Simple Approaches to Minimally-Instrumented, Microfluidic-Based Point-of-Care Nucleic Acid Amplification Tests

Abstract
:1. Introduction and Scope of Review
2. Background: Medical Diagnostics and POC Testing
3. Microfluidic Molecular Diagnostics
4. Isothermal Nucleic Acid Amplification
5. Chip Fabrication
6. Detection of Amplification
7. Multiplexing NAATs
8. Reagent Stabilization and On-Chip Storage
9. Smartphone-Based Detection and Electricity-Free Operation
10. Blood Plasma Extraction Module
11. Summary, Discussion and Outlook
12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Erickson, D.; Li, D. Integrated microfluidic devices. Anal. Chim. Acta 2004, 507, 11–26. [Google Scholar] [CrossRef]
- Mairhofer, J.; Roppert, K.; Ertl, P. Microfluidic systems for pathogen sensing: A review. Sensors 2009, 9, 4804–4823. [Google Scholar] [CrossRef] [PubMed]
- Fu, E.; Yager, P.; Floriano, P.N.; Christodoulides, N.; McDevitt, J. Perspectives on diagnostics for global health. IEEE Pulse 2011, 2, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Niemz, A.; Ferguson, T.M.; Boyle, D.S. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011, 29, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Rivet, C.; Lee, H.; Hirsch, A.; Hamilton, S.; Lu, H. Microfluidics for medical diagnostics and biosensors. Chem. Eng. Sci. 2011, 66, 1490–1507. [Google Scholar] [CrossRef]
- Gubala, V.; Harris, L.F.; Ricco, A.J.; Tan, M.X.; Williams, D.E. Point-of-care diagnostics: Status and future. Anal. Chem. 2012, 84, 487–515. [Google Scholar] [CrossRef] [PubMed]
- Pai, N.P.; Vadnais, C.; Denkinger, C.; Engel, N.; Pai, M. Point-of-care testing for infectious diseases: Diversity, complexity, and barriers in low- and middle-income countries. PLoS Med. 2012, 9, e1001306. [Google Scholar] [CrossRef] [PubMed]
- Foudeh, A.M.; Didar, T.F.; Veres, T.; Tabrizian, M. Microfluidic designs and techniques using lab-on-a-chip devices for pathogen detection for point-of-care diagnostics. Lab Chip 2012, 12, 3249–3266. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.F. Microfluidic systems for diagnostic applications: A review. J. Lab. Autom. 2012, 17, 330–347. [Google Scholar] [CrossRef] [PubMed]
- Olasagasti, F.; Ruiz de Gordoa, J.C. Miniaturized technology for protein and nucleic AID point-of-care testing. Transl. Res. 2012, 160, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.D.; Linder, V.; Sia, S.K. Commercialization of microfluidic point-of-care diagnostic devices. Lab Chip 2012, 12, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.D.; Chin, S.Y.; Lakanasopin, T.; Sia, S.K. Low-Cost Microdevices for Point-of-Care Testing; Issadore, D., Westervelt, R.M., Eds.; Springer: Berlin, Germany, 2013. [Google Scholar]
- Park, S.-M.; Sabour, A.F.; Son, J.H.; Lee, S.H.; Lee, L.P. Toward integrated molecular diagnostic system (iMDX): Principles and applications. IEEE Trans. Biomed. Eng. 2014, 61, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Peeling, R.W.; McNerney, R. Emerging technologies in point-of-care molecular diagnostics for resource-limited settings. Expert Rev. Mol. Diagn. 2014, 14, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Temiz, Y.; Lovchik, R.D.; Kaigala, G.V.; Delamarche, E. Lab-on-a-chip devices: How to close and plug the lab? Microelectron. Eng. 2015, 132, 156–175. [Google Scholar] [CrossRef]
- Jenkins, G.; Mansfield, D. Microfluidic Diagnostics: Methods and Protocols; Springer: Berlin, Germany, 2013. [Google Scholar]
- Becker, H.; Gärtner, C. Microfluidics-Enabled Diagnostic Systems: Markets, Challenges, and Examples. In Microchip Diagnostics: Methods and Protocols, Methods in Molecular Biology; Taly, V., Viovy, J.-L., Descroix, S., Eds.; Springer: Berlin, Germany, 2017. [Google Scholar]
- Drain, P.K.; Hyle, E.P.; Nounary, F.; Freedburg, K.A.; Wilson, D.; Bishai, W.; Rodriguez, W.; Bassett, I.V. Evaluating diagnostic point-of-care tests in resource-limited settings. Lancet Infect. Dis. 2014, 14, 239–249. [Google Scholar] [CrossRef]
- Mims, C.; Dockrell, H.M.; Goering, R.V.; Roitt, I.; Wakelin, D.; Zuckermin, M. Medical Microbiology, 3rd ed.; Mosby: Edinburgh, UK, 2004. [Google Scholar]
- Koczula, K.M.; Gallotta, A. Lateral flow assays. Essays Biochem. 2016, 60, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, S. DNA/RNA preparation for molecular detection. Clin. Chem. 2015, 61, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Boom, R.; Sol, C.J.A.; Salimans, M.M.M.; Jansen, C.L.; Wertheim-van Dillen, P.M.E.; van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [PubMed]
- Borodina, T.A.; Lehrach, H.; Soldatov, A.V. DNA purification on homemade silica spin columns. Anal. Biochem. 2003, 321, 135–137. [Google Scholar] [CrossRef]
- Kim, J.; Heo, J.; Crooks, R.M. Hybridization of DNA to bead-immobilized probes confined within a microfluidic channel. Langmuir 2006, 22, 10130–10134. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Morabito, K.; Tang, J.X.; Tripathi, A. Microfluidic platform for isolating nucleic acid targets using sequence specific hybridization. Biomicrofluidics 2013, 7, 044107. [Google Scholar] [CrossRef] [PubMed]
- Root, B.E.; Agarwal, A.K.; Kelso, D.M.; Barron, A.E. Purification of HIV Rna from serum using a polymer matrix in a microfluidic device. Anal. Chem. 2011, 83, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Lui, C.; Cady, N.C.; Batt, C.A. Nucleic Acid-based detection of bacterial pathogens using integrated microfluidic platform systems. Sensors 2009, 9, 3713–3744. [Google Scholar] [CrossRef] [PubMed]
- Sandetskaya, N.; Moos, D.; Seifert, H.P.S.; Jenerowicz, M.; Becker, H.; Zilch, C.; Kuhlmeier, D. An integrated versatile lab-on-a-chip platform for the isolation and nucleic acid-based detection of pathogens. Future Sci. OA 2017, 3, FS0177. [Google Scholar] [CrossRef] [PubMed]
- Demeke, T.; Jenkins, G.R. Influence of DNA extraction methods, PCR inhibitors and quantification methods on real-time PCR assay of biotechnology-derived traits. Anal. Bioanal. Chem. 2010, 396, 1977–1990. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Legendre, L.A.; Bienvenue, J.M.; Landers, J.P. Purification of nucleic acids in microfluidic devices. Anal. Chem. 2008, 80, 6472–6479. [Google Scholar] [CrossRef] [PubMed]
- Reinholt, S.; Beaumner, A.J. Microfluidic isolation of nucleic acids. Angew. Chem. Int. Ed. Eng. 2014, 53, 13988–14001. [Google Scholar] [CrossRef] [PubMed]
- Breadmore, M.C.; Wolfe, K.A.; Arcibal, I.G.; Leung, W.K.; Dickson, D.; Giordano, B.C.; Power, M.E.; Ferrance, J.P.; Feldman, S.H.; Norris, P.M.; et al. Microchip-based purification of DNA from biological samples. Anal. Chem. 2003, 75, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cady, N.C.; Batt, C.A. A plastic microchip for nucleic acid purification. Biomed. Microdevices 2007, 9, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Price, C.W.; Leslie, D.C.; Landers, J.P. Nucleic acid extraction techniques and application to the microchip. Lab Chip 2009, 9, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Mahalanabis, M.; Do, J.; AlMuayad, H.; Zhang, J.Y.; Klapperich, C.M. An integrated disposable device for DNA extraction and helicase dependent amplification. Biomed. Microdevices 2010, 12, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Johnson, M.; Hill, P.; Sonkul, R.S.; Kim, J.; Gale, B.K. Automated microfluidic DNA/RNA extraction with both disposable and reusable components. J. Micromech. Microeng. 2012, 22, 015007. [Google Scholar] [CrossRef]
- Shaw, K.J.; Hughes, E.M.; Dyer, C.E.; Greenman, J.; Haswell, S.J. Integrated RNA extraction and RT-PCR for semiquantitative gene expression studies on a microfluidic device. Lab. Investig. 2013, 93, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Voelkerding, K.V.; Gale, B. Patterning of a nanoporous membrane for multisample DNA extraction. J. Micromech. MicroEng. 2006, 16, 33–39. [Google Scholar] [CrossRef]
- Witek, M.A.; Llopis, S.D.; Wheatley, A.; McCarely, R.L.; Soper, S.A. Purification and preconcentration of genomic DNA from whole cell lysates using photoactivated polycarbonate (PPC) microfluidic chips. Nucleic Acids Res. 2006, 34, e74. [Google Scholar] [CrossRef] [PubMed]
- Kendall, E.L.; Weinhold, E.; DeVoe, D.L. A chitosan coated monolith for nucleic acid capture in a thermoplastic microfluidic chip. Biomicrofluidics 2014, 8, 044109. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, S.A.; Bishop, J.D.; Lafleur, L.; Buser, J.R.; Lutz, B.; Yager, P. One-step purification and concentration of DNA in porous membranes for point-of-care applications. Lab Chip 2015, 15, 2647–2659. [Google Scholar] [CrossRef] [PubMed]
- Sur, K.; McFall, S.M.; Yeh, E.T.; Jangam, S.R.; Hayden, M.A.; Stroupe, S.D.; Kelso, D.M. Immiscible phase nucleic acid purification eliminates PCR inhibitors with a single pass of paramagnetic particles through a hydrophobic liquid. J. Mol. Diagn. 2010, 12, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.M.; LaVanway, A.J.; Pezzi, H.M.; Guckenberger, D.J.; Anderson, M.A.; Loeb, J.M.; Beebe, D.J. HIV viral RNA extraction in wax immiscible filtration assisted by surface tension (IFAST) devices. J. Mol. Diagn. 2014, 16, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Santini, G.C.; Potrich, C.; Lunelli, L.; Pasquardini, L.; Vaghi, V.; Pederzolli, C. Innovative microRNA purification based on surface properties modification. Colloids Surf. B 2014, 116, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.J.; Thain, L.; Docker, P.T.; Dyer, C.E.; Greenman, J.; Greenway, G.M.; Haswell, S.J. The use of carrier RNA to enhance DNA extraction from microfluidic-based silica monoliths. Anal. Chim. Acta 2009, 652, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Geva, E.; Mauk, M.; Qiu, X.; Abrams, W.R.; Malamud, D.; Curtis, K.; Owen, S.M.; Bau, H.H. An isothermal amplification reactor with an integrated isolation membrane for point-of-care detection of infectious diseases. Analyst 2011, 136, 2069–2976. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Kodzius, R.; Cao, W.; Wen, W. Extraction, amplification and detection of DNA in microfluidic chip-based assays. Microchim. Acta 2014, 181, 1611–1631. [Google Scholar] [CrossRef]
