A Liposomal Platform for Sensing of Extracellular Analytes Near Cells

 , , and
, , and 
Abstract
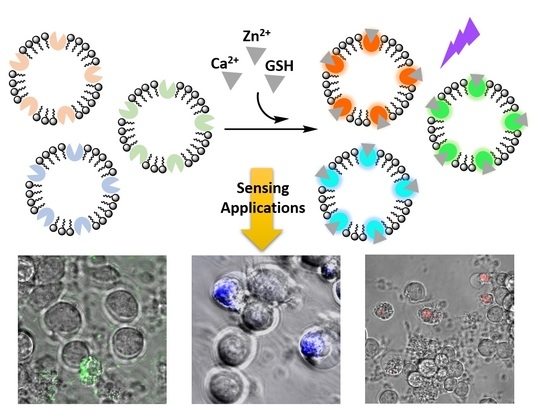
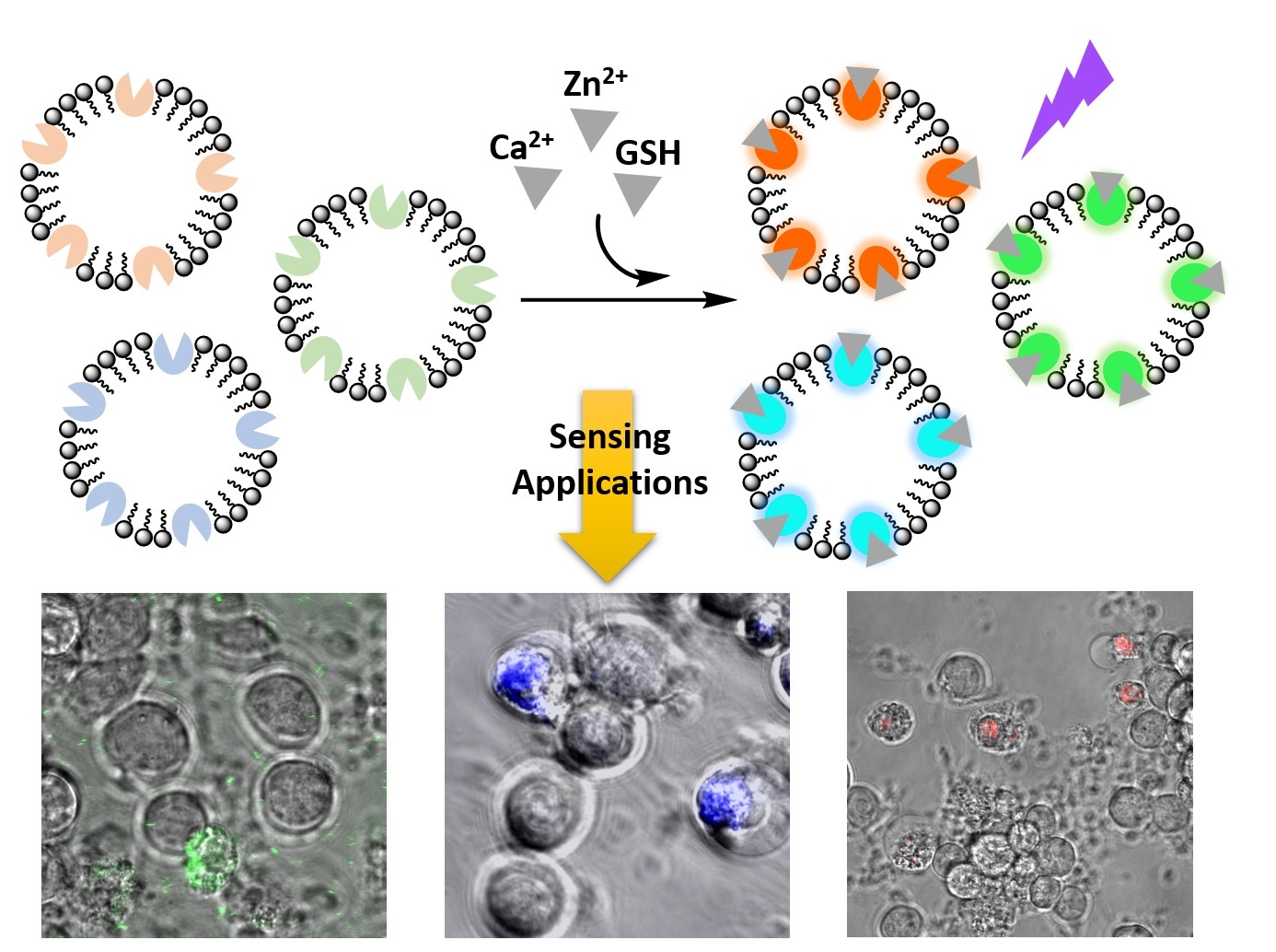
:
1. Introduction
2. Materials and Methods
2.1. General Information
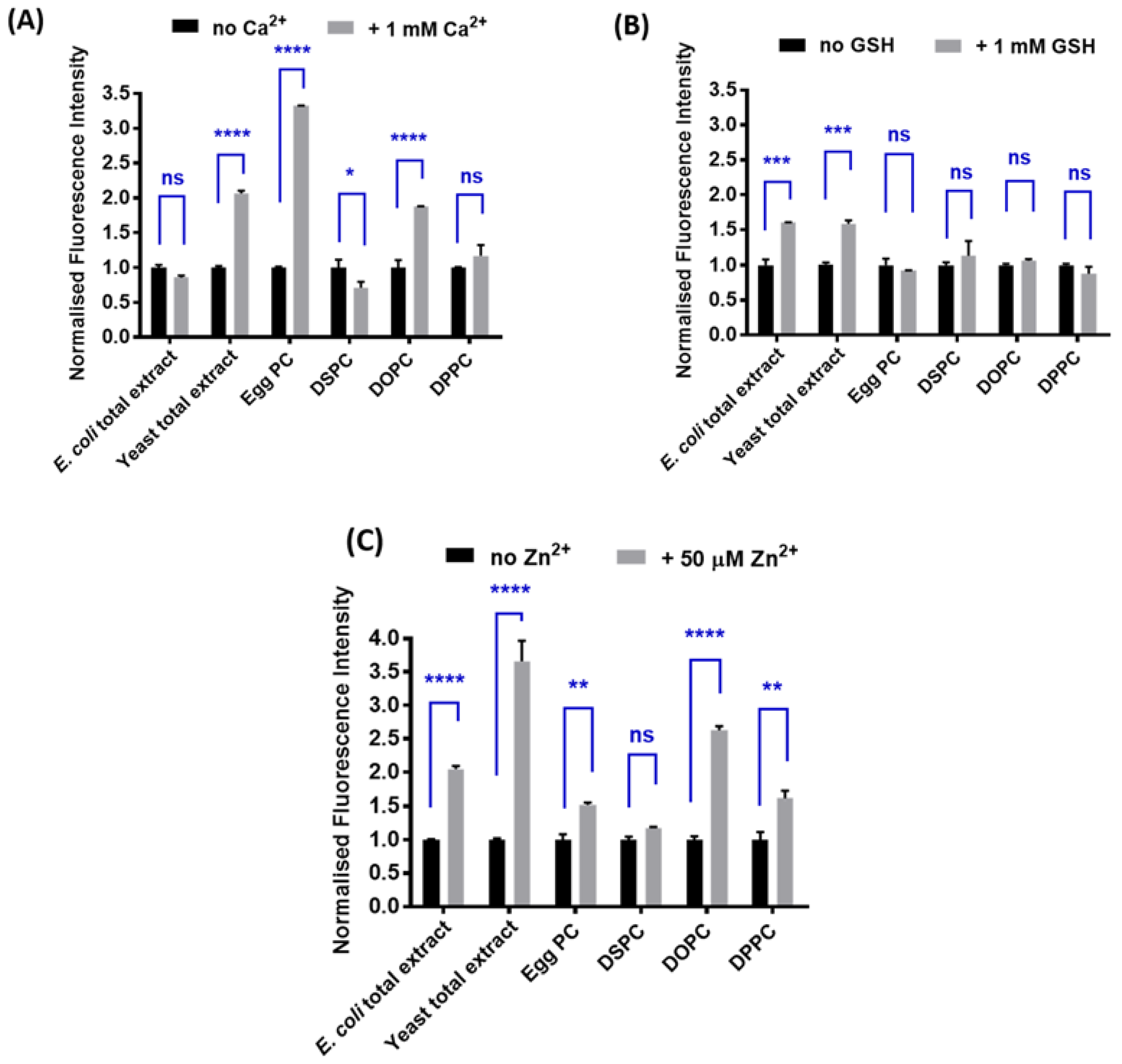
2.2. Determination of the Optimal Lipid Composition
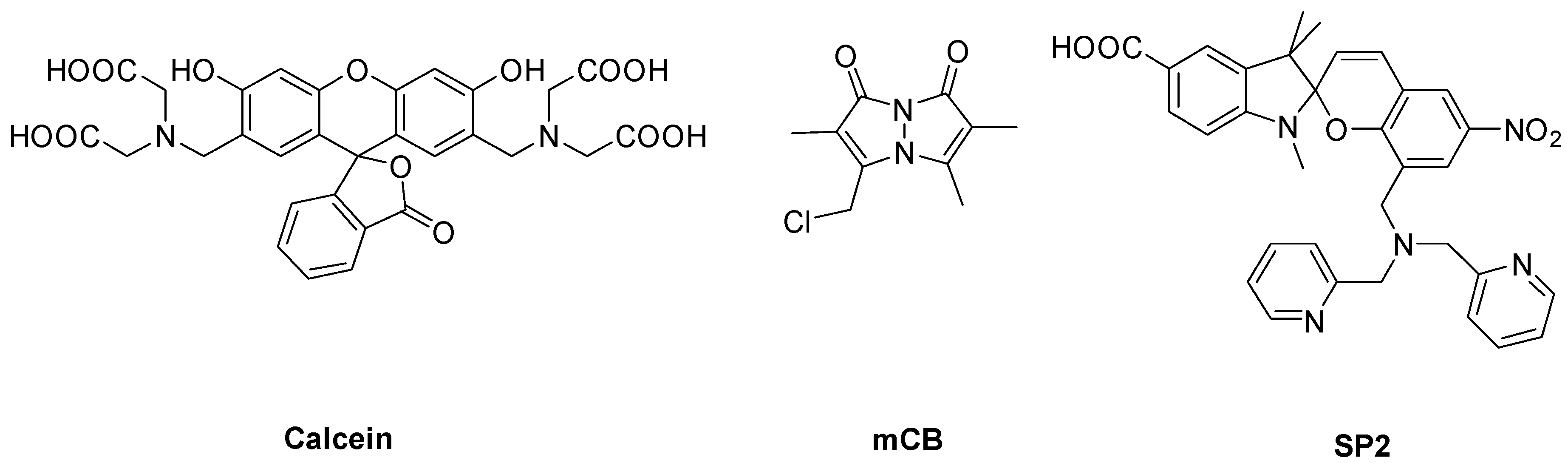
2.3. Spectroscopic Characterization of LP–Cal, LP–mCB, and LP–SP2
2.4. LIVE/DEAD Viability Assay
2.5. Alamar Blue Viability Assay
2.6. Confocal Cell Imaging
3. Results and Discussion
3.1. Design and Preparation of Liposomal Sensors
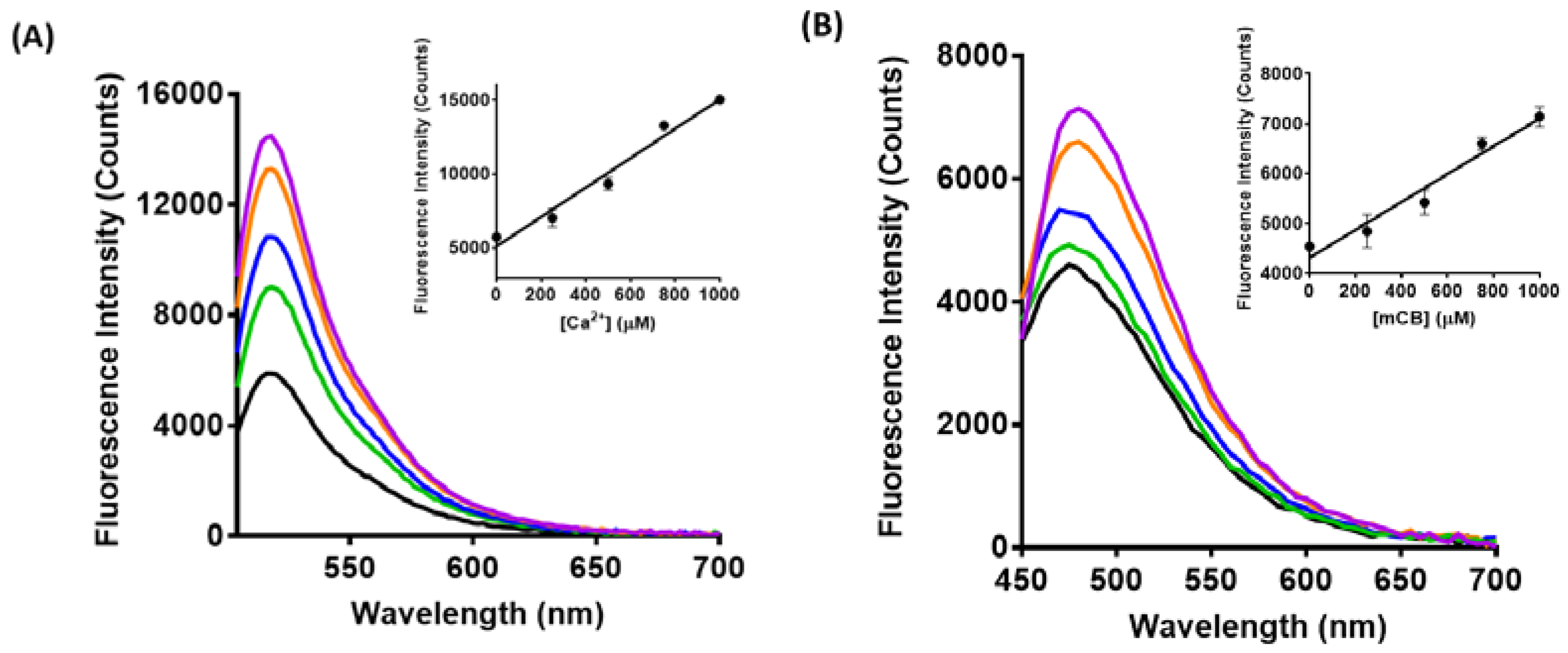
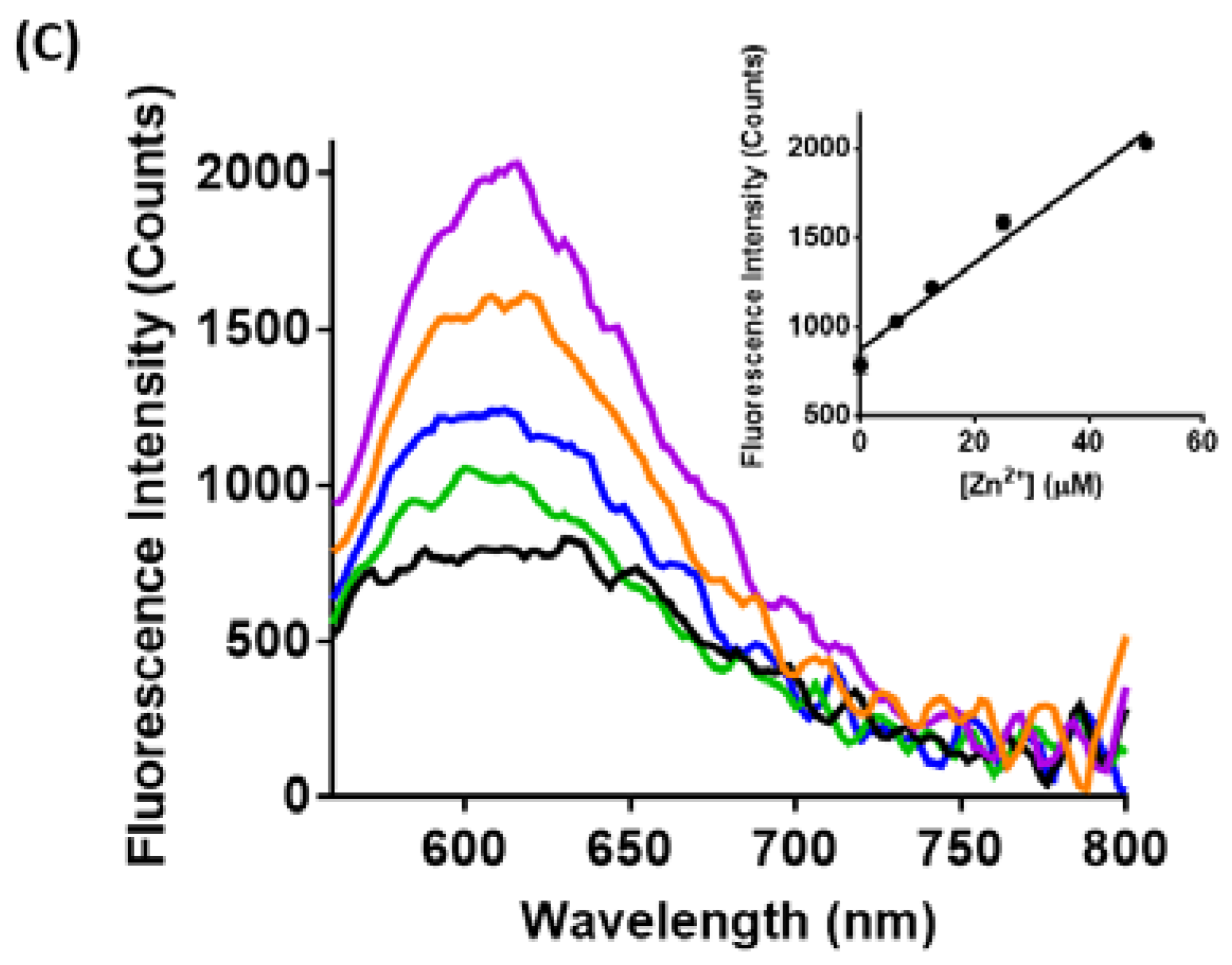
3.2. Spectroscopic Characterization of Yeast Liposomal Sensors
3.3. Imaging Ca2+, Zn2+, and GSH Near Live HEK293 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ward, P.S.; Thompson, C.B. Signaling in Control of Cell Growth and Metabolism. Cold Spring Harb. Perspect. Boil. 2012, 4, a006783. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Fitzgerald, W.; Zimmerberg, J.; Kleinman, H.K.; Margolis, L. Cell-Cell and Cell-Extracellular Matrix Interactions Regulate Embryonic Stem Cell Differentiation. Stem Cells 2007, 25, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daino, H.; Matsumura, I.; Takada, K.; Odajima, J.; Tanaka, H.; Ueda, S.; Shibayama, H.; Ikeda, H.; Hibi, M.; Machii, T.; et al. Induction of apoptosis by extracellular ubiquitin in human hematopoietic cells: Possible involvement of STAT3 degradation by proteasome pathway in interleukin 6-dependent hematopoietic cells. Blood 2000, 95, 2577. [Google Scholar] [PubMed]
