Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance

 , , and
, , and
Abstract
:
1. Introduction
2. Basis of Biofilm-Mediated Antibiotic Survival
2.1. Biogenesis of Biofilms
2.2. Composition of the ECM
2.3. Biofilm Architecture
3. Mechanisms of Biofilm Recalcitrance
3.1. Types of Antibiotic Recalcitrance
3.2. The Protective ECM Barrier
3.3. Physiological Heterogeneity
3.4. Traffic of Substances across the Cell Envelope
3.5. Interbacterial Communication
3.6. HGT in Biofilms
3.7. Mutation and Biofilms
4. Control of Biofilm Infections
4.1. Lessons from Recalcitrant Mechanisms
4.2. Antimicrobial Substances
4.3. Alternative Methods
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; the Review on Antimicrobial Resistance. 2016. Available online: https://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf (accessed on 5 August 2020).
- WHO (World Health Organization). High Levels of Antibiotic Resistance Found Worldwide, New Data Shows. 2018. Available online: https://www.who.int/news-room/detail/29-01-2018-high-levels-of-antibiotic-resistance-found-worldwide-new-data-shows (accessed on 8 August 2020).
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control). The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 2020, 18, e06007. [Google Scholar] [CrossRef] [Green Version]
- Collignon, P. Fluoroquinolone use in food animals. Emerg. Infect. Dis. 2005, 11, 1789–1790. [Google Scholar] [CrossRef] [Green Version]
- Landers, T.F.; Cohen, B.; Wittum, T.E.; Larson, E.L. A review of antibiotic use in food animals: Perspective, policy, and potential. Public Health Rep. 2012, 127, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaux, D.; Ghigo, J.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceri, H.; Olson, M.E.; Stremick, C.; Read, R.R.; Morck, D.; Buret, A. The calgary biofilm device: New technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 1999, 37, 1771–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yassien, M.; Khardori, N.; Ahmedy, A.; Toama, M. Modulation of biofilms of Pseudomonas aeruginosa by quinolones. Antimicrob. Agents Chemother. 1995, 39, 2262–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morck, D.W.; Lam, K.; McKay, S.G.; Olson, M.E.; Prosser, B.; Ellis, B.D.; Cleeland, R.; Costerton, J.W. Comparative evaluation of fleroxacin, ampicillin, trimethoprimsulfamethoxazole, and gentamicin as treatments of catheter-associated urinary tract infection in a rabbit model. Int. J. Antimicrob. Agents 1994, 4, S21–S27. [Google Scholar] [CrossRef]
- Olsen, I. Biofilm-specific antibiotic tolerance and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 877–886. [Google Scholar] [CrossRef]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef]
- Maurice, N.M.; Bedi, B.; Sadikot, R.T. Pseudomonas aeruginosa biofilms: Host response and clinical implications in lung infections. Am. J. Resp. Cell Mol. 2018, 58, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Vuotto, C.; Donelli, G. Biofilm formation in Acinetobacter baumannii. New Microbiol. 2014, 37, 119–127. [Google Scholar] [PubMed]
- Olivares, E.; Badel-Berchoux, S.; Provot, C.; Prévost, G.; Bernardi, T.; Jehl, F. Clinical impact of antibiotics for the treatment of Pseudomonas aeruginosa biofilm infections. Front. Microbiol. 2020, 10, 2894. [Google Scholar] [CrossRef] [PubMed]
- Stickler, D.J. Bacterial biofilms in patients with indwelling urinary catheters. Nat. Clin. Pract. Urol. 2008, 5, 598–608. [Google Scholar] [CrossRef]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [Green Version]
- Falsetta, M.L.; Steichen, C.T.; McEwan, A.G.; Cho, C.; Ketterer, M.; Shao, J.; Hunt, J.; Jennings, M.P.; Apicella, M.A. The composition and metabolic phenotype of Neisseria gonorrhoeae biofilms. Front. Microbiol. 2011, 2, 75. [Google Scholar] [CrossRef] [Green Version]
- WHO (World Health Organization). Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. 2017. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 5 August 2020).