- Kim, J.; Byun, D.; Mauk, M.G.; Bau, H.H. A disposable, self-contained PCR chip. Lab Chip 2009, 9, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Disch, A.; Mueller, C.; Reinecke, H. Low-cost production of disposable microfluidics by blister packaging technology. In Proceedings of the 29th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Lyon, France, 22–26 August 2007; pp. 6322–6325. [Google Scholar]
- Liu, C.; Qiu, X.; Ongagna, S.; Chen, D.; Chen, Z.; Abrams, W.R.; Malamud, D.; Corstjens, P.L.A.M.; Bau, H.H. A timer actuated immunoassay cassette for detecting molecular markers in oral fluids. Lab Chip 2009, 9, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Thompson, J.A.; Chen, Z.; Liu, C.; Chen, D.; Ramprasad, S.; Mauk, M.G.; Ongagna, S.; Barber, C.; Adams, W.R.; et al. PLAM Corstjens, and HH Bau, Finger actuated, self-contained immunoassay cassettes. Biomed. Microdevices 2009, 11, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Mauk, M.; Qiu, X.; Liu, C.; Kim, J.; Ramprasad, S.; Ongagna, S.; Abrams, W.R.; Malamud, D.; Corstjens, P.L.; et al. An integrated, self-contained microfluidic cassette for isolation, amplification, and detection of nucleic acids. Biomed. Microdevices 2010, 12, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Liu, C.; Mauk, M.G.; Hart, R.W.; Chen, D.; Qiu, J.; Keintz, T.; Fiene, J.; Bau, H.H. A portable analyzer for pouch-actuated, immunoassay cassettes. Sens. Actuators B Chem. 2011, 160, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Jangam, S.R.; Agarwal, A.K.; Sur, K.; Kelso, D.M. A point-of-care PCR test for HIV-1 detection in resource-limited settings. Biosens. Bioelectron. 2013, 42, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Sewart, R.; Becker, H.; Roux, P.; Land, K. Blister pouches for effective reagent storage on microfluidic chips for blood cell counting. Microfluid. Nanofluid. 2016, 20, 163. [Google Scholar] [CrossRef]
- Chen, Z.; Abrams, W.R.; Geva, E.; de Dood, C.J.; González, J.M.; Tanke, H.J.; Niedbala, R.S.; Zhou, P.; Malamud, D.; Corstjens, P.L.A.M. Development of a generic microfluidic device for simultaneous detection of antibodies and nucleic acids in oral fluids. BioMed. Res. Int. 2013, 2013, 543294. [Google Scholar] [CrossRef] [PubMed]
- Focke, M.; Kosse, D.; Müller, C.; Reinecke, H.; Zengerle, R.; von Stetten, F. Lab-on-a-foil: Microfluidics on thin and flexible films. Lab Chip 2010, 10, 1365–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xing, D.; Li, Y. Micropumps, microvalves, and micromixers within PCR microfluidic chips: Advances and trends. Biotechnol. Adv. 2007, 25, 483–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Cheng, L.; Wang, C.-H.; Ling, W.-S.; Wang, S.-S.; Lee, G.-B. An integrated chip capable of performing sample pretreatment and nucleic acid amplification for HIV-1 detection. Biosens. Bioelectron. 2013, 41, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, C.; Mauk, M.G.; Peng, J.; Schoenfeld, T.; Bau, H.H. A multifunctional reactor with dry-stored reagents for enzymatic amplification. Anal. Chem. 2018, 90, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Al-Soud, W.A.; Rådström, P. Purification and characterization of PCR-inhibitory components in blood cells. J. Clin. Microbiol. 2001, 39, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Opel, K.L.; Chung, D.; McCord, B.R. A study of PCR inhibition mechansims using real-time PCR. J. Forensics Sci. 2010, 55, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.T.; Fan, A.; Klapperich, C.M.; Cabodi, M. Sample concentration and purification for point-of-care diagnostics. Eng. Med. Biol. Soc. 2012, 2012, 2396–2399. [Google Scholar]
- Park, S.; Zhang, Y.; Lin, S.; Wang, T.-H.; Yang, S. Advances in microfluidic PCR for point-of-care infectious disease diagnostics. Biotechnol. Adv. 2011, 29, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Venka, V. Novel fluorescence detection for non-contat temperature sensing in microchip PCR. J. Biochem. Biophys. Methods 2007, 70, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Seyrig, G.; Tourlousse, D.M.; Stedtfeld, R.D.; Tiedje, J.M.; Harsham, S.A. A CCD-based fluorescence imaging system for real-time loop-mediated isothermal amplification-based rapid and sensitive detection of waterborne pathogens on microchips. Biomed. Microdevices 2011, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Almassian, D.R.; Cockrell, L.M.; Nelson, W.M. Portable nucleic acid thermocyclers. Chem. Soc. Rev. 2013, 42, 8469–8798. [Google Scholar] [CrossRef] [PubMed]
- Miralles, V.; Huerre, A.; Malloggi, F.; Jullien, M.C. A review of heating and temperature control in microfluidic systems: Techniques and application. Diagnostics 2013, 3, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Asiello, P.J.; Baeumner, A.J. Miniaturized isothermal nucleic acid amplification, a review. Lab Chip 2011, 11, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Hashsham, S.A. Miniaturized nucleic acid amplification systems for rapid and point-of-care diagnostics: A review. Anal. Chem. Acta 2012, 733, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Craw, P.; Balachandran, W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: A critical review. Lab Chip 2012, 12, 2469–2486. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Chen, C.-C.; Wei, S.-C.; Lu, H.-H.; Liang, Y.-H.; Lin, C.-W. Diagnostic devices for isothermal nucleic acid amplification. Sensors 2012, 12, 8319–8337. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-M.; Chang, W.-H.; Wang, C.-H.; Wang, J.-H.; Mai, J.D.; Lee, G.-B. Nucleic acid amplification using microfluidic systems. Lab Chip 2013, 13, 1125–1242. [Google Scholar] [CrossRef] [PubMed]
- Tröger, V.; Niemann, K.; Gärtig, C.; Kuhlmeier, D. Isothermal amplification and quanitification of nucleic acids and its use in microsystems. J. Nanomed. Nanotechnol. 2015, 6, 1000282. [Google Scholar]
- Safavieh, M.; Kanakasabapthy, M.K.; Tarlan, F.; Ahmed, M.U.; Zourob, M.; Ashghar, W.; Safiee, H. Emerging loop-mediated isothermal amplification-based microchip and microdevice technologies for nucleic acid detection. ACS Biomater. Sci. Eng. 2016, 2, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Kawana, T.; Fukishima, E.; Suzutani, T. Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J. Biochem. Biophys. Methods 2007, 70, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Francois, P.; Tangomo, M.; Hibbs, J.; Bonetti, E.-J.; Boehme, C.C.; Notomi, T.; Perkins, M.D.; Schrenzel, J. Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Microbiol. 2011, 62, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Nkouawa, A.; Sako, Y.; Li, T.; Wandra, X.C.T.; Swastika, I.K.; Nakao, M.; Yanagida, T.; Nakaya, K.; Qiu, D.; Ito, A. Evaluation of a loop-mediated isothermal amplification method using fecal specimens for differential detection of Taenia species from humans. J. Clin. Microbiol. 2010, 48, 3330–3352. [Google Scholar] [CrossRef] [PubMed]
- García, M.E.; Blanco, J.L.; Caballero, J.; Gargallo-Viola, D. Anticoagulants interfere with PCR used to diagnose invasive aspergillosis. J. Clin. Microbiol. 2002, 40, 1567. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, V.; Stankovic, M.; Nikolic, A.; Antonijevic, N.; Rakicevic, L.J.; Divac, A.; Radoojkovic, M. PCR amplification on whole blood samples treated with different commonly used anticoagulants. Pediatr. Hematol. Oncol. 2006, 23, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Chander, Y.; Koelbl, J.; Puckett, J.; Moser, M.J.; Klingele, A.J.; Liles, M.R.; Carrias, A.; Mead, D.A.; Schoenfeld, T.W. A novel thermostable polymerase for RNA and DAN loop-mediated isothermal amplification (LAMP). Front. Microbiol. 2014, 5, 395. [Google Scholar] [CrossRef] [PubMed]
- Rebrikov, D.V.; Trofimov, D.Y. Real-time PCR: A review of approaches to data analysis. Appl. Biochem. Microbiol. 2006, 42, 455–463. [Google Scholar] [CrossRef]
- Ren, K.; Zhou, J.; Wu, H. Materials for microfabrication. Acc. Chem. Res. 2013, 46, 2396–2406. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kwok, Y.C. Polymeric microfluidic system for DNA analysis. Anal. Chim. Acta 2006, 556, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.; Gärtner, C. Polymer microfabrication technologies for microfluidic systems. Anal. Bioanal. Chem. 2008, 390, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Ogończyk, D.; Węgrzyn, J.; Jankowski, P.; Dabrowski, B.; Garstecki, P. Bonding of microfluidic devices fabricated in polycarbonate. Lab Chip 2010, 10, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Kistrup, K.; Poulsen, C.E.; Hansen, M.F.; Wolff, A. Ultrasonic welding for fast bonding of self-aligned structures in lab-on-a-chip systems. Lab Chip 2015, 9, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, G.; Guo, Y.; Tian, Y. Study of PMMA thermal bonding. Microsyst. Technol. 2007, 13, 403–407. [Google Scholar] [CrossRef]
- Gong, X.; Yi, X.; Xiao, K.; Li, S.; Kodzius, R.; Qin, J.; Wen, W. Wax-bonding 3D microfluidic chips. Lab Chip 2010, 10, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Lee, D.L. Bonding of thermoplastic polymer microfluidics. Microfluid. Nanofluid. 2009, 6, 1–16. [Google Scholar] [CrossRef]
- Nath, P.; Maity, T.S.; Pettersson, F.; Resnick, J.; Kunde, Y.; Kraus, N.; Castano, N. Polymerase chain compatibility of adhesive transfer tape based microfluidic platforms. Microsyst. Technol. 2014, 20, 1187–1193. [Google Scholar] [CrossRef]
- Gonzalez, A.; Grimes, R.; Walsh, E.J.; Dalton, T.; Davies, M. Interaction of quantitative PCR components with polymeric surfaces. Biomed. Microdevices 2007, 9, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T.B.; Pedersen, C.M.; Gröndahl, K.G.; Jensen, T.G.; Sekulovic, A.; Bang, D.D.; Wolff, A. PCR biocompatibility of lab-on-a-chip and MEMS materials. J. Micromech. Microeng. 2007, 17, 1527–1532. [Google Scholar] [CrossRef]
