Investigating Colorimetric Protein Array Assay Schemes for Detection of Recurrence of Bladder Cancer



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chip Fabrication
2.3. Preparation of Immunogold Probes
2.4. Oxidation of Carbon Nanoparticles
2.5. Antibody Coupling to oCNPs
2.6. Modification of Carbon NPs with Neutravidin
2.7. Chip Processing
2.7.1. Detection Antibody Labeled with Carbon-and Gold NPs
2.7.2. Detection via Streptavidin/Neutravidin-Biotin Binding
2.8. Silver Staining
2.9. Data Analysis
3. Results and Discussion
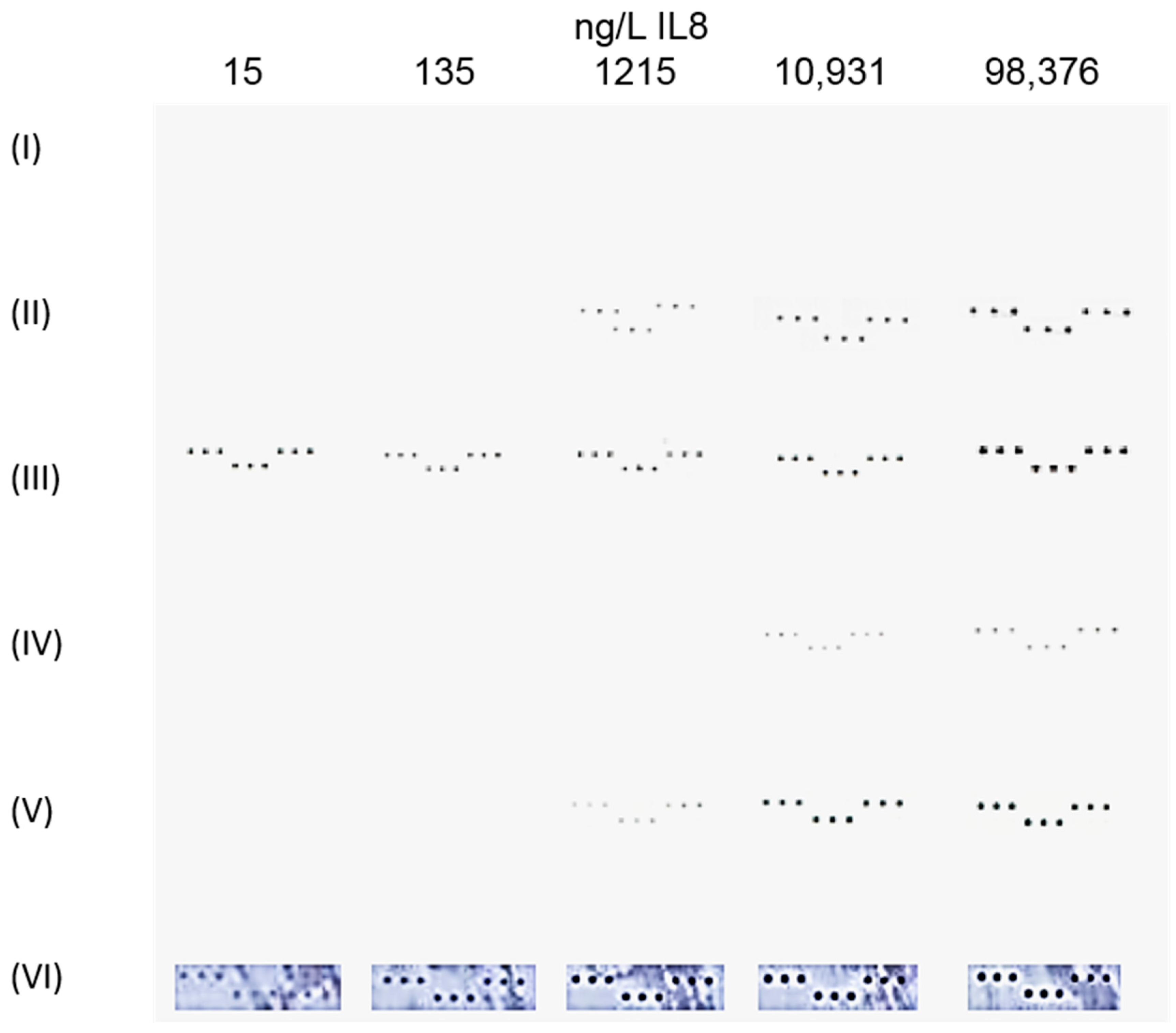
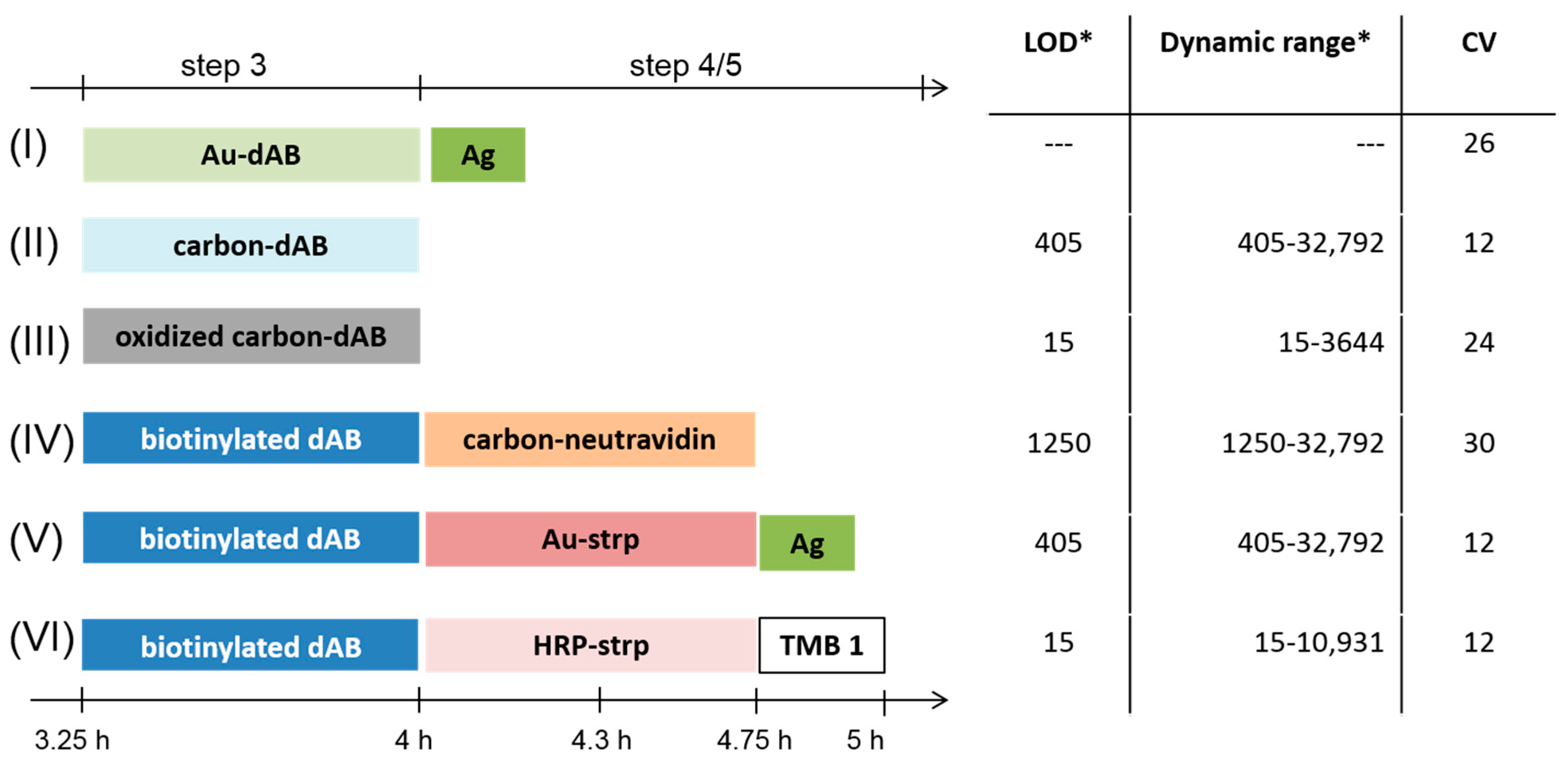