- Charras, G.; Sahai, E. Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell. Biol. 2014, 15, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Pockley, A.G.; Henderson, B.; Multhoff, G. Extracellular cell stress proteins as biomarkers of human disease. Biochem. Soc. Trans. 2014, 42, 1744. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell. Boil. 2012, 196, 395. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.M.; Bernhardt, M.L.; Kong, B.Y.; Ahn, R.W.; Vogt, S.; Woodruff, T.K.; O’Halloran, T.V. Zinc Sparks Are Triggered by Fertilization and Facilitate Cell Cycle Resumption in Mammalian Eggs. ACS Chem. Boil. 2011, 6, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.L.; Wessel, G.M. Extracellular matrix modifications at fertilization: Regulation of dityrosine crosslinking by transamidation. Development 2009, 136, 1835–1847. [Google Scholar] [CrossRef] [PubMed]
- Wessel, G.M.; Wong, J.L. Cell Surface Changes in the Egg at Fertilization. Mol. Reprod. Dev. 2009, 76, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Rozario, T.; DeSimone, D.W. The Extracellular Matrix In Development and Morphogenesis: A Dynamic View. Dev. Boil. 2010, 341, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.E.; Miller, A.L. Calcium signalling during embryonic development. Nat. Rev. Mol. Cell. Biol. 2003, 4, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Duncan, F.E.; Que, E.L.; O’Halloran, T.V.; Woodruff, T.K. The fertilization-induced zinc spark is a novel biomarker of mouse embryo quality and early development. Sci. Rep. 2016, 6, 22772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoine, A.F.; Faure, J.