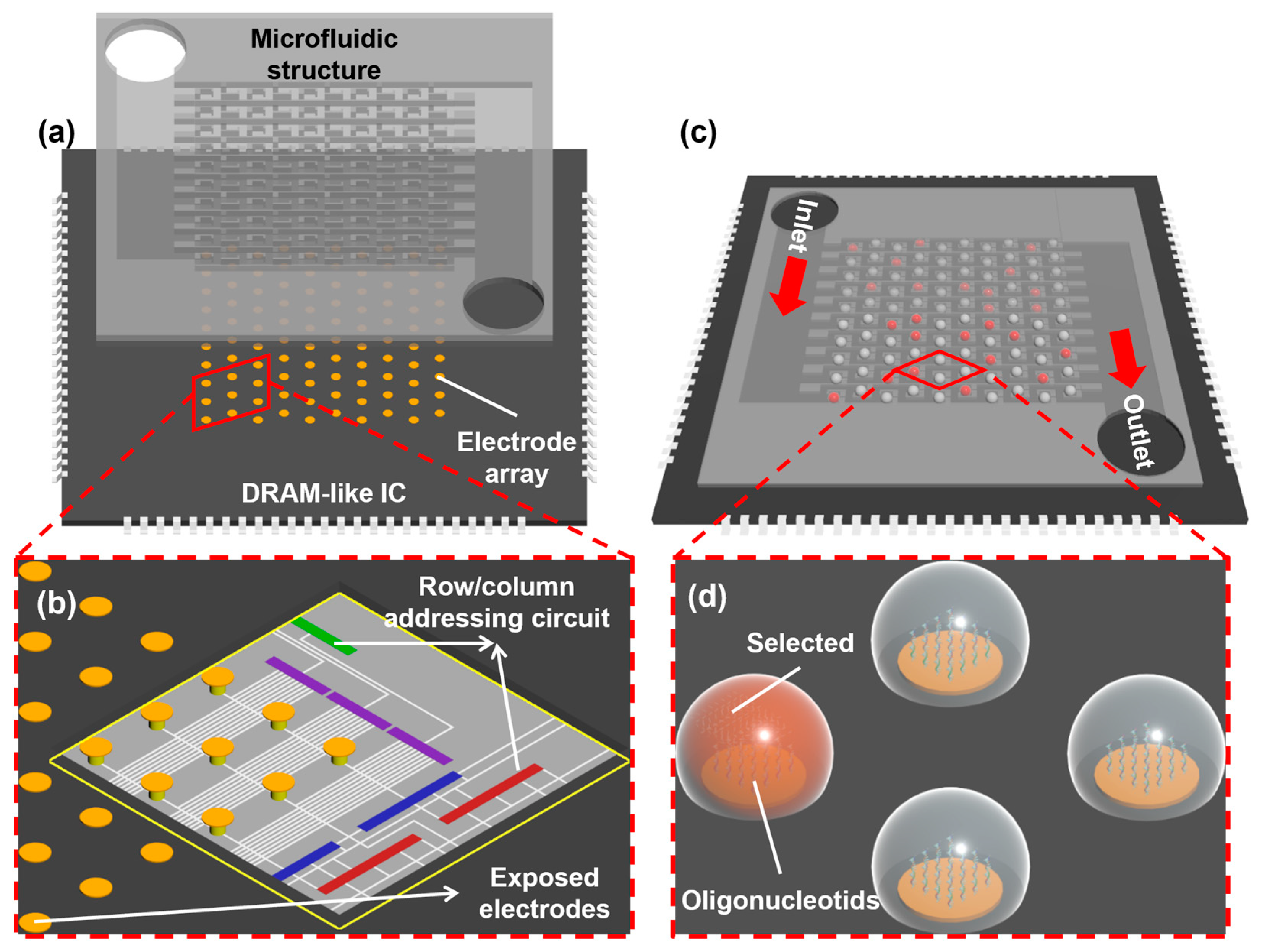
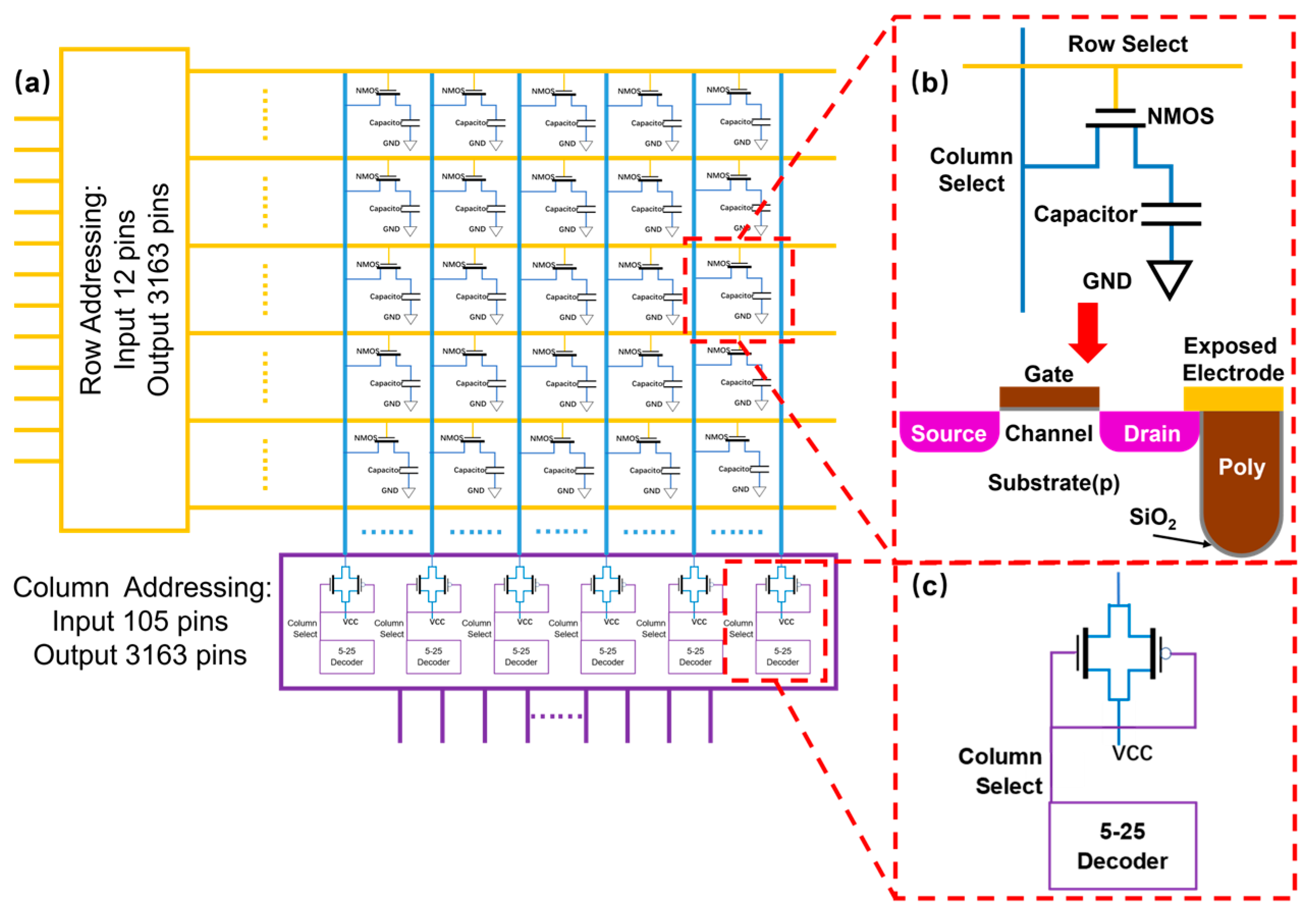
Among existing platforms, electrochemically directed DNA synthesis has achieved the highest density of synthesis spots. With the advancement of modern microelectronics design and fabrication processes, this method holds tremendous potential for even greater improvement in density and throughput. It could become the best candidate for high-speed data writing in future DNA data storage. However, as the number of individually addressable reaction sites expands (and thus the synthesis throughput), a more sophisticated circuit design is needed to implement a precise logic for controlling each electrochemical reaction. Moreover, as the density of sites increases, crosstalk caused by proton diffusion among neighboring sites continues to hinder the further development of this technology. In this study, we seek to explore the design of a novel high-density, high-throughput, electrochemically directed DNA synthesis platform (
Figure 1). The presented platform utilizes a dynamic random-access memory (DRAM)-like integrated circuit (IC) to achieve individual addressing control of electrodes in high-throughput DNA synthesis. Simultaneously, it incorporates a static droplet array (SDA)-based microfluidic structure [
19,
20] to restrict proton diffusion, thereby avoiding strategies involving organic bases and cathodic reduction. Our design registers a throughput of 10 million possible synthesis spots with the density in the platform reaching 10 million/cm
2, a combination that exceeds all reported high-throughput platforms to date [
5]. In the following, we show the integrated circuit design for implementing an independent selection of electrodes and confirm its functionality in accurate addressability and freedom for voltage adjustment through simulation. We also demonstrate a static droplet formation using a two-phase flow microfluidic model. Finally, we prepared a microfabricated electrode array on which a preliminary oligonucleotide synthesis and characterization experiment was performed to test the feasibility of our design.
