Homotransfer FRET Reporters for Live Cell Imaging

 ,
,
Abstract
:1. Introduction
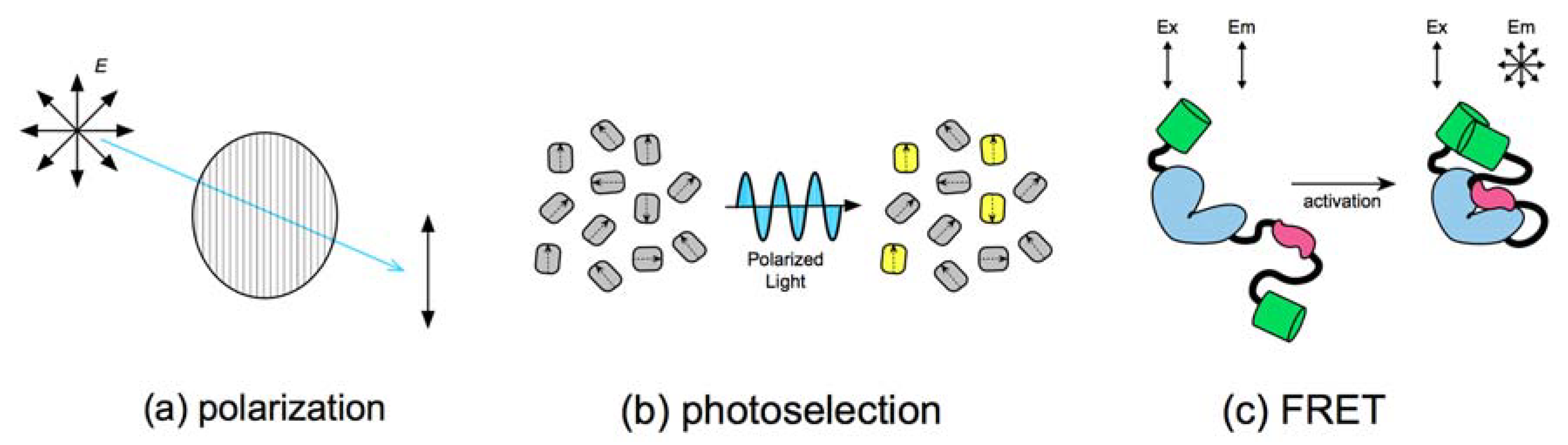
2. Fluorescence Polarization and FRET
2.1. Photoselection during Fluorescence Illumination
2.2. FRET Efficiency and Depolarization
2.3. Calculating FRET Efficiency from Time-Resolved Anisotropy Measurements
3. Fluorescence Polarization Microscopy
3.1. Steady-State Polarization Measurements
3.2. Time-Resolved Measurements
3.3. Optical-Sectioning
4. Biosensor Design
4.1. Double-Fluorophore Biosensors
4.2. Single-Fluorophore Biosensors
5. Comparison to Heterotransfer Reporters
5.1. Multisensor Applications
5.2. Compatibility with Other Optical Tools
5.3. Quantitative Analysis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Akerboom, J.; Carreras Calderon, N.; Tian, L.; Wabnig, S.; Prigge, M.; Tolo, J.; Gordus, A.; Orger, M.B.; Severi, K.E.; Macklin, J.J.; et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 2013, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauban, J.R.; Fairfax, S.T.; Rizzo, M.A.; Zhang, J.; Wier, W.G. A method for noninvasive longitudinal measurements of [Ca2+] in arterioles of hypertensive optical biosensor mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H173–H181. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Zhang, J. Reporting from the field: Genetically encoded fluorescent reporters uncover signaling dynamics in living biological systems. Annu. Rev. Biochem. 2011, 80, 375–401. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.W.; Kremers, G.J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Pietraszewska-Bogiel, A.; Gadella, T.W. FRET microscopy: From principle to routine technology in cell biology. J. Microsc. 2011, 241, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Okumoto, S.; Jones, A.; Frommer, W.B. Quantitative imaging with fluorescent biosensors. Annu. Rev. Plant Biol. 2012, 63, 663–706. [Google Scholar] [CrossRef] [PubMed]
- Spiering, D.; Bravo-Cordero, J.J.; Moshfegh, Y.; Miskolci, V.; Hodgson, L. Quantitative ratiometric imaging of FRET-biosensors in living cells. Methods Cell Biol. 2013, 114, 593–609. [Google Scholar] [PubMed]
- Dale, R.E.; Eisinger, J.; Blumberg, W.E. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 1979, 26, 161–193. [Google Scholar] [CrossRef] [Green Version]
- Clegg, R.M. FRET tells us about proximities, distances, orientations and dynamic properties. J. Biotechnol. 2002, 82, 177–179. [Google Scholar] [PubMed]
- Iqbal, A.; Arslan, S.; Okumus, B.; Wilson, T.J.; Giraud, G.; Norman, D.G.; Ha, T.; Lilly, D.M.J. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nucleic acids. Proc. Natl. Acad. Sci. USA 2008, 105, 11176–11181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szöllősi, J. Understanding FRET as a research tool for cellular studies. Int. J. Mol. Sci. 2015, 16, 6718–6756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Campbell, R.E.; Ting, A.Y.; Tsien, R.Y. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A Guide to Fluorescent Protein FRET Pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Springer, G.; Segawa, K.; Zipfel, W.R.; Piston, D.W. Optimization of pairings and detection conditions for measurement of FRET between cyan and yellow fluorescent proteins. Microsc. Microanal. 2006, 12, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar] [CrossRef]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 2001, 31, 973–985. [Google Scholar] [CrossRef]