3.1. Colorimetric Assay Schemes
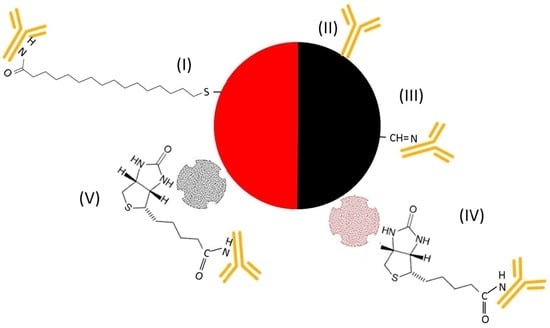
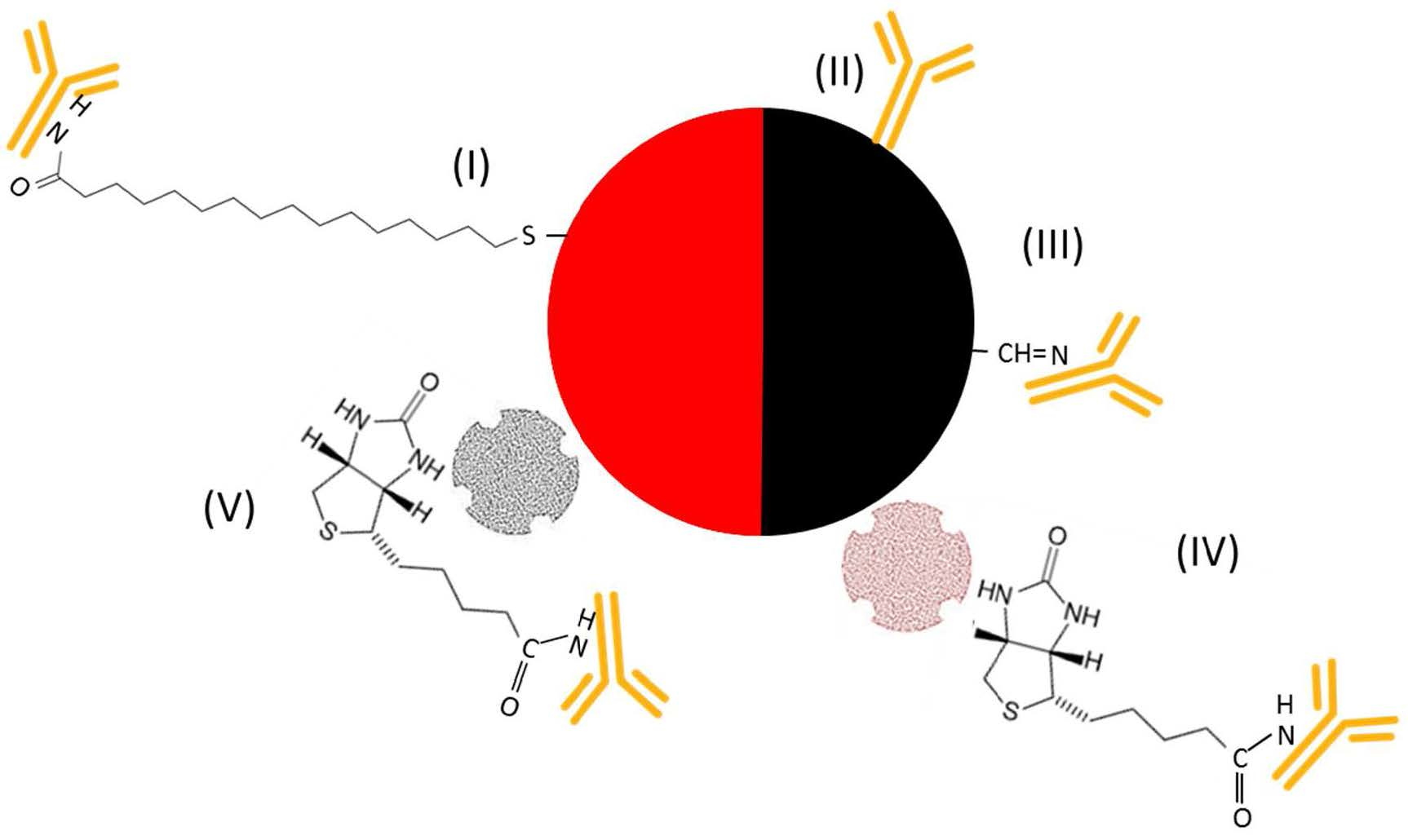
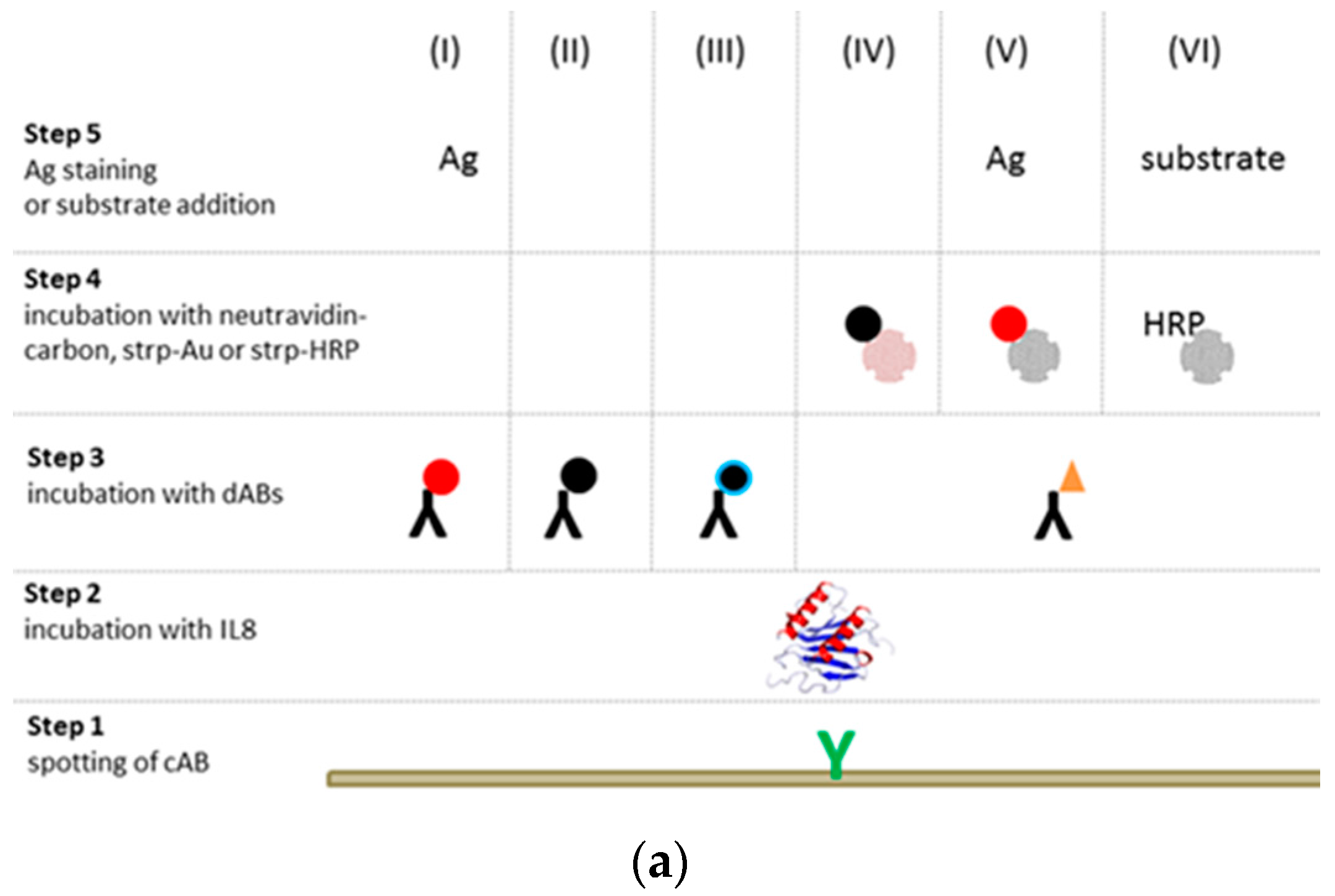
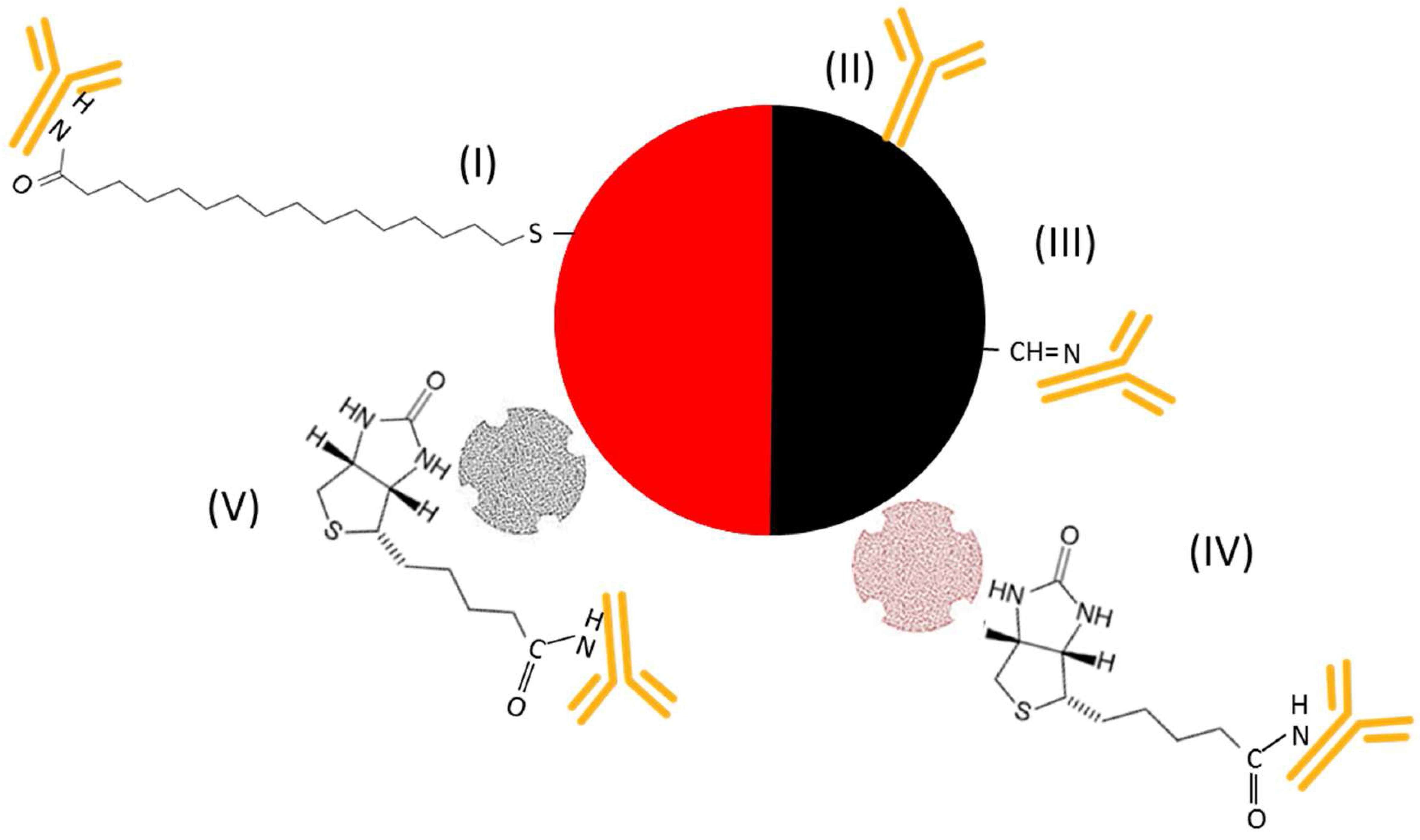
 ) were spotted onto the chip to bind IL8 (
) were spotted onto the chip to bind IL8 (  ) present in the sample solution (Step 2). For detection of the binding, the chip was, in a 3rd step, incubated with a detection antibody that was either labeled with NPs (I–III) or biotinylated (IV–VI
) present in the sample solution (Step 2). For detection of the binding, the chip was, in a 3rd step, incubated with a detection antibody that was either labeled with NPs (I–III) or biotinylated (IV–VI  ). While incubation with an antibody labeled with (I) gold (AuNPs
). While incubation with an antibody labeled with (I) gold (AuNPs  ), (II) carbon (CNPs
), (II) carbon (CNPs  ), or (III) oxidized carbon nanoparticles (oCNPs
), or (III) oxidized carbon nanoparticles (oCNPs  ) allows direct detection of the binding, the use of a biotinylated antibody requires additional incubation steps for signal detection. In Format (IV), Step 4, the biotinylated detection antibody reacts with neutravidin adsorbed on carbon nanoparticles (
) allows direct detection of the binding, the use of a biotinylated antibody requires additional incubation steps for signal detection. In Format (IV), Step 4, the biotinylated detection antibody reacts with neutravidin adsorbed on carbon nanoparticles (  ); in Format (V), streptavidin is coupled with Au (
); in Format (V), streptavidin is coupled with Au (  ); in Format (VI), streptavidin is linked to HRP (
); in Format (VI), streptavidin is linked to HRP (  ). Additionally, Formats (I) and (V) are treated with silver (Ag), while Format (VI) is further processed with different enzyme substrates (Step 5).