-E.; Cordeiro, S.; Dumas, C.; Rougier, M.; Feijó, J.A. A calcium influx is triggered and propagates in the zygote as a wavefront during in vitro fertilization of flowering plants. Proc. Natl. Acad. Sci. USA 2000, 97, 10643–10648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alasmari, W.; Barratt, C.L.R.; Publicover, S.J.; Whalley, K.M.; Foster, E.; Kay, V.; da Silva, S.M.; Oxenham, S.K. The clinical significance of calcium signalling pathways mediating human sperm hyperactivation. Hum. Reprod. 2013, 28, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Truong, J.; Reeves, W.B.; Hahm, J.-i. Emerging Cytokine Biosensors with Optical Detection Modalities and Nanomaterial-Enabled Signal Enhancement. Sensors 2017, 17, 428. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-B.; Li, M.; Wang, W.-P.; Wang, X.-L. High throughput screening technologies for ion channels. Acta Pharmacol. Sin. 2016, 37, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hong, Y.; Liu, Y.; Liu, J.; Leung, C.W.T.; Li, M.; Kwok, R.T.K.; Zhao, E.; Lam, J.W.Y.; Yu, Y.; et al. Full-Range Intracellular pH Sensing by an Aggregation-Induced Emission-Active Two-Channel Ratiometric Fluorogen. J. Am. Chem. Soc. 2013, 135, 4926–4929. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, K.; Yoshida, M.; Kamiya, M.; Yamasoba, T.; Urano, Y. Rational design of reversible fluorescent probes for live-cell imaging and quantification of fast glutathione dynamics. Nat. Chem. 2017, 9, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.T.; Johnstone, T.C.; Deliz Liang, A.; Lippard, S.J.; Rivera-Fuentes, P. A Fast and Selective Near-Infrared Fluorescent Sensor for Multicolor Imaging of Biological Nitroxyl (HNO). J. Am. Chem. Soc. 2014, 136, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Orellana, G. Fluorescence-Based Sensors. In Optical Chemical Sensors; Baldini, F., Chester, A.N., Homola, J., Martellucci, S., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 99–116. [Google Scholar] [CrossRef]
- Heng, S.; McDevitt, C.A.; Stubing, D.B.; Whittall, J.J.; Thompson, J.G.; Engler, T.K.; Abell, A.D.; Monro, T.M. Microstructured Optical Fibers and Live Cells: A Water-Soluble, Photochromic Zinc Sensor. Biomacromolecules 2013, 14, 3376–3379. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.L.; Mayfield, B.L.; Duray, A.M.; Tembo, M.; Beleny, D.O.; Napolitano, M.A.; Sauer, M.L.; Wisner, B.W.; Carlson, A.E. Extracellular Ca2+ Is Required for Fertilization in the African Clawed Frog, Xenopus laevis. PLoS ONE 2017, 12, e0170405. [Google Scholar] [CrossRef] [PubMed]
- Duncan, F.E.; Que, E.L.; Zhang, N.; Feinberg, E.C.; O’Halloran, T.V.; Woodruff, T.K. The zinc spark is an inorganic signature of human egg activation. Sci. Rep. 2016, 6, 24737. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, Y.; Nishimura, M.; Matsumoto, K.; Miyashita, M.; Takeo, T.; Nakagata, N.; Hosoi, Y.; Anzai, M. The influence of reduced glutathione in fertilization medium on the fertility of in vitro–matured C57BL/6 mouse oocytes. Theriogenology 2013, 80, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.V.; Kourghi, M.; De Ieso, M.L.; Campbell, E.M.; Dorward, H.S.; Hardingham, J.E.; Yool, A.J. Differential Inhibition of Water and Ion Channel Activities of Mammalian Aquaporin-1 by Two Structurally Related Bacopaside Compounds Derived from the Medicinal Plant Bacopa monnieri. Mol. Pharmacol. 2016, 90, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Barhoumi, R.; Bailey, R.H.; Burghardt, R.C. Kinetic analysis of glutathione in anchored cells with monochlorobimane. Cytometry 1995, 19, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamencic, H.; Lyon, A.; Paterson, P.G.; Juurlink, B.H.J. Monochlorobimane Fluorometric Method to Measure Tissue Glutathione. Anal. Biochem. 2000, 286, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Ntailianas, H.A.; Whitney, R.M. Calcein as an Indicator for the Determination of Total Calcium and Magnesium and Calcium Alone in the Same Aliquot of Milk. J. Dairy Sci. 1964, 47, 19–27. [Google Scholar] [CrossRef]
- Toribara, T.Y.; Koval, L. Determination of calcium in biological material: The use of calcein as an indicator in the edta titration. Talanta 1961, 7, 248–252. [Google Scholar] [CrossRef]
- Du, S.J.; Frenkel, V.; Kindschi, G.; Zohar, Y. Visualizing Normal and Defective Bone Development in Zebrafish Embryos Using the Fluorescent Chromophore Calcein. Dev. Boil. 2001, 238, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Zhang, Z.; Lv, J.; Cheng, H. Flow-injection analysis–fluorescence detection for the in vivo on-line determination of calcium in blood with microdialysis sampling. Anal. Chim. Acta 2000, 419, 175–184. [Google Scholar] [CrossRef]