- Thaler, C.; Koushik, S.V.; Puhl, H.L.; Blank, P.S.; Vogel, S.S. Structural rearrangement of CaMKIIalpha catalytic domains encodes activation. Proc. Natl. Acad. Sci. USA 2009, 106, 6369–6374. [Google Scholar] [CrossRef] [PubMed]
- Weigert, F. Über polarisiertes fluoreszenzlicht. In Verhandlungen der Deutschen Physikalischen Gesellschaft; Physik-Verlag: Weinheim, Germany, 1920; Volume 23, p. 100. [Google Scholar]
- Gaviola, E.; Pringsheim, P. Über den Einfluß der Konzentration auf die Polarisation der Fluoreszenz von Farbstofflösungen. Z. Phys. 1924, 24, 24–36. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare energiewanderung und fluo-reszenz., English translation. Ann. Phys. 1948, 6, 54–75. [Google Scholar]
- Clegg, R.M. The history of FRET. In Reviews in Fluorescence 2006; Springer: Berlin, Germany, 2006; pp. 1–45. [Google Scholar]
- Axelrod, D. Fluorescence polarization microscopy. Methods Cell Biol. 1989, 30, 333–352. [Google Scholar] [PubMed]
- Clayton, A.H.; Hanley, Q.S.; Arndt-Jovin, D.J.; Subramaniam, V.; Jovin, T.M. Dynamic fluorescence anisotropy imaging microscopy in the frequency domain (rFLIM). Biophys. J. 2002, 83, 1631–1649. [Google Scholar] [CrossRef]
- Inoué, S.; Shimomura, O.; Goda, M.; Shribak, M.; Tran, P.T. Fluorescence polarization of green fluorescence protein. Proc. Natl. Acad. Sci. USA 2002, 99, 4272–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattheyses, A.L.; Kampmann, M.; Atkinson, C.E.; Simon, S.M. Fluorescence anisotropy reveals order and disorder of protein domains in the nuclear pore complex. Biophys. J. 2010, 99, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.W.; Rizzo, M.A. FRET by fluorescence polarization microscopy. Methods Cell Biol. 2008, 85, 415–430. [Google Scholar] [PubMed]
- Rizzo, M.A.; Piston, D.W. High-contrast imaging of fluorescent protein FRET by fluorescence polarization microscopy. Biophys. J. 2005, 88, L14–L16. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Enderle, T.; Chemla, S.; Selvin, R.; Weiss, S. Single Molecule Dynamics Studied by Polarization Modulation. Phys. Rev. Lett. 1996, 77, 3979–3982. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A.C. Polarizations and assignments of transitions: The method of photoselection. J. Mol. Spectrosc. 1961, 6, 84–108. [Google Scholar] [CrossRef]
- Swaminathan, R.; Hoang, C.P.; Verkman, A.S. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: Cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 1997, 72, 1900–1907. [Google Scholar] [CrossRef]
- Bader, A.N.; Hofman, E.G.; van Bergen En Henegouwen, P.M.; Gerritsen, H.C. Imaging of protein cluster sizes by means of confocal time-gated fluorescence anisotropy microscopy. Opt. Express 2007, 15, 6934–6945. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.W.; Hink, M.A.; van Hoek, A.; Visser, A.J. Effects of refractive index and viscosity on fluorescence and anisotropy decays of enhanced cyan and yellow fluorescent proteins. J. Fluoresc. 2005, 15, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.L.; Tenner, B.; Markwardt, M.L.; Zviman, A.; Shi, G.; Kerr, J.P.; Snell, N.E.; McFarland, J.J.; Mauban, J.R.; Ward, C.W.; et al. Single-color, ratiometric biosensors for detecting signaling activities in live cells. eLife 2018, 7, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Wouters, F.S.; Bastiaens, P.I. Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells. Curr. Biol. 1999, 9, 1127–1130. [Google Scholar] [CrossRef]
- Hoppe, A.; Christensen, K.; Swanson, J.A. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 2002, 83, 3652–3664. [Google Scholar] [CrossRef]