- Kodzius, R.; Xiao, K.; Wu, J.; Yi, X.; Gong, X.; Foulds, I.G.; Wen, W. Inhibitory effect of common microfluidic materials on PCR outcome. Sens. Actuators B Chem. 2012, 161, 349–358. [Google Scholar] [CrossRef]
- Lou, X.J.; Panaro, N.J.; Wilding, P.; Fortina, P.; Kricka, L. Increased amplification efficiency of microchip-based PCR by dynamic surface passivation. BioTechniques 2004, 36, 248–251. [Google Scholar] [PubMed]
- Piruska, A.; Nikcevic, I.; Lee, S.H.; Ahn, C.; Heineman, W.R.; Limbach, P.A.; Seliskar, C.J. The autofluorescence of plastic materials and chips measured under laser irradiation. Lab Chip 2005, 5, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Comina, G.; Suska, A.; Filippini, D. Low cost lab-on-a-chip prototyping with a consumer grade 3D printer. Lab Chip 2014, 14, 2978–2982. [Google Scholar] [CrossRef] [PubMed]
- Shallan, A.I.; Smeikal, P.; Corbant, M.; Guiit, R.M.; Breadmore, M.C. Cost-effective printing of three-dimensional printing of visibly transparent microchips within minutes. Anal. Chem. 2014, 86, 3124–3130. [Google Scholar] [CrossRef] [PubMed]
- Au, A.K.; Bhattacharjee, N.; Horowitz, L.F.; Chang, T.C.; Folch, A. 3D-printed microfluidic automation. Lab Chip 2015, 15, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.M.B.; Ng, S.H.; Li, K.H.H.; Yoon, Y.-J. 3D printed microfluidics for biological applications. Lab Chip 2015, 15, 3627. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Hsieh, T.-M.; Lee, D.Y.S.; Ali, E.M.; Xie, H.; Looi, X.L.; Koay, E.S.; Li, M.H.; Ying, J.Y. A self-contained all-in-one cartridge for sample preparation and real-time PCR in rapid influenza diagnosis. Lab Chip 2010, 10, 3103–3111. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Sano, S.; Takahashi, K.; Jikihara, T. Method for colormetric detection of double-stranded nucleic acid using leuco triphenylmethane dyes. Anal. Biochem. 2015, 473, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Mauk, M.G.; Hackett, B.A.; Cherry, S.; Bau, H.H.; Liu, C. Instrument-free point-of-care molecular detection of Zika virus. Anal. Chem. 2016, 88, 7289–7294. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.J.; Shu, Y.L.; Qin, M.; Wang, D.Y.; Gao, R.B.; Wang, M.; Wen, L.Y.; Han, F.; Zhou, S.M.; Zhao, X.; et al. Visual detection of pandemic influenza AH1N1 virus 2009 by reverse-transcription loop-mediated isothermal amplification with hydroxynaphthol blue dye. J. Virol. Meth. 2010, 167, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.Y.; Jin, W.; Zhu, Q.Y.; Yang, W.X.; Wu, Q.Q.; Jin, Q.H.; Mu, Y. Real-time detection of loop-mediated isothermal amplification reaction on microfluidic chip. Bioinform. Biomed. Eng. 2011. [Google Scholar] [CrossRef]
- Kivlehan, F.; Mavré, F.; Talini, L.; Limoges, B.; Marchal, D. Real-time electrochemical monitoring of isothermal helicase-dependent amplification of nucleic acids. Analyst 2011. [Google Scholar] [CrossRef] [PubMed]
- Nagatani, N.; Yamanaka, K.; Saito, M.; Koketsu, R.; Sasaki, T.; Ikuta, K.; Miyahara, T.; Tamiya, E. Semi-real time electrochemical monitoring for influenza virus RNA by reverse transcription loop-mediated isothermal amplification using a USB powered portable poteniostat. Analyst 2011, 136, 5143. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, K.; Patterson, A.S.; Ferguson, B.S.; Plaxco, K.W.; Soh, H.T. Rapid, sensitive, and quantitative detection of pathogenic DNA at the point of care through microfluidic electrochemical quantitative loop-mediated isothermal amplification. Angew. Chem. Int. 2012, 51, 4896–4900. [Google Scholar] [CrossRef] [PubMed]
- Safavieh, M.; Ahmed, M.U.; Ng, A.; Zourob, M. High-throughput real-time electrochemical monitoring of LAMP for pathogenic bacteria detection. Biosens. Bioelectron. 2014, 58, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Fang, X.; Ye, D.; Li, H.; Chen, H.; Zhang, S.; Kong, J. A real-time microfluidic multiplex electrochemical loop-mediated isothermal amplification chip for differentiating bacteria. Biosens. Bioelectron. 2014, 60, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mauk, M.G.; Wang, J.; Abrams, W.R.; Corstjens, P.L.; Niedbala, R.S.; Malamud, D.; Bau, H.H. A Microfluidic System for Saliva-Based Detection of Infectious Diseases. Ann. N. Y. Acad. Sci. 2007, 1098, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Kersting, S.; Rausch, V.; Bier, F.F.; von Nickisch-Rosenegk, M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malaria J. 2014, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Roskos, K.; Hickerson, A.I.; Lu, H.-W.; Ferguson, T.M.; Shinde, D.N.; Klaue, Y.; Niemz, A. Simple system for isothermal DNA amplification coupled to lateral flow detection. PLoS ONE 2013, 8, e69355. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Oh, S.J.; Park, B.H.; Seo, T.S. Combination of multiplex isothermal amplification with an immunochromatographic strip for subtyping influenza A virus. In Proceedings of the 18th International Conference Miniaturized Systems for Chemistry and Life Sciences, San Antonio, TX, USA, 26–30 October 2014; pp. 1578–1580. [Google Scholar]
- Liu, C.; Sadik, M.M.; Mauk, M.G.; Edelstein, P.H.; Bushman, F.D.; Gross, R.; Bau, H.H. Nuclemeter: A reaction-diffusion based method for quantifying nucleic acids undergoing enzymatic amplification. Sci. Rep. 2014, 4, 7335. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, C.; Mauk, M.G.; Rankin, S.C.; Lok, J.B.; Greenberg, R.M.; Bau, H.H. Two-stage isothermal enzymatic amplification for concurrent multiplex molecular detection. Clin. Chem. 2016, 63, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.B.; Atwood, S.M.; Bergmeyer, L.; Chemelli, J.; Christi, K.; Cummins, T.; Donish, W.; Ekeze, T.; Falvo, J.; Patterson, D.; et al. Automated closed-vessel system for in vitro diagnostics based on polymerase chain reaction. Clin. Chem. 1993, 39, 1927–1933. [Google Scholar] [PubMed]
- Hitzbleck, M.; Delamarche, E. Reagents in microfluidics: An ‘in’ and ‘out’ challenge. Chem. Soc. Rev. 2013, 42, 8494–8516. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.H.; Yang, J.; Lenigk, R.; Bonanno, J.; Grodzinski, P. Self-contained, fully integrated biochip for sample preparation, polymerase chain reaction amplification, and DNA microarray detection. Anal. Chem. 2004, 76, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.Y.; Petri, C.R.; Yager, P. On-card dry reagent storage for disposable microfluidic immunoassays. In Proceedings of the Twelfth International Conference on Miniaturized Systems for Chemistry and Life Sciences, San Diego, CA, USA, 12–16 October 2008; pp. 188–190. [Google Scholar]
- Lutz, S.; Weber, P.; Focke, M.; Faltin, B.; Hoffmann, J.; Müller, C.; Mark, D.; Roth, G.; Munday, P.; Armes, N.; et al. Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification (RPA). Lab Chip 2010, 10, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Hitzbleck, M.; Gervais, L.; Delamarche, E. Controlled release of reagents in capillary-driven microfluidics using reagent integrators. Lab Chip 2011, 11, 2680–2685. [Google Scholar] [CrossRef] [PubMed]
- Fridley, G.; Le, H.Q.; Fu, E.; Yager, P. Controlled release of dry reagents in porous media for tunable temporal and spatial distribution upon rehydration. Lab Chip 2012, 12, 4321–4327. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Høgberg, J.; Christine, T.; Florian, L.; Monsalve, L.G.; Rodriguez, S.; Cao, C.; Wolff, A.; Ruano-Lopez, J.M.; Bang, D.D. Pre-storage of gelified reagents in lab-on-a-foil system for rapid nucleic acid analysis. Lab Chip 2013, 13, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Manage, D.P.; Lauzon, J.; Zahariadis, G.; Pilarski, L.M. Storing self-contained gel capillary cassettes for POC medical diagnostics. Lab Chip 2013, 13, 4087–4095. [Google Scholar] [CrossRef] [PubMed]
- Lenk, G.; Stemme, G.; Roxhed, N. Dry reagent storage in dissolvable films and liquid triggered release for programmed multi-step lab-on-achip diagnostics. In Proceedings of the 28th IEEE International Conference on Micro Electro Mechanical Systems (MEMS), Estoril, Portugal, 18–22 January 2015; pp. 451–454. [Google Scholar]
- Rahmanian, O.D.; DeVoe, D.L. Single-use thermoplastic microfluidic burst valves enabling on-chip reagent storage. Microfluid. Nanofluid. 2014. [Google Scholar] [CrossRef] [PubMed]
- Czurratis, D.; Beyl, Y.; Zinober, S.; Zengerle, R.; Lärmer, F. Long-term on-chip storage and release of liquid reagents for diagnostic lab-on-a-chip applications. Int. J. Math. Comput. Phys. Electr. Comput. Eng. 2013, 7, 1266–1269. [Google Scholar]
- Czurratis, D.; Beyl, Y.; Zinober, S.; Lärmer, F.; Zengerle, R. A novel concept for long-term pre-storage and release of liquids for pressure-drive lab-on-a-chip devices. J. Micromech. Microeng. 2015, 25, 045002. [Google Scholar] [CrossRef]
- Czurratis, D.; Beyl, Y.; Grimm, A.; Brettschneider, T.; Zinober, S.; Lärmer, F.; Zengerle, R. Liquids on-chip: Direct storage and release employing micro-perforated vapor barrier films. Lab Chip 2015, 15, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Matsuo, T.; Kawamura, Y.; Ohashi, M.; Yonekawa, T.; Kanda, H.; Notomi, T.; Ihira, M. Direct detection of human herpesvirus 6B by the LAMP method using newly developed dry reagents. J. Virol. Methods 2014, 201, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Wong Tzeling, J.M.; Yean, C.Y. A shelf-stable fluorogenic isothermal amplification assay for the detection of Burkholderia pseudomallei. Analyst 2016, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-W.; Weissenberger, G.; Ching, W.-M. Development of lyophilized loop-mediated isothermal amplification reagents for the detection of Leptospira. Mil. Med. 2016, 181, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Biolyph Innovative Lyophilization Services, Life Sciences and Diagnostic Reagents. Available online: http://www.biolyph.com/Innovative-Lyophilization-Services.php (accessed on 20 February 2018).