2. Methods
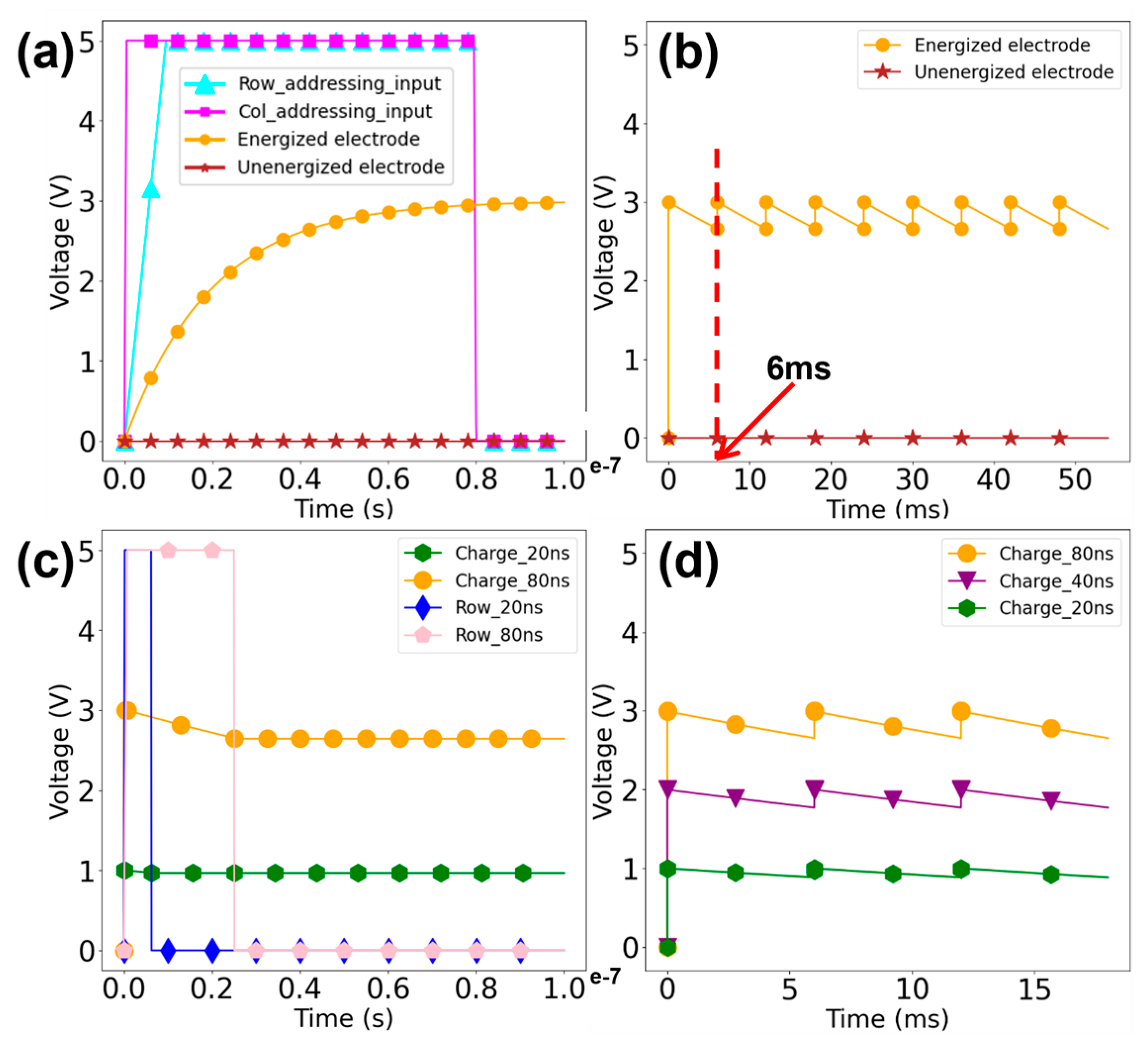
2.1. Integrated Circuit Simulation
The procedure for designing the integrated circuit unfolds as follows: initially, we crafted the Verilog code in line with the addressing specifications, subjecting it to pre-simulation for functional validation. Subsequently, we undertook a compilation phase to produce a netlist and scrutinized the logical consistency between the netlist and the code. Following this, the layout was crafted based on the generated netlist. Ultimately, post-simulation was executed post-extraction of the layout, subsequent to the clearance of DRC and LVS checks.
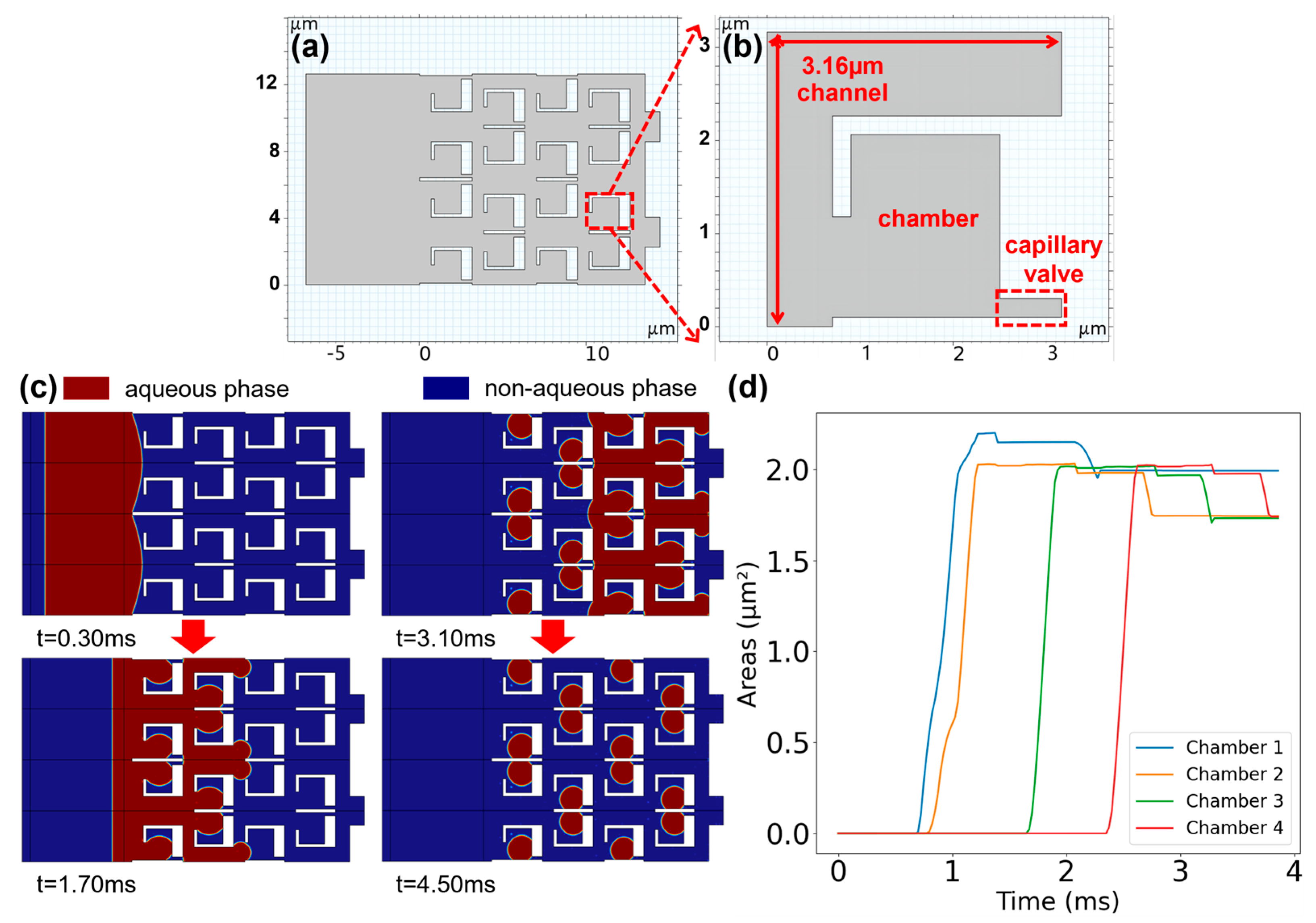
2.2. Microfluidic Numerical Simulation
The generation of droplets within the SDA-based microfluidic structure was simulated using the CFD module of COMSOL Multiphysics software (version 6.1), the interaction between