). Additionally, Formats (I) and (V) are treated with silver (Ag), while Format (VI) is further processed with different enzyme substrates (Step 5).3.2. Assay Schemes with Nanoparticle-Labeled Detection Antibody
3.3. Assay Schemes with Enzymatic and Nanoparticles Labeled Avidin
3.3.1. Evaluation of Enzyme Substrate
3.3.2. Detection Schemes Using the Streptavidin–Biotin Bridge
3.4. Comparison of the Six Protein Array Assay Schemes
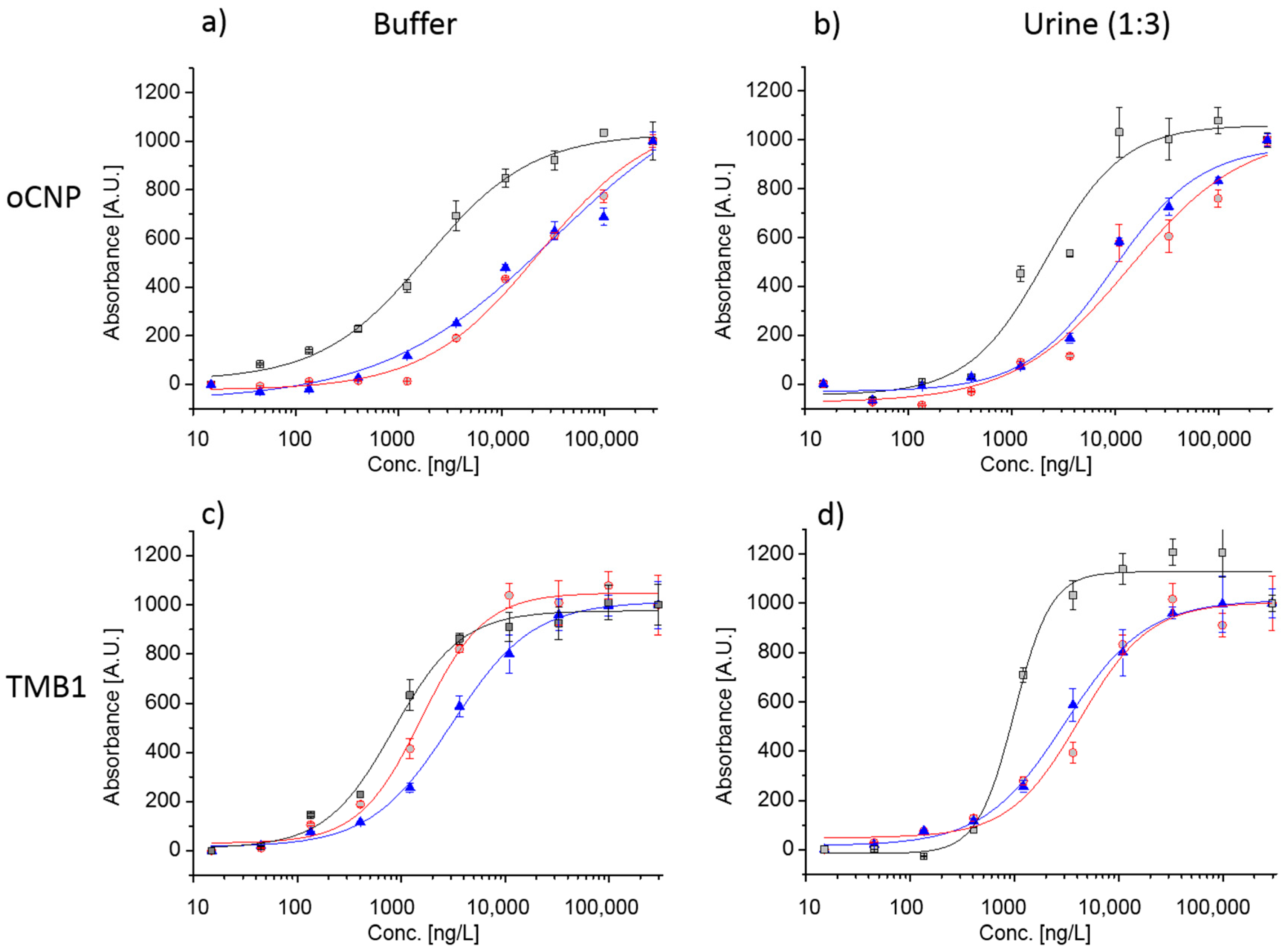
3.5. Multiplexed Detection of IL8, VEGF and DCN in Assay Buffer and Synthetic Urine (1:3)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Budman, L.I.; Kassouf, W.; Steinberg, J.R. Biomarkers for detection and surveillance of bladder cancer. Can. Urol. Assoc. J. 2008, 2, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Tilki, D.; Burger, M.; Dalbagni, G.; Grossman, H.B.; Hakenberg, O.W.; Palou, J.; Reich, O.; Rouprêt, M.; Shariat, S.F.; Zlotta, A.R. Urine markers for detection and surveillance of non-muscle-invasive bladder cancer. Eur. Urol. 2011, 60, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Gogalic, S.; Sauer, U.; Doppler, S.; Preininger, C. Bladder cancer biomarker array to detect aberrant levels of proteins in urine. Analyst 2015, 140, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Gogalic, S.; Sauer, U.; Doppler, S.; Heinzel, A.; Perco, P.; Lukas, A.; Simpson, G.; Pandha, H.; Horvath, A.; Preininger, C. Validation of a protein panel for the non-invasive detection of recurrent non-muscle invasive bladder cancer. Biomarkers 2017, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Satija, J.; Punjabi, N.; Mishraac, D.; Mukherji, S. Plasmonic-ELISA: Expanding horizons. RSC Adv. 2016, 6, 85440–85456. [Google Scholar] [CrossRef]
- Lan, J.; Xu, W.; Wan, Q.; Zhang, X.; Lin, J.; Chen, J.; Chen, J. Colorimetric determination of sarcosine in urine samples of prostatic carcinoma by mimic enzyme palladium nanoparticles. Anal. Chim. Acta 2014, 825, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhang, C.; Qian, J.; Liu, S. Multianalyte immunoassay chip for detection of tumor markers by chemiluminescent and colorimetric methods. Anal. Bioanal. Chem. 2011, 401, 3269–3274. [Google Scholar] [CrossRef] [PubMed]
- Geckeler, K.E.; Eckstein, H. (Eds.) Bioanalytische und Biochemische Labormethoden; Vieweg & Sohn Verlagsgesellschaft mbH: Wiesbaden, Germany, 2008. [Google Scholar]