- Lee, D.; Chou, W.P.; Yeh, S.H.; Chen, P.J.; Chen, P.H. DNA detection using commercial mobile phones. Biosens. Bioelectron. 2011, 26, 4349–4354. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, S.; MacCraith, B.D.; Burke, C.S. A novel camera phone-based platform for quantitative fluorescence sensing. Anal. Methods 2013, 5, 1904–1908. [Google Scholar] [CrossRef]
- Preechaburana, P.; Suska, A.; Filippini, D. Biosensing with cell phones. Trends Biotechnol. 2014, 32, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Vashist, S.K.; Schneider, E.M.; Luong, J.H.T. Commercial Smartphone-based devices and smart applications for personalized healthcare monitoring and management. Diagnostics 2014, 4, 104–128. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.; Zumla, A. Rapid infectious diseases diagnostics using Smartphones. Ann. Transl. Med. 2015, 3, 215. [Google Scholar] [PubMed]
- Xu, X.; Akay, A.; Wei, H.; Wang, S.Q.; Pingguan-Murphy, B.; Erlandsson, B.-E.; Li, X.J.; Lee, W.G.; Hu, J.; Wang, L.; et al. Advances in Smartphone-based point-of-care diagnostics. Proc. IEEE 2015, 103, 236–246. [Google Scholar] [CrossRef]
- Laksanasopin, T.; Guo, T.W.; Nayak, S.; Sridhara, A.A.; Xie, S.; Olowookere, O.O.; Cadinu, P.; Meng, F.; Chee, N.H.; Kim, J.; et al. A smartphone dongle for diagnosis of infectious diseases at the point of care. Sci. Transl. Med. 2015, 7, 272. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Zhang, C.; Xing, D. A handheld flow genetic analysis system (FGAS): Towards rapid, sensitive, quantitative and multiplex molecular diagnosis at the point-of-care level. Lab Chip 2015, 15, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Damhorst, G.L.; Duarte-Guevara, C.; Chen, W.; Ghonge, T.; Cunningham, B.T.; Bashir, R. Smartphone-imaged HIV-1 reverse-transcription loop-miediated isothermal amplification (RT-LAMP) on a chip from whole blood. Engineering 2015, 1, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.C.; Peng, J.; Mauk, M.G.; Awasthi, S.; Song, J.; Friedman, H.; Bau, H.H.; Liu, C. Smart Cup: A minimally-instrumented, Smartphone-based point-of-care molecular diagnostic device. Sens. Actuators B Chem. 2016, 229, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Weigl, B.; Domingo, G.; LaBarre, P.; Gerlach, J. Towards non and minimally instrumented, microfluidics-based diagnostic devices. Lab Chip 2008, 8, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- LaBarre, P.; Hawkins, K.R.; Gerlach, J.; Wilmoth, J.; Beddoe, A.; Singleton, J.; Boyle, D.; Weigl, B. A simple, inexpensive device for nucleic acid amplification without electricity-Toward instrument-free molecular diagnostics in low-resource settings. PLoS ONE 2011, 6, e19738. [Google Scholar] [CrossRef] [PubMed]
- Lafleur, L.K.; Bishop, J.D.; Heininger, E.K.; Gallagher, R.P.; Wheeler, M.D.; Kauffman, P.; Zhang, X.; Kline, E.C.; Buser, J.R.; Kumar, S.; et al. A rapid, instrument-free, sample-to-result nucleic acid amplification test. Lab Chip 2016, 16, 3777–3787. [Google Scholar] [CrossRef] [PubMed]
- Pardy, T.; Rang, T.; Tulp, I. Development of temperature control solutions for non-instrumented nucleic acid amplification tests (NINAAT). Micromachines 2017, 8, 180. [Google Scholar] [CrossRef]
- Curtis, K.A.; Rudolph, D.L.; Nejad, I.; Singleton, J.; Beddoe, A.; Weigl, B.; Labarre, P.; Owen, S.M. Isothermal amplification using a chemical heating device for point-of-care detetion of HIV-1. PLoS ONE 2012, 7, e31432. [Google Scholar] [CrossRef] [PubMed]