dispersed and continuous phases in the two-phase flow was modeled using the level-set physics field. To reduce computational complexity and time consumption, simulations were conducted using a two-dimensional model. The continuous phase selected was FC-3283, with a dynamic viscosity of 1.365 × 10−3 Pa s and a density of 1.82 × 103 kg/m3; while the dispersed phase was water from the COMSOL material library, with a contact angle set to 135° to simulate hydrophobic materials. To ensure accurate numerical simulation results in structures with significant size variations, a mapped mesh was employed to uniformly partition the simulation structure. The degree to which the mesh elements resemble the ideal geometric shapes—square in our 2D mapped mesh—is reflected in the average mesh element quality of 0.9948. A value closer to 1 indicates that the elements are well-shaped and size-consistent, which is essential for minimizing numerical errors and ensuring the reliability of the simulation outcomes. To mimic sequential fluid injection in experiments, an injection channel was placed before the SDA structure, with the SDA structure and the front inlet of injection channel pre-filled with the continuous phase, while the remaining portion of the injection channel was filled with the dispersed phase. The continuous phase was injected at a speed of 3 mm/s to ensure that when the array contained ten million capillary valve units with a density of 107/cm2, the solution throughout the entire structure could be replenished within 10 s.
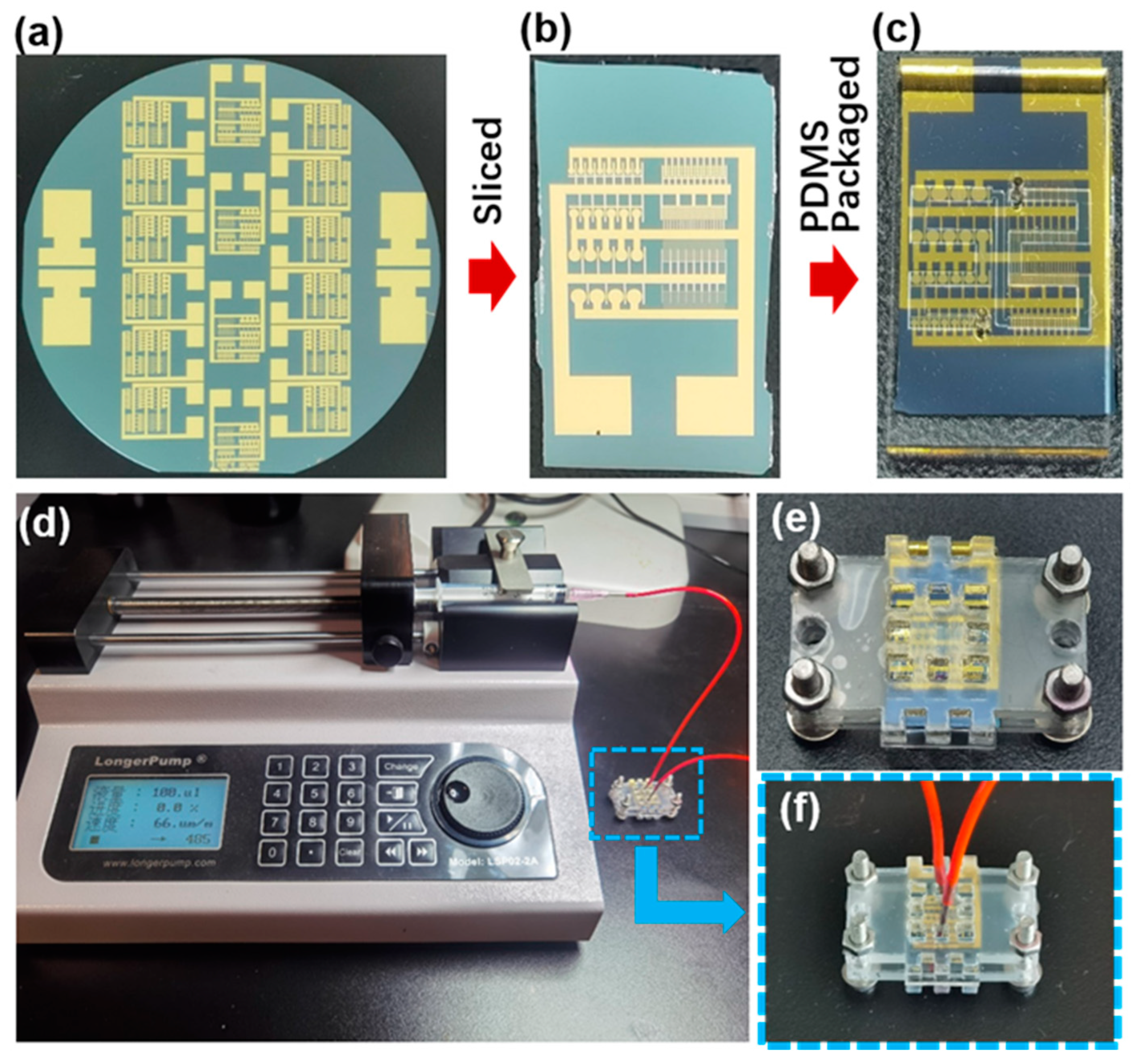
2.3. Microelectrode Array Fabrication
Electrodes were prepared on 4-inch silicon wafers with a silicon dioxide layer using a lift-off process, as shown in
Supplementary Information (Supplementary Materials Figure S1). First, the wafers were baked at 150 °C for 5 min to remove moisture. Then, hexamethyldisilazane (HMDS) was spin-coated on the wafer at 2000 rpm for 10 s, followed by spin-coating of AZ NLOF 2020 negative photoresist at 500 rpm for 30 s and again at 2000 rpm for 10 s. After leveling, soft baking was performed at 100 °C for 90 s. The wafers were exposed for 10 s in the MA6B Double Sided Mask Aligner System, then post-baked at 100 °C for 90 s. Subsequently, the exposed wafers were immersed in ZX-238 developer for 2 min, then thoroughly rinsed with ultrapure water. A 20 nm titanium (Ti) adhesion layer and 70 nm gold (Au) layer were then deposited onto the wafer surfaces using a BC1800 Electron Beam Precise Nanoscale Deposition System. Finally, the patterned Ti/Au microelectrode arrays were obtained by lifting off the photoresist in acetone with ultrasonication.