- Penn, S.G.; He, L.; Natan, M.J. Nanoparticles for bioanalysis. Curr. Opin. Chem. Biol. 2003, 7, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Esimbekova, E.N.; Kratasyuk, V.A. Rapid biosensing tools for cancer biomarkers. Biosens. Bioelectron. 2017, 87, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yu, Z.; Du, W.; Tang, Z.; Jiang, T.; Zhang, C.; Lu, Z. Development of a fluorescent and colorimetric detection methods-based protein microarray for serodiagnosis of TORCH infections. Biosens. Bioelectron. 2008, 24, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Goryacheva, I.Y.; Lenain, P.; De Saeger, S. Nanosized labels for rapid immunotests. TrAC Trends Anal. Chem. 2013, 46, 30–43. [Google Scholar] [CrossRef]
- Posthuma-Trumpie, G.A.; Wichers, J.H.; Koets, M.; Berendsen, L.B.J.M.; Amerongen, A. Amorphous carbon nanoparticles: A versatile label for rapid diagnostic (immuno)assays. Anal. Bioanal. Chem. 2011, 402, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, A.; Wichers, J.H.; Berendsen, L.B.; Timmermans, A.J.; Keizer, G.D.; van Doorn, A.W.; Bantjes, A.; van Gelder, W.M. Colloidal carbon particles as a new label for rapid immunochemical test methods: Quantitative computer image analysis of results. J. Biotechnol. 1993, 30, 185–195. [Google Scholar] [CrossRef]
- Noguera, P.S.; Posthuma-Trumpie, G.A.; van Tuil, M.; van der Wal, F.J.; Boer, A.D.; Moers, A.P.H.A.; van Amerongen, A. Carbon nanoparticles as detection labels in antibody microarrays. Detection of genes encoding virulence factors in Shiga toxin-producing Escherichia coli. Anal. Chem. 2011, 83, 8531–8536. [Google Scholar] [CrossRef] [PubMed]
- Lavorgna, M.; Romeoa, V.; Martonea, A.; Zarrellia, M.; Giordanoa, M.; Buonocorea, G.G.; Qub, M.Z.; Feic, G.X.; Xiac, H.S. Silanization and silica enrichment of multiwalled carbon nanotubes: Synergistic effects on the thermal-mechanical properties of epoxy nanocomposites. Eur. Polym. J. 2013, 49, 428–438. [Google Scholar] [CrossRef]
- Mattsson, L.; Jungmann, C.; Lieberzeit, P.A.; Preininger, C. Modified carbon black as label in a colorimetric on-chip immunoassay for histamine. Sens. Actuators B 2017, 246, 1092–1099. [Google Scholar] [CrossRef]
- Noguera, P.; Posthuma-Trumpie, G.A.; van Tuil, M.; van der Wal, F.J.; de Boer, A.; Moers, A.P.H.A.; van Amerongen, A. Carbon nanoparticles in lateral flow methods to detect genes encoding virulence factors of Shiga toxin-producing Escherichia coli. Anal. Bioanal. Chem. 2011, 399, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Seydack, M. Nanoparticle labels in immunosensing using optical detection methods. Biosens. Bioelectron. 2005, 20, 2454–2469. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Chilkoti, A. A colorimetric gold nanoparticle biosensor: Effect of particle size on sensitivity. In Proceedings of the Second Joint 24th Annual Conference and the Annual Fall Meeting of the Biomedical Engineering Society EMBS/BMES Conference, Houston, TX, USA, 23–26 October 2002; Volume 1, pp. 574–575. [Google Scholar]