- Singleton, J.; Osborn, J.L.; Lillis, L.; Hawkins, K.; Guelig, D.; Price, W.; Johns, R.; Ebels, K.; Boyle, D.; Weigl, B.; et al. Electricity-free amplification and detection for molecular point-of-care diagnosis of HIV-1. PLoS ONE 2014, 9, e113693. [Google Scholar] [CrossRef] [PubMed]
- Singleton, J.; Gueling, D.; Buser, J.; Burton, R.; Edeh, O.; Hawkins, K.; Weigl, B.; LaBarre, P. Advancing electricity-free molecular diagnostics at the point-of-care: Optimizing the NINA platform for a malaria LAMP assay. In Proceedings of the 2014 IEEE Global Humanitarian Technology Conference (GHTC), San Jose, CA, USA, 10–13 October 2014; pp. 721–725. [Google Scholar]
- Mielczarek, W.S.; Obaje, E.A.; Bachmann, T.T.; Kersaudy-Kerhoas, M. Microfluidic blood plasma separation for medical diagnostics: Is it worth it? Lab Chip 2016, 16, 3441. [Google Scholar] [CrossRef] [PubMed]
- Guidance for Industry and FDA Staff, Recommendations for Clinical Laboroatory Improvement Amendments of 1988 (CLIA) Waiver Applications for Manufacturers of In Vitro Diagnostic Devices. Available online: https://www.fda.gov/RegulatoryInformation/Guidances/ucm079632.htm#3 (accessed on 20 February 2018).
- Sollier, E.; Cubizolles, M.; Faiver, M.; Fouillet, Y.; Achard, J. A passive microfluidic device for plasma extraction from whole human blood. In Proceedings of the 2009 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Minneapolis, MN, USA, 3–6 September 2009; pp. 7030–7033. [Google Scholar]
- Shim, J.S.; Browne, A.W.; Ahn, C.H. An on-chip whole blood/plasma separator with bead-packed microchannel on COC polymer. Biomed. Microdevices 2010, 12, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Bercich, R.; Bernhard, J.; Larson, K.; Lindsey, J. Hand-held plasma isolation device for point-of-care testing. IEEE Trans. Biomed. Eng. 2011, 58, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Nabatiyan, A.; Parpia, Z.A.; Elgnanian, R.; Kelso, D.M. Membrane-based plasma collection device for point-of-care diagnosis of HIV. J. Virol. Methods 2011, 173, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Sarenac, D.; Chen, M.H.; Huang, S.-H.; Giguel, F.F.; Kuritzkes, D.R.; Demirci, U. Simple filter microchip for rapid separation of plasma and viruses from whole blood. Int. J. Nanomed. 2012, 7, 5019–5028. [Google Scholar] [Green Version]
- Yang, X.; Forouzan, O.; Brow, T.P.; Shevkoplyas, S.S. Integrated separation of blood plasma from whole blood for microfluidic paper-based analytical devices. Lab Chip 2012, 12, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.W.; Bhagat, A.A.S.; Lee, W.C.; Huang, S.; Han, J.; Lim, C.T. Microfluidic devices for blood fractionation. Micromachines 2011, 2, 319–343. [Google Scholar] [CrossRef]
- Tripathi, S.; Kumar, Y.V.B.V.; Proabhakar, A.; Joshi, S.S.; Agrawal, A. Performance study of microfluidic devices for plasma separation—A designer’s perspective. J. Micromech. Microeng. 2015, 25, 084004. [Google Scholar] [CrossRef]
- Tripathi, S.; Kumar, Y.V.N.V.; Prabhakar, A.; Joshi, S.S.; Agrawal, A. Passive blood plasma separation at the microscale: A review of design principles and microdevices. J. Micromech. Microeng. 2015, 25, 083001. [Google Scholar] [CrossRef]