- Fernández-Checa, J.; Kaplowitz, N. The use of monochlorobimane to determine hepatic GSH levels and synthesis. Anal. Biochem. 1990, 190, 212–219. [Google Scholar] [CrossRef]
- Wolfgang, S. Light Scattering from Polymer Solutions and Nanoparticle Dispersions; Springer Science & Business Media: Berlin, Germany, 2007. [Google Scholar]
- Ge, J.; Wood, D.K.; Weingeist, D.M.; Prasongtanakij, S.; Navasumrit, P.; Ruchirawat, M.; Engelward, B.P. Standard Fluorescent Imaging of Live Cells is Highly Genotoxic. Cytom. Part A J. Int. Soc. Anal. Cytol. 2013, 83, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Checker, R.; Sandur, S.K.; Sharma, D.; Patwardhan, R.S.; Jayakumar, S.; Kohli, V.; Sethi, G.; Aggarwal, B.B.; Sainis, K.B. Potent Anti-Inflammatory Activity of Ursolic Acid, a Triterpenoid Antioxidant, Is Mediated through Suppression of NF-κB, AP-1 and NF-AT. PLoS ONE 2012, 7, e31318. [Google Scholar] [CrossRef] [PubMed]
- Heng, S.; McDevitt, C.A.; Kostecki, R.; Morey, J.R.; Eijkelkamp, B.A.; Ebendorff-Heidepriem, H.; Monro, T.M.; Abell, A.D. Microstructured Optical Fiber-based Biosensors: Reversible and Nanoliter-Scale Measurement of Zinc Ions. ACS Appl. Mater. Interfaces 2016, 8, 12727–12732. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.D. Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies; Life Technologies Corporation: Carlsbad, CA, USA, 2010. [Google Scholar]