- Wouters, F.S.; Bastiaens, P.I. Imaging protein-protein interactions by fluorescence resonance energy transfer (FRET) microscopy. Curr. Protoc. Cell Biol. 2001. [Google Scholar] [CrossRef]
- Zimmermann, T.; Rietdorf, J.; Girod, A.; Georget, V.; Pepperkok, R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett. 2002, 531, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Pathway, S.; Pisterzi, L.F.; Biener, G.; Holz, J.D.; Oliver, J.A.; Wells, J.W.; Raicu, V. Experimental verification of the kinetic theory of FRET using optical microspectroscopy and obligate oligomers. Biophys. J. 2015, 108, 1613–1622. [Google Scholar]
- Gautier, I.; Tramier, M.; Durieux, C.; Coppey, J.; Pansu, R.B.; Nicolas, J.C.; Kemnitz, K.; Coppey-Moisan, M. Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins. Biophys. J. 2001, 80, 3000–3008. [Google Scholar] [CrossRef]
- Sharma, P.; Varma, R.; Sarasij, R.C.; Ira; Gousset, K.; Krishnamoorthy, G.; Rao, M.; Mayor, S. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell 2004, 116, 577–589. [Google Scholar] [CrossRef]
- Runnels, L.W.; Scarlata, S.F. Theory and application of fluorescence homotransfer to melittin oligomerization. Biophys. J. 1995, 69, 1569–1583. [Google Scholar] [CrossRef] [Green Version]
- Bader, A.N.; Hoetzl, S.; Hofman, E.G.; Voortman, J.; van Bergen en Henegouwen, P.M.; van Meer, G.; Gerritsen, H.C. Homo-FRET imaging as a tool to quantify protein and lipid clustering. ChemPhysChem 2011, 12, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Lidke, K.A.; Rieger, B.; Lidke, D.S.; Jovin, T.M. The role of photon statistics in fluorescence anisotropy imaging. IEEE Trans. Image Process. 2005, 14, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, M.L.; Snell, N.E.; Guo, M.; Wu, Y.; Christensen, R.; Liu, H.; Shroff, H.; Rizzo, M.A. A Genetically Encoded Biosensor Strategy for Quantifying Non-Muscle Myosin II Phosphorylation Dynamics in Living Cells and Organisms. Cell Rep. 2018, 24, 1060–1070.E4. [Google Scholar] [CrossRef] [PubMed]
- Tramier, M.; Piolot, T.; Gautier, I.; Mignotte, V.; Coppey, J.; Kemnitz, K.; Durieux, C.; Coppey-Moisan, M. Homo-FRET versus hetero-FRET to probe homodimers in living cells. Methods Enzymol. 2003, 360, 580–597. [Google Scholar] [PubMed]
- Nguyen, T.A.; Sarkar, P.; Veetil, J.V.; Koushik, S.V.; Vogel, S.S. Fluorescence polarization and fluctuation analysis monitors subunit proximity, stoichiometry, and protein complex hydrodynamics. PLoS ONE 2012, 7, e38209. [Google Scholar] [CrossRef] [PubMed]
- Volkmer, A.; Subramaniam, V.; Birch, D.J.; Jovin, T.M. One- and two-photon excited fluorescence lifetimes and anisotropy decays of green fluorescent proteins. Biophys. J. 2000, 78, 1589–1598. [Google Scholar] [CrossRef]
- Zhao, Q.; Young, I.T.; De Jong, J.G.S. Photon budget analysis for fluorescence lifetime imaging microscopy. J. Biomed. Opt. 2011, 16, 086007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, W.D.; Bui, C.V.; Hutchinson, A.; Loppnau, P.; Gräslund, S.; Rocheleau, J.V. Apollo-NADP(+): A spectrally tunable family of genetically encoded sensors for NADP(+). Nat. Methods 2016, 13, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Vishwasrao, H.D.; Trifilieff, P.; Kandel, E.R. In vivo imaging of the actin polymerization state with two-photon fluorescence anisotropy. Biophys. J. 2012, 102, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Tramier, M. FRET microscopy in the living cell: Different approaches, strengths and weaknesses. BioEssays 2012, 34, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Ghitani, A.; Christensen, R.; Santella, A.; Du, Z.; Rondeau, G.; Bao, Z.; Colón-Ramos, D.; Shroff, H. Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2011, 108, 17708–17713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedde, P.N.; Ranjit, S.; Gratton, E. 3D fluorescence anisotropy imaging using selective plane illumination microscopy. Opt. Express 2015, 23, 22308–22317. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Arai, Y.; Tani, T.; Takatsuka, H.; Saito, Y.; Kawashima, T.; Kawakami, S.; Miyawaki, A.; Nagai, T. Simultaneous imaging of multiple cellular events using high-accuracy fluorescence polarization microscopy. Microscopy 2017, 66, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Seckinger, K.M.; Rao, V.P.; Snell, N.E.; Mancini, A.E.; Markwardt, M.L.; Rizzo, M.A. Nitric Oxide Activates β-Cell Glucokinase by Promoting Formation of the “Glucose-Activated” State. Biochemistry 2018, 57, 5136–5144. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.A.; Puhl, H.L.; Pham, A.K.; Vogel, S.S. Auto-FPFA: An Automated Microscope for Characterizing Genetically Encoded Biosensors. Sci. Rep. 2018, 8, 7374. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, M.L.; Seckinger, K.M.; Rizzo, M.A. Regulation of Glucokinase by Intracellular Calcium Levels in Pancreatic β Cells. J. Biol. Chem. 2016, 291, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Patterson, G.H.; Davidson, M.W. Advances in fluorescent protein technology. J. Cell Sci. 2007, 120, 4247–4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varma, R.; Mayor, S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 1998, 394, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Warren, S.C.; Margineanu, A.; Katan, M.; Dunsby, C.; French, P.M. Homo-FRET Based Biosensors and Their Application to Multiplexed Imaging of Signalling Events in Live Cells. Int. J. Mol. Sci. 2015, 16, 14695–14716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeow, E.K.; Clayton, A.H. Enumeration of oligomerization states of membrane proteins in living cells by homo-FRET spectroscopy and microscopy: Theory and application. Biophys. J. 2007, 92, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.N.; Hofman, E.G.; Voortman, J.; en Henegouwen, P.M.; Gerritsen, H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009, 97, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, D.M.; Cox Cammer, N.; Huisman, T.M.; Verkhusha, V.V.; Hodgson, L. Direct multiplex imaging and optogenetics of Rho GTPases enabled by near-infrared FRET. Nat. Chem. Biol. 2018, 14, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Drobizhev, M.; Makarov, N.S.; Tillo, S.E.; Hughes, T.E.; Rebane, A. Two-photon absorption properties of fluorescent proteins. Nat. Methods 2011, 8, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, G.; Szellas, T.; Huhn, W.; Kateriya, S.; Adeishvili, N.; Berthold, P.; Ollig, D.; Hegemann, P.; Bamberg, E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möglich, A.; Moffat, K. Engineered photoreceptors as novel optogenetic tools. Photochem. Photobiol. Sci. 2010, 9, 1286–1300. [Google Scholar] [CrossRef] [PubMed]
- Strickland, D.; Moffat, K.; Sosnick, T.R. Light-activated DNA binding in a designed allosteric protein. Proc. Natl. Acad. Sci. USA 2008, 105, 10709–10714. [Google Scholar] [CrossRef] [PubMed]