- Bond, M.M.; Richards-Kortum, R.R. Drop-to-drop variation in the cellular components of fingerprick blood. Am. J. Clin. Pathol. 2015, 144, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mauk, M.; Gross, R.; Bushman, F.D.; Edelstein, P.H.; Collman, R.G.; Bau, H.H. Membrane-based, sedimentation-assisted plasma separator for point-of-care applications. Anal. Chem. 2013, 85, 10463–10470. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liao, S.-C.; Song, J.; Mauk, M.G.; Li, X.; Wu, G.; Ge, D.; Greenberg, R.M.; Yang, S.; Bau, H.H. A high-efficiency superhydrophobic plasma separator. Lab Chip 2016, 16, 553. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Park, J.; Kim, C.J.; Cho, Y.K. Fully integrated lab-on-a-disc for nucleic acid analysis for food-borne pathogens. Anal. Chem. 2014, 86, 3841–3848. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Song, D.; Shrestha, S.; Miao, J.; Cui, L.; Guan, W. A field-deployable mobile molecular diagnostic system for malaria at the point of need. Lab Chip 2016, 16, 4341–4349. [Google Scholar] [CrossRef] [PubMed]
- Myers, F.B.; Henrikson, R.H.; Bone, J.; Lee, L.P. A handheld point-of-care genomic diagnostic system. PLoS ONE 2013, 8, e70266. [Google Scholar] [CrossRef]
- Yetisen, A.K.; Akram, M.S.; Lowe, C.R. Paper-based microfluidic point-of-care diagnostic devices. Lab Chip 2013, 13, 2210–2251. [Google Scholar] [CrossRef] [PubMed]
- Linnes, J.C.; Rodriguez, N.M.; Liu, L.; Klapperich, C.M. Polyethersulfone improves isothermal nucleic acid amplification compared to current paper-based diagnostics. Biomed. Microdevices 2016, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, N.M.; Wong, W.S.; Liu, L.; Dewar, R.; Klapperich, C.M. A fully integrated paperfluidic molecular diagnostics chip for the extraction, amplification, and detection of nucleic acids from clinical samples. Lab Chip 2016, 16, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Li, L.; Nichols, K.P.; Ismagilov, R.F. SlipChip. Lab Chip 2009, 9, 2286–2292. [Google Scholar] [CrossRef] [PubMed]
- Varapula, D.; Gutta, S.; Mauk, M.G.; Liu, C. A highly simplified nucleic acid microfluidic test device based on capillary wicking action for point-of-care diagnostics. In Proceedings of the Biomedical Engineering Conference, Troy, NY, USA, 17–19 April 2015. [Google Scholar]
- Connelly, J.T.; Rolland, J.P.; Whitesides, G.M. ’Paper machine’ for molecular diagnostics. Anal. Chem. 2015, 87, 7595–7601. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Du, B.; Zhang, P.; Haleyurgirisetty, M.; Zhao, J.; Ragupathy, V.; Lee, S.; DeVoe, D.L.; Hewlett, I.K. Development of a microchip Europium nanoparticle immunoassay for sensitive point-of-care HIV detection. Biosens. Bioelectron. 2014, 61, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Wang, J.-H.; Wu, H.-W.; Lee, G.-B. Microfluidic immunoassays. J. Assoc. Lab. Autom. 2010, 15, 253–272. [Google Scholar] [CrossRef]
- Gervais, L.; de Rooij, N.; Delamarche, E. Microfluidic chips for point-of-care immunodiagnostics. Adv. Mater. 2011, 23, H151–H176. [Google Scholar] [CrossRef] [PubMed]
- Corstjens, P.L.A.M.; Chen, Z.; Zuiderwijk, M.; Bau, H.H.; Abrams, W.R.; Malamud, D.; Niedbala, S.; Tanke, H.J. Rapid assay format for multiplex detection of humoral immune responses to infectious disease pathogens (HIV, HCV, and TB). Ann. N. Y. Acad. Sci. 2007, 1098, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, V.; Leonard, P. Next-Generation Immunoassays. In Immunoassays: Development, Applications and Future Trends; O’Kennedy, R., Murphy, C., Eds.; Pan Stanford: Boca Raton, FL, USA, 2017. [Google Scholar]