- Shi, H.; Yuan, L.; Wu, Y.; Liu, S. Colorimetric immunosensing via protein functionalized gold nanoparticle probe combined with atom transfer radical polymerization. Biosens. Bioelectr. 2011, 26, 3788–3793. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Liu, B.H.; Hsu, Y.T.; Yu, F.Y. Sensitive competitive direct enzyme-linked immunosorbent assay and gold nanoparticle immunochromatographic strip for detecting aflatoxin M1 in milk. Food Control 2011, 22, 964–969. [Google Scholar] [CrossRef]
- Gordon, J.; Michel, G. Analytical sensitivity limits for lateral flow immunoassays. Clin. Chem. 2008, 54, 1250–1251. [Google Scholar] [CrossRef] [PubMed]
- Josephy, P.D.; Eling, T.; Mason, R.P. The horseradish peroxidase-catalyzed oxidation of 3,5,3′,5′-tetramethylbenzidine. Free radical and charge-transfer complex intermediates. J. Biol. Chem. 1982, 257, 3669–3675. [Google Scholar] [PubMed]
- Liang, R.-Q.; Tan, C.-Y.; Ruan, K.-C. Colorimetric detection of protein microarrays based on nanogold probe coupled with silver enhancement. J. Immunol. Methods 2004, 285, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, C. Label-free immunosensor based on gold nanoparticle silver enhancement. Anal. Biochem. 2009, 385, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Xu, Z.; Zhou, L.; Qin, H.; Wang, Y.; Wang, H. Rapid and simultaneous detection of Ureaplasma parvum and Chlamydia trachomatis antibodies based on visual protein microarray using gold nanoparticles and silver enhancement. Diagn. Microbiol. Infect. Dis. 2010, 67, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Cretich, C.; Sedini, V.; Damin, F.; Pelliccia, M.; Sola, L.; Chiari, M. Coating of nitrocellulose for colorimetric DNA microarrays. Anal. Biochem. 2010, 397, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, G.C.; Corgier, B.P.; Mandona, C.A.; De Crozals, G.; Chaix, C.; Loïc, J.; Blum, L.J.; Marquette, C.A. Impact of immobilization support on colorimetric microarrays performances. Biosens. Bioelectr. 2012, 35, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Guo, Y.; Zhao, M.; Li, H.; Zhang, Z. Ring-oven washing technique integrated paper-based immunodevice for sensitive detection of cancer biomarker. Anal. Chem. 2015, 87, 7951–7957. [Google Scholar] [CrossRef] [PubMed]
- Lönnberg, M.; Carlsson, J. Quantitative detection in the attomole range for immunochromatographic tests by means of a flatbed scanner. Anal. Biochem. 2001, 293, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, G.J.; Wichers, J.H.; Ferreira, T.M.F.; Ghati, D.; van Amerongen, A.; Deelder, A.M. Diagnosis of schistosomiasis by reagent strip test for detection of circulating cathodic antigen. J. Clin. Microbiol. 2004, 42, 5458–5461. [Google Scholar] [CrossRef] [PubMed]