- Lee, J.S.; Hwang, S.Y.; Lee, E.K. Imaging-based analysis of liposome internalization to macrophage cells: Effects of liposome size and surface modification with PEG moiety. Colloids Surf. B Biointerfaces 2015, 136, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Maurer, N.; Fenske, D.B.; Cullis, P.R. Developments in liposomal drug delivery systems. Expert Opin. Boil. Ther. 2001, 1, 923–947. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Z-Average 1 (d.nm) | PDI |
|---|---|---|
| LP–SP2 | 166 ± 2 | 0.21 ± 0.01 |
| LP–Cal | 165 ± 2 | 0.11 ± 0.04 |
| LP–mCB | 175 ± 1 | 0.19 ± 0.03 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Heng, S.; Pei, J.; Morey, J.R.; McDevitt, C.A.; Abell, A.D. A Liposomal Platform for Sensing of Extracellular Analytes Near Cells. Biosensors 2018, 8, 117. https://doi.org/10.3390/bios8040117
Zhang X, Heng S, Pei J, Morey JR, McDevitt CA, Abell AD. A Liposomal Platform for Sensing of Extracellular Analytes Near Cells. Biosensors. 2018; 8(4):117. https://doi.org/10.3390/bios8040117
Chicago/Turabian StyleZhang, Xiaozhou, Sabrina Heng, Jinxin Pei, Jacqueline R. Morey, Christopher A. McDevitt, and Andrew D. Abell. 2018. "A Liposomal Platform for Sensing of Extracellular Analytes Near Cells" Biosensors 8, no. 4: 117. https://doi.org/10.3390/bios8040117
APA StyleZhang, X., Heng, S., Pei, J., Morey, J. R., McDevitt, C. A., & Abell, A. D. (2018). A Liposomal Platform for Sensing of Extracellular Analytes Near Cells. Biosensors, 8(4), 117. https://doi.org/10.3390/bios8040117