- Möglich, A.; Ayers, R.A.; Moffat, K. Design and signaling mechanism of light-regulated histidine kinases. J. Mol. Biol. 2009, 385, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Sadaghiani, A.M.; Hsueh, B.; Dolmetsch, R.E. Induction of protein-protein interactions in live cells using light. Nat. Biotechnol. 2009, 27, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Niopek, D.; Benzinger, D.; Roensch, J.; Draebing, T.; Wehler, P.; Eils, R.; Di Ventura, B. Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat. Commun. 2014, 5, 4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoltowski, B.D.; Vaccaro, B.; Crane, B.R. Mechanism-based tuning of a LOV domain photoreceptor. Nat. Chem. Biol. 2009, 5, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Pudasaini, A.; El-Arab, K.K.; Zoltowski, B.D. LOV-based optogenetic devices: Light-driven modules to impart photoregulated control of cellular signaling. Front. Mol. Biosci. 2015, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Bhupathy, P.; Babu, G.J. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc. Res. 2008, 77, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Desmond, P.F.; Labuza, A.; Muriel, J.; Markwardt, M.L.; Mancini, A.E.; Rizzo, M.A.; Bloch, R.J. Interactions between small ankyrin 1 and sarcolipin coordinately regulate activity of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA1). J. Biol. Chem. 2017, 292, 10961–10972. [Google Scholar] [CrossRef] [PubMed]
- Pau, G.; Fuchs, F.; Sklyar, O.; Boutros, M.; Huber, W. EBImage—An R package for image processing with applications to cellular phenotypes. Bioinformatics 2010, 26, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Liu, Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001, 81, 2395–2402. [Google Scholar] [CrossRef]
- Zal, T.; Gascoigne, N.R. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 2004, 86, 3923–3939. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sensor | Color | Reference |
|---|---|---|
| Protein Kinase A (AKAR) | cyan fluorescent protein (CFP), yellow fluorescent protein (YFP), green fluorescent protein (GFP), mCherry | [34] |
| Cyclic adenosine monophosphate (ICUE3) | YFP | [34] |
| Calcium (Cameleon) | CFP, YFP, mCherry | [34,55] |
| Calcium (Twitch-4) | YFP | [58] |
| ER Calcium (D1) | CFP | [34] |
| Myosin Light Chain Kinase | YFP | [34] |
| Protein Kinase C (CKAR) | CFP, YFP, mCherry | [34] |
| Mitogen-Activated Protein Kinase (EKAR) | CFP, YFP, mCherry | [34] |
| Glucokinase | YFP | [56] |
| Sensor | Color | Reference |
|---|---|---|
| Actin | GFP | [51] |
| Non-muscle myosin II | CFP, GFP, mCherry | [45] |
| Ca2+/calmodulin-dependent protein kinase II | YFP | [18,47] |
| Akt pleckstrin homology domain | mCherry | [63] |
| glycophosphatidylinositol | GFP | [32] |
| Epidermal growth factor receptor | GFP | [65] |
| Nicotinamide adenine dinucleotide phosphate (Apollo) | CFP, YFP | [50] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snell, N.E.; Rao, V.P.; Seckinger, K.M.; Liang, J.; Leser, J.; Mancini, A.E.; Rizzo, M.A. Homotransfer FRET Reporters for Live Cell Imaging. Biosensors 2018, 8, 89. https://doi.org/10.3390/bios8040089
Snell NE, Rao VP, Seckinger KM, Liang J, Leser J, Mancini AE, Rizzo MA. Homotransfer FRET Reporters for Live Cell Imaging. Biosensors. 2018; 8(4):89. https://doi.org/10.3390/bios8040089
Chicago/Turabian StyleSnell, Nicole E., Vishnu P. Rao, Kendra M. Seckinger, Junyi Liang, Jenna Leser, Allison E. Mancini, and M. A. Rizzo. 2018. "Homotransfer FRET Reporters for Live Cell Imaging" Biosensors 8, no. 4: 89. https://doi.org/10.3390/bios8040089
APA StyleSnell, N. E., Rao, V. P., Seckinger, K. M., Liang, J., Leser, J., Mancini, A. E., & Rizzo, M. A. (2018). Homotransfer FRET Reporters for Live Cell Imaging. Biosensors, 8(4), 89. https://doi.org/10.3390/bios8040089