- Linares, E.M.; Kubota, L.T.; Michaelis, J.; Thalhammer, S. Enhancement of the detection limit for lateral flow immunoassays: Evaluation and comparison of bioconjugates. J. Immunol. Methods 2012, 375, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Goodison, S.; Ogawa, O.; Matsui, Y.; Kobayashi, T.; Miyake, M.; Ohnishi, S.; Fujimoto, K.; Dai, Y.; Shimizu, Y.; Tsukikawa, K.; et al. A multiplex urinary immunoassay for bladder cancer detection: Analysis of a Japanese cohort. J. Transl. Med. 2016, 14, 287–295. [Google Scholar] [CrossRef] [PubMed]



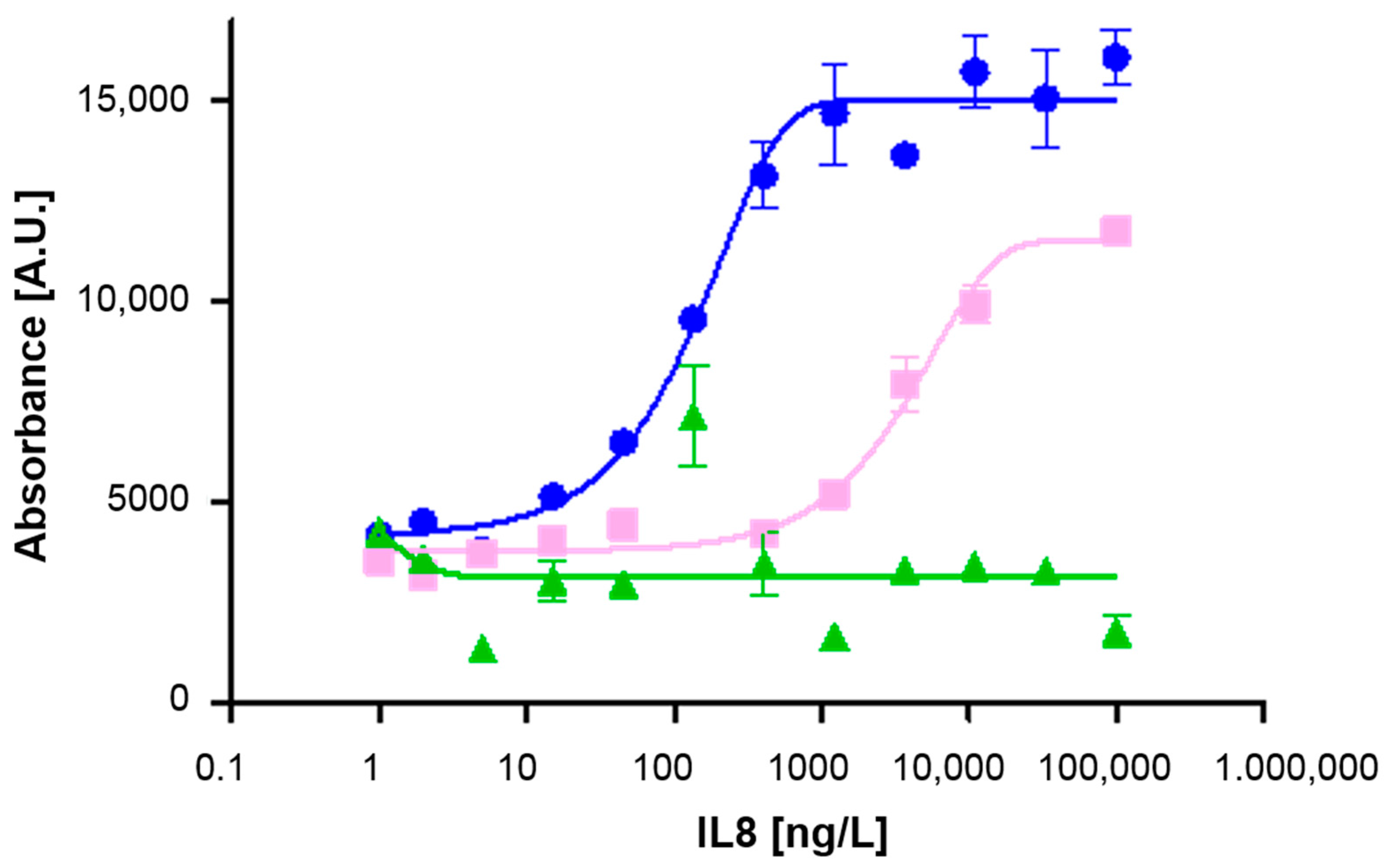
 ), TMB2 (
), TMB2 (  ), and ODI
(
), and ODI
(  ). The
blank (0 ng/L) is represented by 1 ng/L on the log scale.
), TMB2 ( ), and ODI
( ). The
blank (0 ng/L) is represented by 1 ng/L on the log scale.
). The
blank (0 ng/L) is represented by 1 ng/L on the log scale.
), TMB2 ( ), and ODI
( ). The
blank (0 ng/L) is represented by 1 ng/L on the log scale.

 IL8,
IL8,  VEGF, and
VEGF, and  DCN; including standard deviations of three replicates. The blank signal (0 ng/L) is represented by 15 ng/L on the log scale.
IL8, VEGF, and DCN; including standard deviations of three replicates. The blank signal (0 ng/L) is represented by 15 ng/L on the log scale.
DCN; including standard deviations of three replicates. The blank signal (0 ng/L) is represented by 15 ng/L on the log scale.
IL8, VEGF, and DCN; including standard deviations of three replicates. The blank signal (0 ng/L) is represented by 15 ng/L on the log scale.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogalic, S.; Sauer, U.; Doppler, S.; Preininger, C. Investigating Colorimetric Protein Array Assay Schemes for Detection of Recurrence of Bladder Cancer. Biosensors 2018, 8, 10. https://doi.org/10.3390/bios8010010
Gogalic S, Sauer U, Doppler S, Preininger C. Investigating Colorimetric Protein Array Assay Schemes for Detection of Recurrence of Bladder Cancer. Biosensors. 2018; 8(1):10. https://doi.org/10.3390/bios8010010
Chicago/Turabian StyleGogalic, Selma, Ursula Sauer, Sara Doppler, and Claudia Preininger. 2018. "Investigating Colorimetric Protein Array Assay Schemes for Detection of Recurrence of Bladder Cancer" Biosensors 8, no. 1: 10. https://doi.org/10.3390/bios8010010
APA StyleGogalic, S., Sauer, U., Doppler, S., & Preininger, C. (2018). Investigating Colorimetric Protein Array Assay Schemes for Detection of Recurrence of Bladder Cancer. Biosensors, 8(1), 10. https://doi.org/10.3390/bios8010010