2.4. Microelectrode Array Functionalization
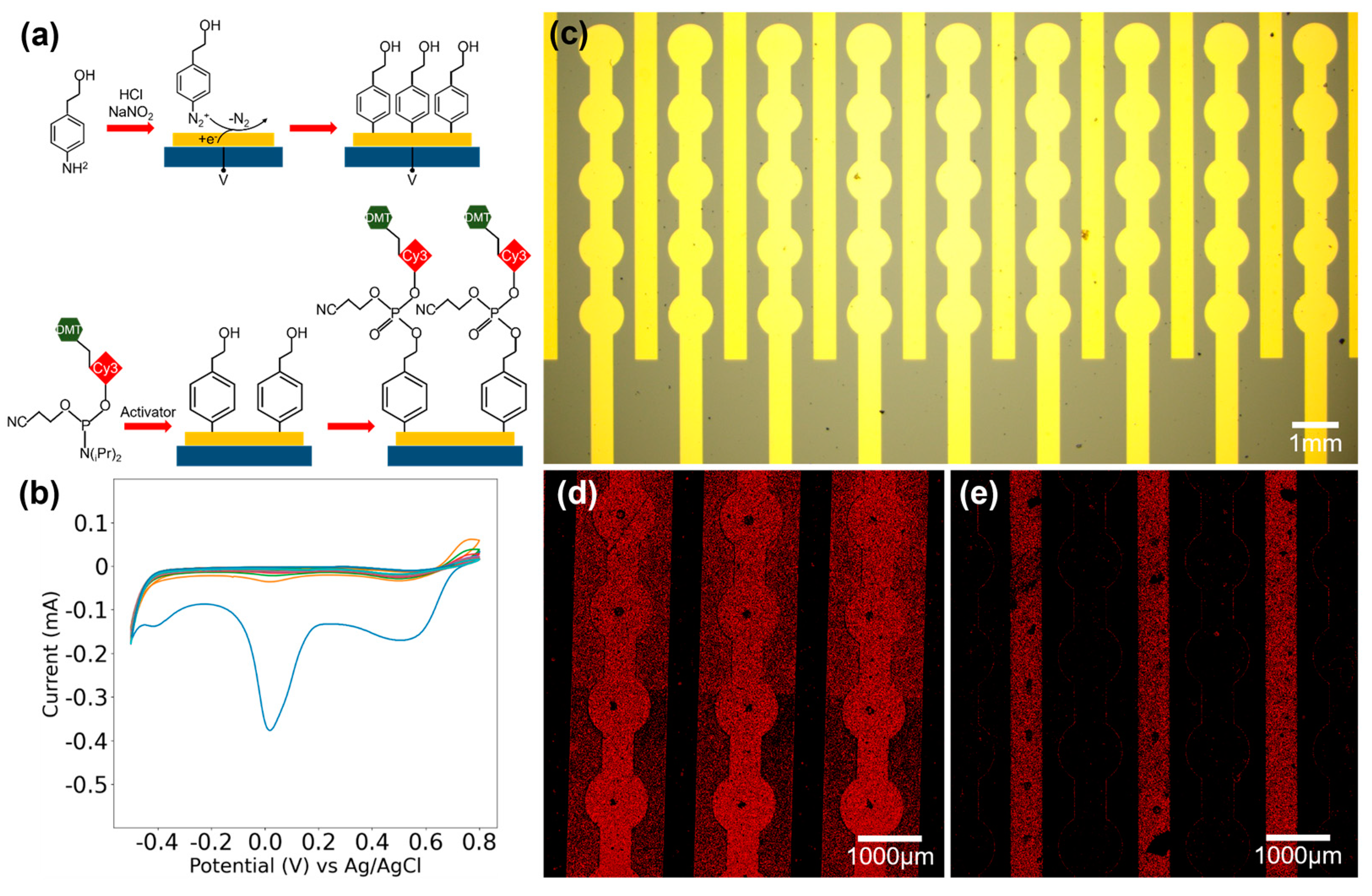
The silicon wafer is initially sliced into chips, followed by a sonication process in acetone and IPA and thorough rinsing with ultrapure water. The microelectrode array chips undergo a 10 min cleaning process in a PDC-MG plasma cleaner at 150 W power. Hydroxyl groups are grafted onto the microelectrode array via electrografting of 4-aminophenylethanol. Initially, a 60 mL solution of 0.2M HCl is prepared at 0 °C and degassed in a vacuum pump for 5 min. Then, 41.1 mg of 4-aminophenylethanol is dissolved in the HCl solution, stirred thoroughly with a glass rod, and degassed for an additional 5 min. Next, 99.4 mg of sodium nitrite is added, ensuring complete dissolution. Then, degassing for an additional 5 min is carried out using a vacuum pump. The temperature of the solution is maintained at around 0 °C throughout the above procedures. The cleaned chips were immersed in a prepared solution, with the working electrode and working sense electrode of the Gamry 1010E electrochemical workstation clamped onto the pad of the chip. Functionalization of the electrode surface was performed using cyclic voltammetry, with a scan rate of 50 mV/s, starting and ending at the open-circuit potential (OCP), cycling 30 times between −0.5 and 0.8 V vs. Ag/AgCl. Finally, the electrodes were thoroughly rinsed with deionized water, acetone, and IPA and then dried for later use.