- Jordan, J.A.; Ibe, C.O.; Moore, M.S.; Host, C.; Simon, G.L. Evaluation of a manual DNA extraction protocol and an isothermal amplification assay for detecting HIV-1 DNA from dried blood spots for use in resource-limited settings. J. Clin. Virol. 2012, 54, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Leuthold, L.A.; Heudi, O.; Déglon, J.; Raccuglia, M.; Augsburger, M.; Picard, F.; Kretz, O.; Thomas, A. New microfluidic-based sampling procedure for overcoming the hematocrit problem associated with dried blood spot analysis. Anal. Chem. 2015, 87, 2068–2071. [Google Scholar] [CrossRef] [PubMed]
- Begolo, S.; Shen, F.; Ismagilov, R.F. A microfluidic device for dry sample preservation in remote settings. Lab Chip 2013, 13, 4331–4342. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, S.; Fan, A.; Trueb, J.; Jareczek, F.; Mazzochette, M.; Sharon, A.; Sauer-Budge, A.F.; Klapperich, C.M. A portable, pressure driven, room temperature nucleic acid extraction and storage system for point of care molecular diagnostics. Anal. Methods 2013, 5, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Liga, A.; Vleigenthart, A.D.B.; Oosthuyzen, W.; Dear, J.W.; Kersaudy-Kerhoas, M. Exosome isolation: A microfluidic perspective. Lab Chip 2015, 15, 2388. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.R.; Norton, S.E.; Luna, K.K.; Lechner, J.M.; Qin, J. Stabilization of cell-free RNA in blood samples using a new collection device. Clin. Biochem. 2012, 45, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Moturi, S.; Angkachatchai, V.; Mueller, R.; DeSantis, G.; van den Boom, D.; Ehrich, M. Optimizing blood collection, transport and storage conditions for cell free DNA increases access to prenatal testing. Clin. Biochem. 2013, 46, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Alt, J.R.; Hunsley, B.A.; Williams, T.L.; Rohan Fernando, M. Stabilization of circulating tumor cells in blood using a collection device with a preservative reagent. Cancer Cell Int. 2014, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, S.K.; Mallick, R.; Yendamuri, S. Detection of microRNAs in dried serum plots. Anal. Biochem. 2010, 407, 147–149. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
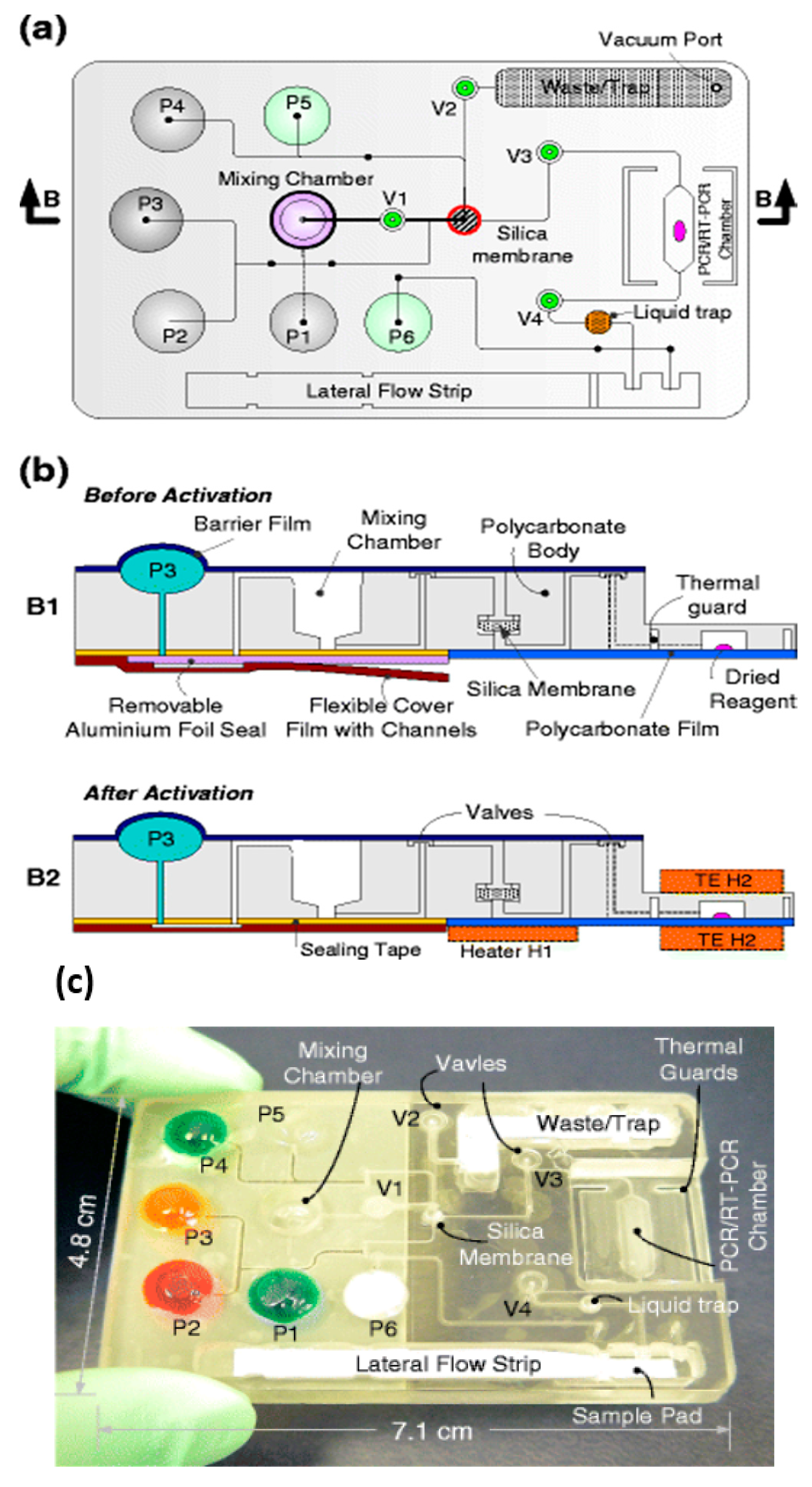{kind=link}
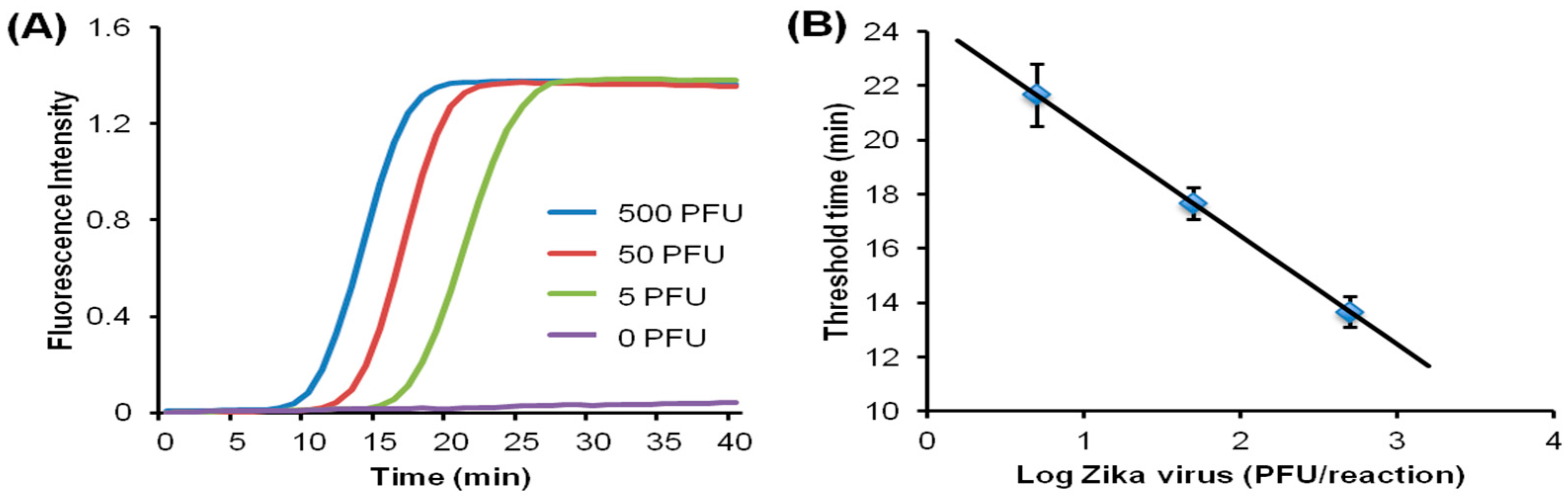{kind=link}
{kind=link}
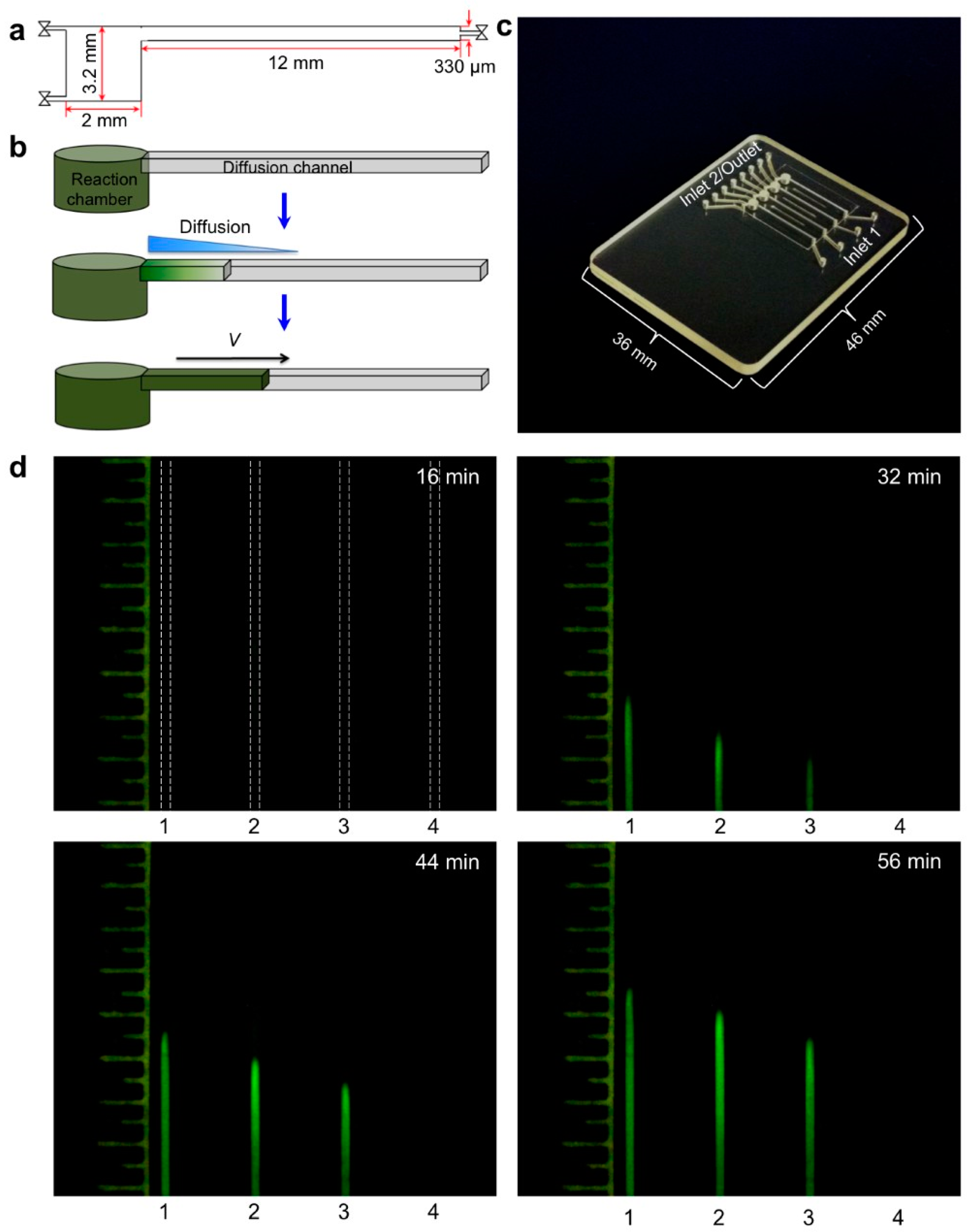{kind=link}
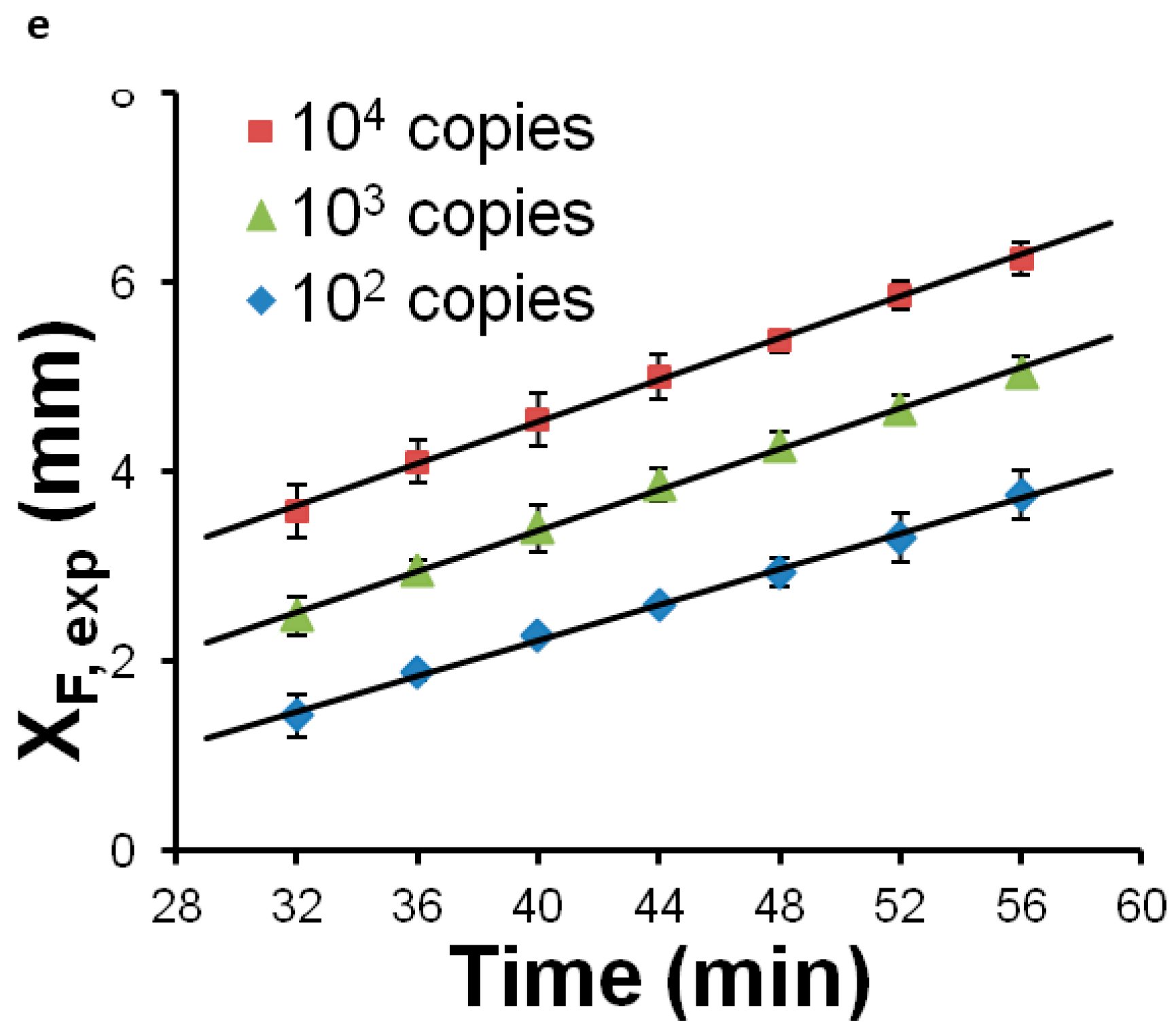{kind=link}
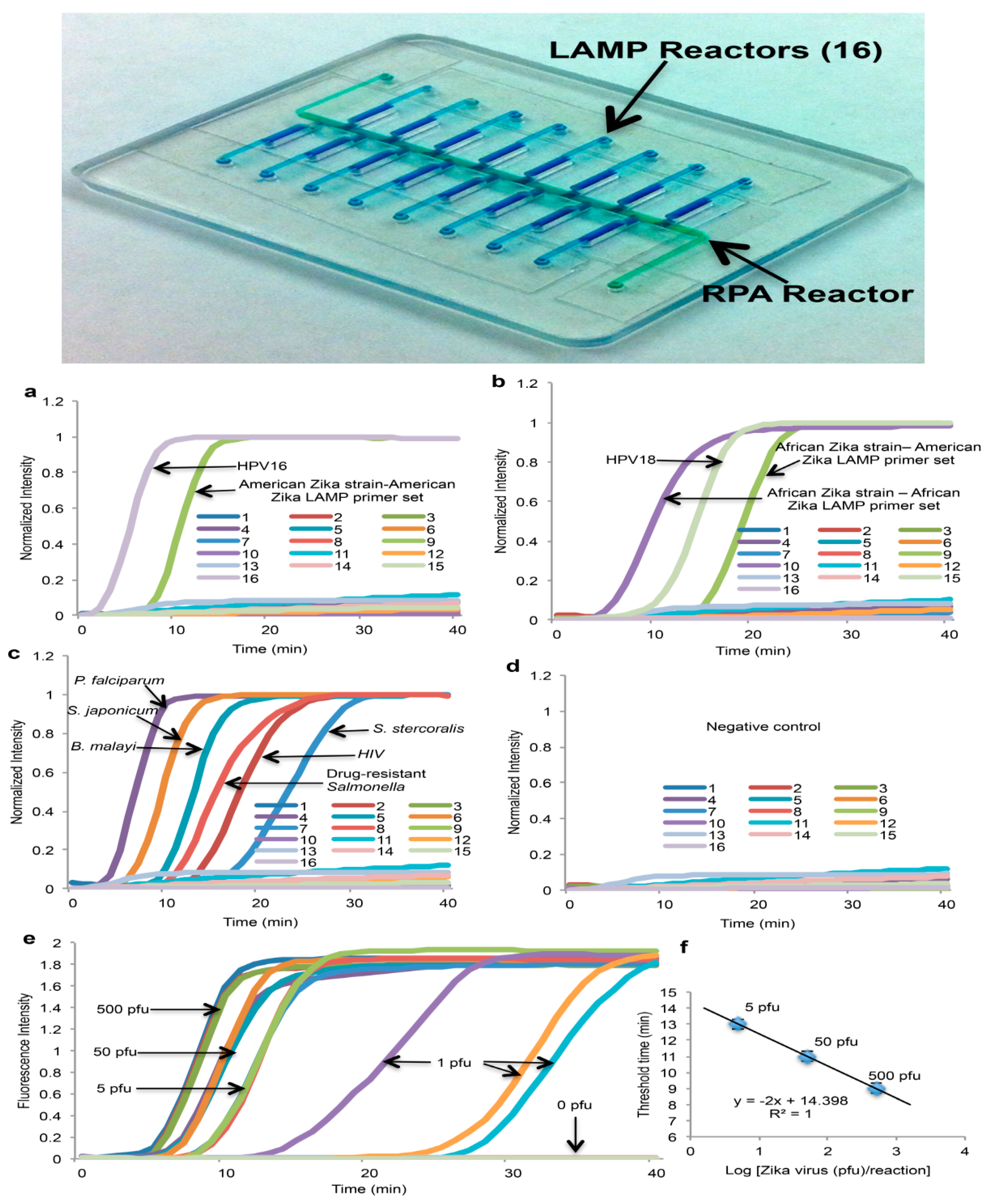{kind=link}
{kind=link}
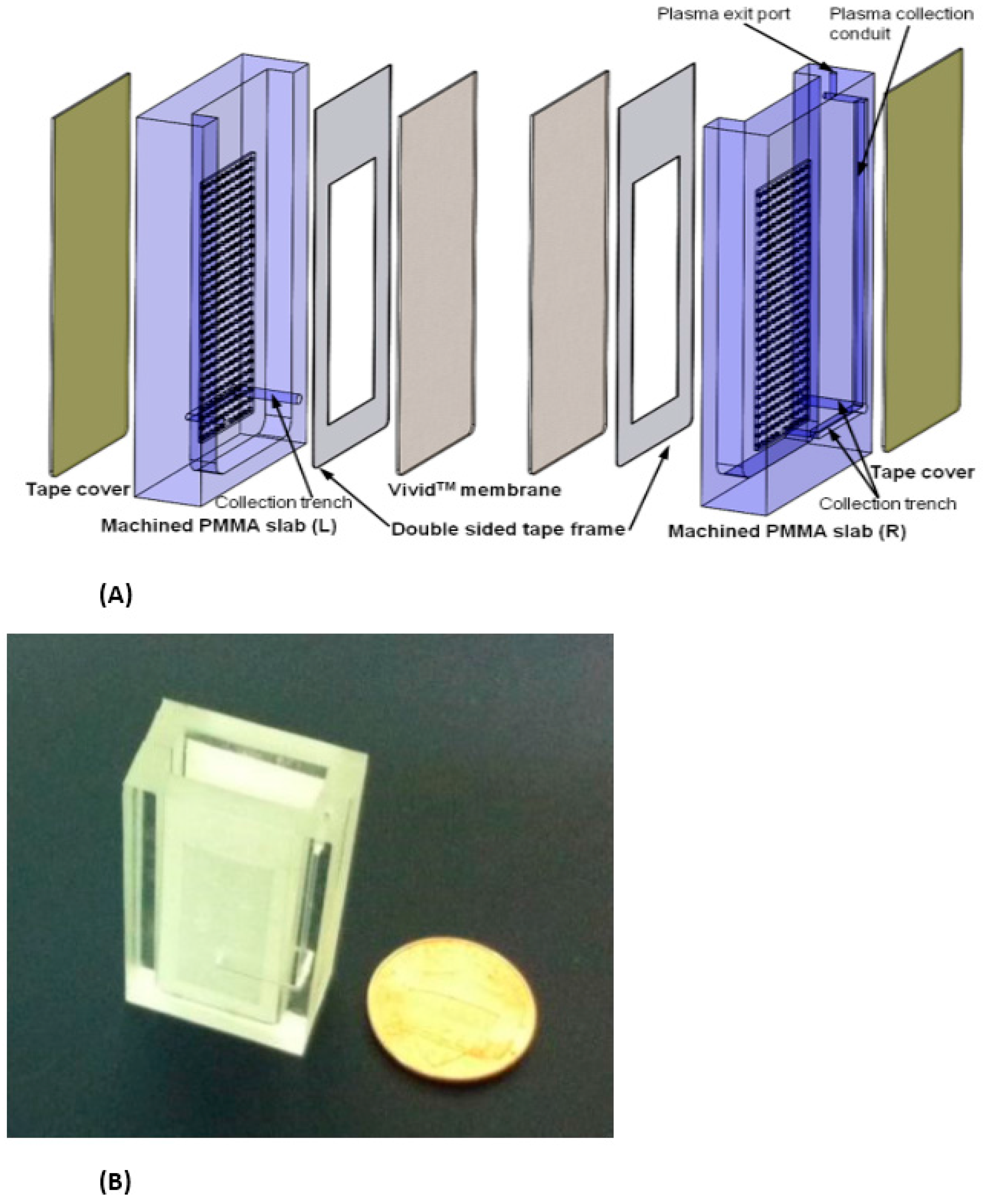{kind=link}
{kind=link}
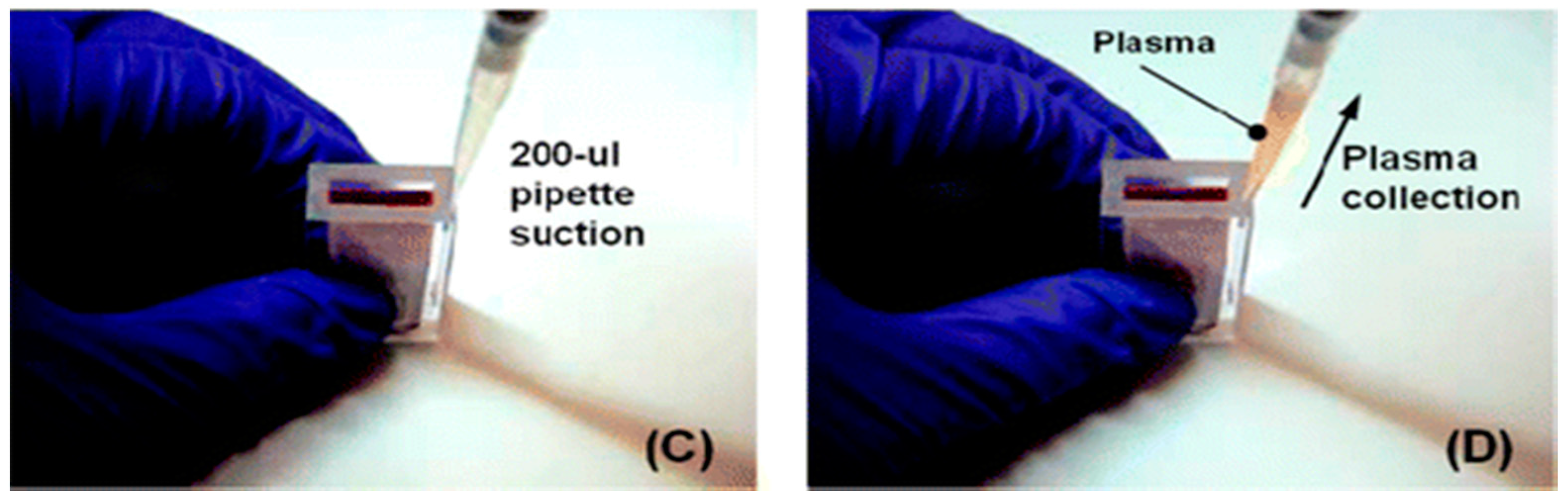{kind=link}
{kind=link}
| Criteria | Aim | Comments |
|---|---|---|
| Test Cost | $5–10 consumables per test | Disposable, single-use plastic cartridge, pre-loaded with reagents Compatible with injection molding, |
| Instrument Cost | <$500 | Portable, handheld or desktop, providing temperature regulation, stable optical platform for cellphone or other detector, limited actuation; made with off-the-shelf components |
| Performance: Sensitivity, Limit of Detection (LOD); Specificity | Comparable to laboratory tests | e.g., HIV viral load testing LOD: 1–100 virons/mL plasma; Screening tests LOD: 1000 genome copies per sample. Sensitivity/Specificity (false negatives/ false positives): 98 to 99%. |
| Time for testing | 30 to 60 min | Time spanning sample-in to report-out should be about 30 min, primarily due to time required for amplification of low-concentration target |
| Operator skill level | minimal, non-professional, semi-skilled | Training in less than 1 h. No pipetting, sample collection and loading raw sample into cartridge, no sample transfer after sample loading (e.g., no pre-sample processing with centrifuge), no addition of reagents at time of use. |
| Power | battery or chemical heating/cellphone | Ideally, should operate independent of grid electric power (Many resource-limited areas of the world do not have reliable electric power.) |
| Shelf life | test cartridge: 1 year @ 40 °C | No cold-chain, hermetically sealed cartridge can be stored in tropical climates for a year without refrigeration. |
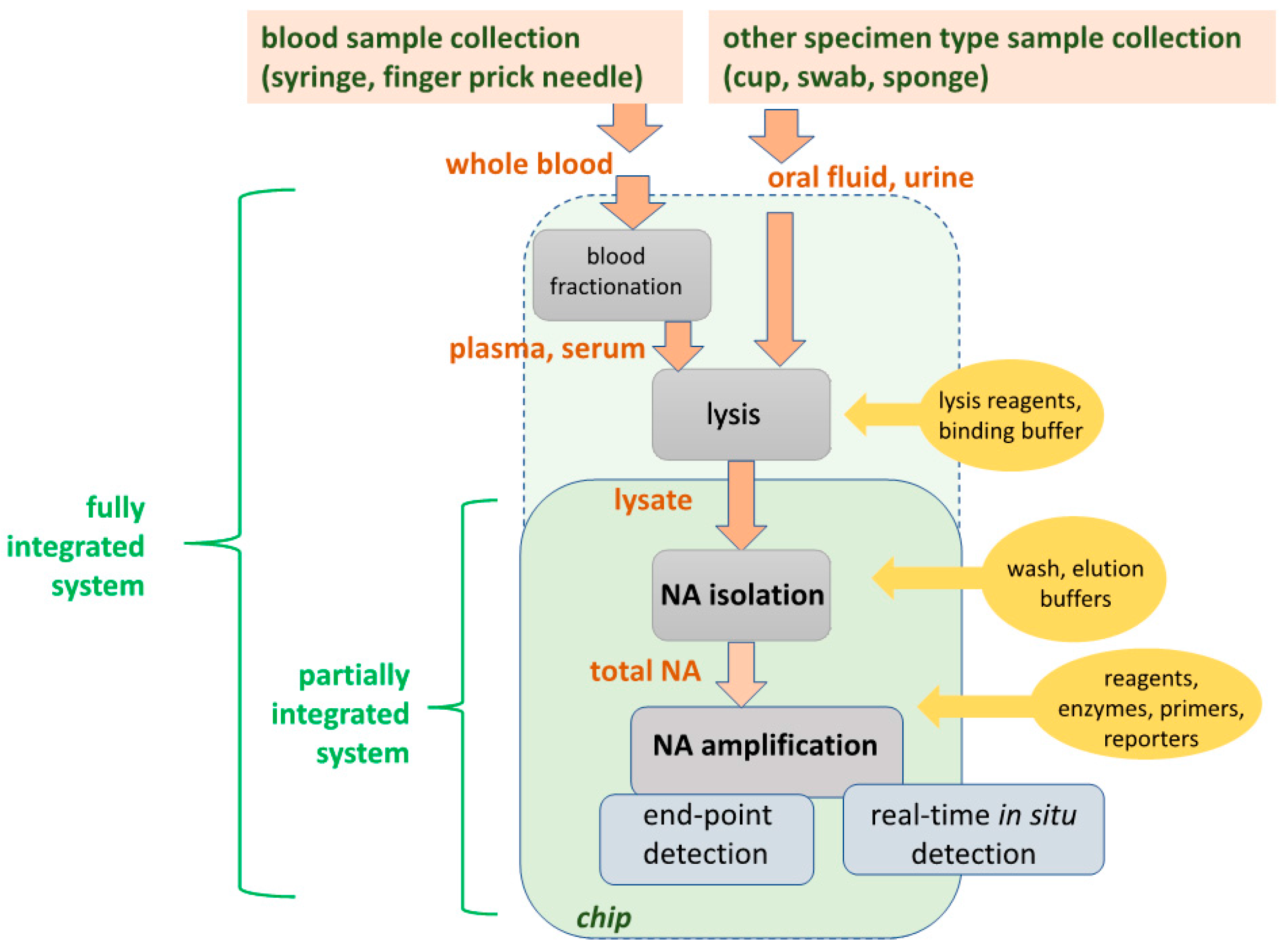
| Unit Operation | Options | POC Issues |
|---|---|---|
| Blood Fractionation |
| Centrifugation is difficult to integrate into the chip, and a separate centrifugation step may be needed. Most LOC filtration/sedimentation and fluid flow fractionation is typically limited to small sample volumes. |
| Lysis |
| Viruses are comparatively easy to lyse with detergents and chaotropic salts; vegetative bacterial cells are somewhat more difficult; and spores most difficult, requiring enzymes and/or mechanical disruption. |
| Nucleic Acid (NA) Isolation (Extraction, Purification, Concentration) |
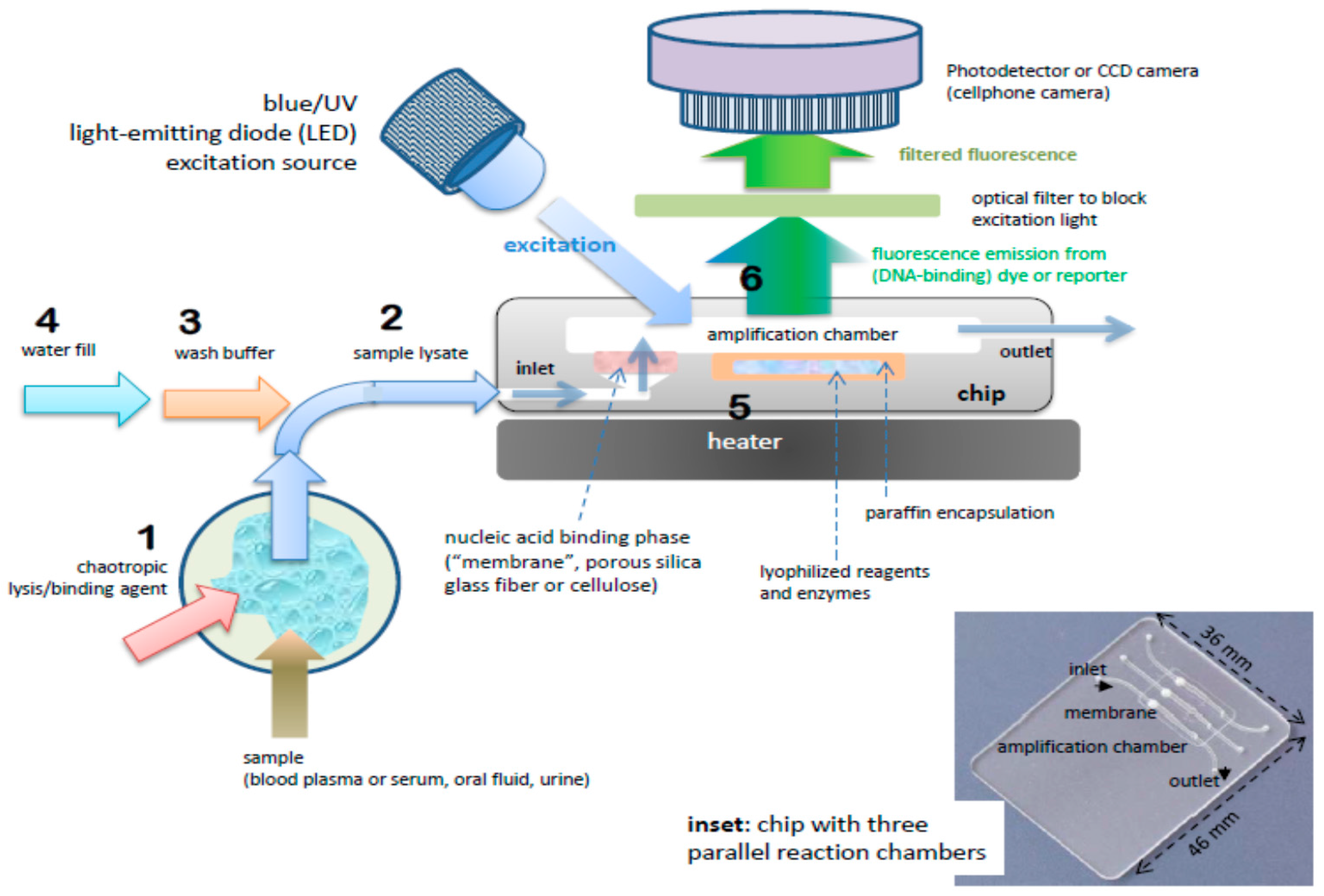
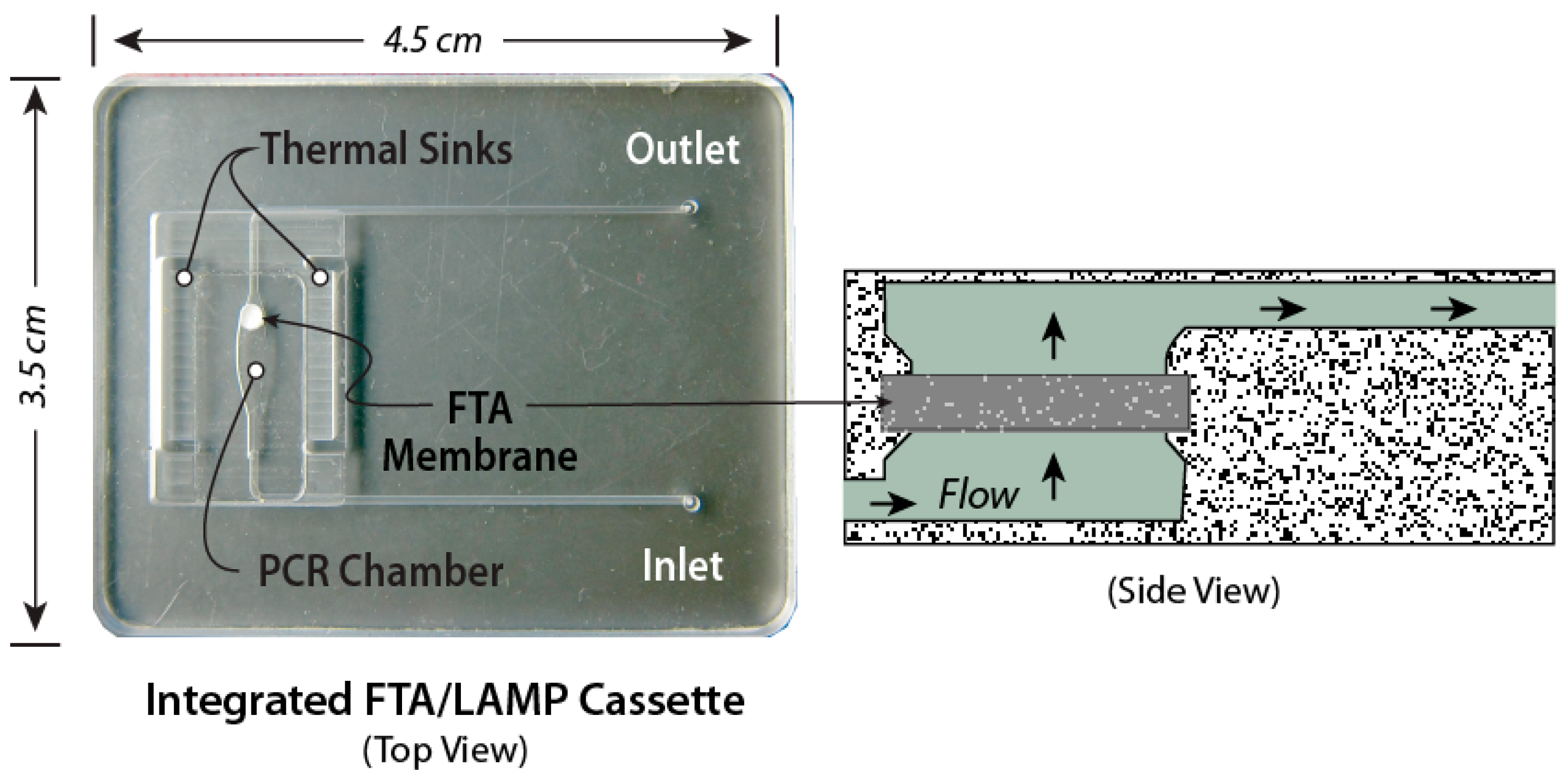
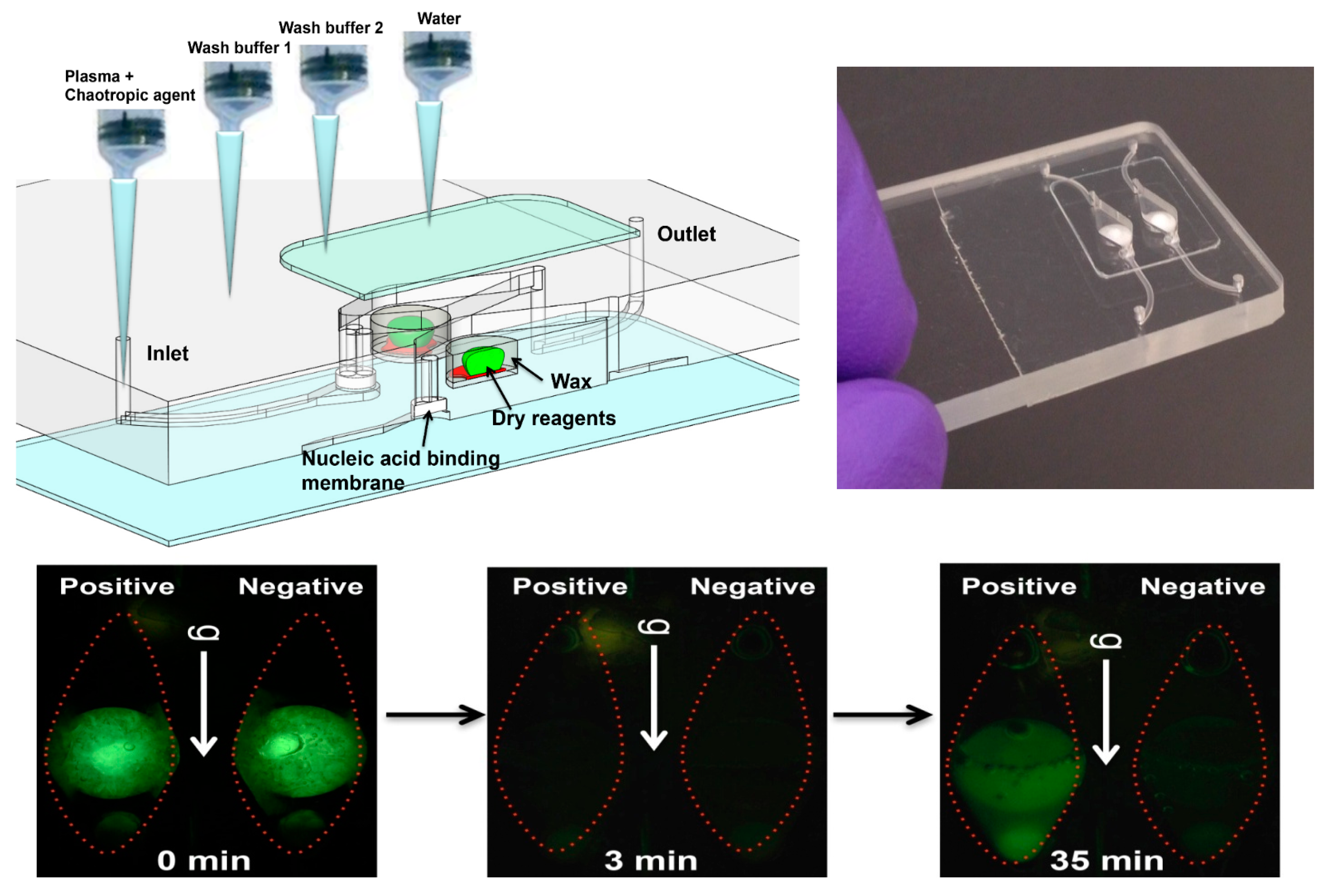
| Solid-phase extraction using a NA-binding phase (e.g., silica) and binding, wash, and elution buffers is readily implemented with microfluidics. |
| Nucleic Acid (NA) Amplification |
| PCR is well developed, but requires instrumentation for precise thermal cycling, and has relatively high power consumption. Isothermal methods require much less instrumentation. LAMP (65 °C constant temperature incubation) appears to be the most used method. |
| Amplicon Detection |
| Fluorescent dyes are very sensitive. Bioluminescent reporters do not require light sources or optical filters. Colorimetric dyes can be read by eye for instrument-free operation. Electrochemical sensors are more compact, but generally less sensitive and difficult to interface with a disposable chip. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauk, M.G.; Song, J.; Liu, C.; Bau, H.H. Simple Approaches to Minimally-Instrumented, Microfluidic-Based Point-of-Care Nucleic Acid Amplification Tests. Biosensors 2018, 8, 17. https://doi.org/10.3390/bios8010017
Mauk MG, Song J, Liu C, Bau HH. Simple Approaches to Minimally-Instrumented, Microfluidic-Based Point-of-Care Nucleic Acid Amplification Tests. Biosensors. 2018; 8(1):17. https://doi.org/10.3390/bios8010017
Chicago/Turabian StyleMauk, Michael G., Jinzhao Song, Changchun Liu, and Haim H. Bau. 2018. "Simple Approaches to Minimally-Instrumented, Microfluidic-Based Point-of-Care Nucleic Acid Amplification Tests" Biosensors 8, no. 1: 17. https://doi.org/10.3390/bios8010017
APA StyleMauk, M. G., Song, J., Liu, C., & Bau, H. H. (2018). Simple Approaches to Minimally-Instrumented, Microfluidic-Based Point-of-Care Nucleic Acid Amplification Tests. Biosensors, 8(1), 17. https://doi.org/10.3390/bios8010017