2.5. Oligonucleotide Synthesis and Characterization
First, 9.1 mg Cy3 was dissolved in 100 μL of activator to achieve a concentration of 0.1 M. The solution was injected into the microfluidic channel for 30 min by a syringe pump, ensuring thorough reaction of the activated Cy3 with the hydroxyl groups on the functionalized electrode surface. Subsequently, the channel was flushed with sufficient acetonitrile, followed by the injection of 1 mL of oxidizing agent for 2 min. Then, another round of acetonitrile flushing was carried out. The microelectrode array chip was immersed in acetonitrile, undergoing ultrasonic cleaning for 15 min to remove physical adsorption. The chip was subsequently removed, dried, and observed using the Zeiss LSM 900 laser scanning confocal microscope obtained from Oberkochen Germany.
4. Discussion
The electrochemically directed synthesis strategy offers great potential for achieving the highest density and throughput due to the advancement of microelectronics design and fabrication processes. Yet tremendous challenges still remain in pursuing high-throughput DNA synthesis, mainly in terms of individual voltage addressability with a growing number of synthesis spots and the proton interference among adjacent spots with increasing density. The number of independent input pins a chip packaging could offer is extremely limited compared to the amount that is needed for high-throughput DNA synthesis. We here repurposed the DRAM integrated circuit design previously used for memory chips to achieve the goal of independent addressability. Similar to memory chips, the electrochemical deprotection requires a steady voltage level to be maintained while being selected. As detailed in the previous section, the number of addressable electrodes scales exponentially with the available controlling input, leading to fairly manageable design constraints (i.e., 145 pins to control 10 million spots).
Another contribution in our design is the incorporation of the static droplet microfluidic structure as the restriction mechanism for preventing interference. Physical confinement is the most straightforward strategy in these circumstances. The major difficulty with this method is the fact that different reaction sites within the same chip cannot be completely isolated from each other. They need to share the same set of reagents that facilitate processes like deprotection, activation, oxidation, and flushing. Hence, as the size of each reaction site continues to shrink in order to pursue even higher density, it is becoming extremely difficult to rely only on partial physical confinement to prevent interference [
23]. Two additional methods have been proposed, one deploying counter microelectrodes around each working electrode to act as a proton sink [
11], and the other relying on dissolving organic base as a proton neutralizing agent [
13]. Yet both methods could still become less effective as the density increases. The incorporation of static droplets aims to completely overcome such difficulties by using the oil phase to completely isolate each reaction site. In addition, even though the highest oligonucleotide synthesis density has been achieved by the method of providing both physical and chemical barriers by microwells and counter microelectrodes, more complicated IC design and processing are required to realize the individual addressing control of electrodes based on this mechanism in the future. It is also possible to use air as the non-aqueous phase and thus further simplify the reagent administration process during the synthesis experiment [
24].
For the platform we designed, there are still some crucial issues that need to be addressed in platform development and implementation. Firstly, even though the state-of-the-art microelectronic manufacturing process can realize our design of a DRAM-like IC with 10 million electrodes on a 1 cm
2 chip, the unconventional packaging is the most crucial component since it must be compatible with the electronic driving system and allow the IC chip and the microfluidic structure to bond together. Secondly, the PDMS surface has a serious adsorption issue, which can affect the efficiency of the synthesis. It is possible to further coat PDMS walls with SiO2 which has been recently reported to be able to eliminate non-specific adsorption and ensure maintenance of the synthesis [
25]. In addition, because the minimum width of the microchannel (the width of the capillary valve) is 0.2 μm and bonding between the IC chip and the microfluidic chip needs to be considered, our microfluidic design cannot be easily realized with PDMS materials. On the other hand, the utilization of glass as the structural material for microfluidics can potentially address both issues and result in further improvement of the device performance.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