- López, D.; Vlamakis, H.; Kolter, R. Biofilms. CSH Perspect. Biol. 2010, 2, a000398. [Google Scholar] [CrossRef]
- Davies, D.G.; Parsek, M.R.; Pearson, J.P.; Iglewski, B.H.; Costerton, J.W.; Greenberg, E.P. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 1998, 280, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Prigent-Combaret, C.; Prensier, G.; Le Thi, T.T.; Vidal, O.; Lejeune, P.; Dorel, C. Developmental pathway for biofilm formation in curli-producing Escherichia coli strains: Role of flagella, curli and colanic acid. Environ. Microbiol. 2000, 2, 450–464. [Google Scholar] [CrossRef]
- Tormo, M.A.; Knecht, E.; Götz, F.; Lasa, I.; Penades, J.R. Bap-dependent biofilm formation by pathogenic species of Staphylococcus: Evidence of horizontal gene transfer? Microbiology 2005, 151, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Borlee, B.R.; Goldman, A.D.; Murakami, K.; Samudrala, R.; Wozniak, D.J.; Parsek, M.R. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 2010, 75, 827–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grijpstra, J.; Arenas, J.; Rutten, L.; Tommassen, J. Autotransporter secretion: Varying on a theme. Res. Microbiol. 2013, 164, 562–582. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, G.A.; Kolter, R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 1998, 30, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Tommassen, J.; Arenas, J. Biological functions of the secretome of Neisseria meningitidis. Front. Cell. Infect. Microbiol. 2017, 7, 256. [Google Scholar] [CrossRef]
- Muñoz-Egea, M.C.; García-Pedrazuela, M.; Mahillo-Fernandez, I.; Esteban, J. Effect of antibiotics and antibiofilm agents in the ultrastructure and development of biofilms developed by non pigmented rapidly growing Mycobacteria. Microb. Drug Resist. 2016, 22, 1–6. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Das, T.; Sharma, P.K.; Busscher, H.J.; van der Mei, H.C. Role of extracellular DNA in initial bacterial adhesion and surface aggregation. Appl. Environ. Microbiol. 2010, 76, 3405–3408. [Google Scholar] [CrossRef] [Green Version]
- Das, T.; Sharma, P.K.; Krom, B.P.; van der Mei, H.C.; Busscher, H.J. Role of eDNA on the adhesion forces between Streptococcus mutans and substratum surfaces: Influence of ionic strength and substratum hydrophobicity. Langmuir 2011, 27, 10113–10118. [Google Scholar] [CrossRef]
- Qin, Z.; Ou, Y.; Yang, L.; Zhu, Y.; Tolker-Nielsen, T.; Molin, S.; Qu, D. Role of autolysin-mediated DNA release in biofilm formation of Staphylococcus epidermidis. Microbiology 2007, 153, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Arenas, J.; Nijland, R.; Rodriguez, F.J.; Bosma, T.N.; Tommassen, J. Involvement of three meningococcal surface-exposed proteins, the heparin-binding protein NhbA, the α-peptide of IgA protease and the autotransporter protease NalP, in initiation of biofilm formation. Mol. Microbiol. 2013, 87, 254–268. [Google Scholar] [CrossRef]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- Arenas, J.; Tommassen, J. Meningococcal biofilm formation: Let’s stick together. Trends Microbiol. 2017, 25, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Castillo Pedraza, M.C.; Novais, T.F.; Faustoferri, R.C.; Quivey, R.G., Jr.; Terekhov, A.; Hamaker, B.R.; Klein, M.I. Extracellular DNA and lipoteichoic acids interact with exopolysaccharides in the extracellular matrix of Streptococcus mutans biofilms. Biofouling 2017, 33, 722–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Preston, J.F.; Romeo, T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 2004, 186, 2724–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izano, E.A.; Amarante, M.A.; Kher, W.B.; Kaplan, J.B. Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl. Environ. Microbiol. 2008, 74, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Da Re, S.; Ghigo, J.M. A CsgD-independent pathway for cellulose production and biofilm formation in Escherichia coli. J. Bacteriol. 2006, 188, 3073–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solano, C.; García, B.; Valle, J.; Berasain, C.; Ghigo, J.M.; Gamazo, C.; Lasa, I. Genetic analysis of Salmonella enteritidis biofilm formation: Critical role of cellulose. Mol. Microbiol. 2002, 43, 793–808. [Google Scholar] [CrossRef]
- Ude, S.; Arnold, D.L.; Moon, C.D.; Timms-Wilson, T.; Spiers, A.J. Biofilm formation and cellulose expression among diverse environmental Pseudomonas isolates. Environ. Microbiol. 2006, 8, 1997–2011. [Google Scholar] [CrossRef]
- Stevenson, G.; Andrianopoulos, K.; Hobbs, M.; Reeves, P.R. Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J. Bacteriol. 1996, 178, 4885–4893. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.; Chan, R.; Lam, K.; Costerton, J.W. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect. Immun. 1980, 28, 546–556. [Google Scholar]
- Hentzer, M.; Teitzel, G.M.; Balzer, G.J.; Heydorn, A.; Molin, S.; Givskov, M.; Parsek, M.R. Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J. Bacteriol. 2001, 183, 5395–5401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulcrano, G.; Iula, D.V.; Raia, V.; Rossano, F.; Catania, M.R. Different mutations in mucA gene of Pseudomonas aeruginosa mucoid strains in cystic fibrosis patients and their effect on algU gene expression. New Microbiol. 2012, 35, 295–305. [Google Scholar] [PubMed]
- Jennings, L.K.; Storek, K.M.; Ledvina, H.E.; Coulon, C.; Marmont, L.S.; Sadovskaya, I.; Secor, P.R.; Tseng, B.S.; Scian, M.; Filloux, A.; et al. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl. Acad. Sci. USA 2015, 112, 11353–11358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, M.S.; Sadovskaya, I.; Vinogradov, E.; Lu, H.; Sprinkle, A.B.; Richardson, S.H.; Ma, L.; Ralston, B.; Parsek, M.R.; Anderson, E.M.; et al. Genetic and biochemical analyses of the Pseudomonas aeruginosa Psl exopolysaccharide reveal overlapping roles for polysaccharide synthesis enzymes in Psl and LPS production. Mol. Microbiol. 2009, 73, 622–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Jackson, K.D.; Landry, R.M.; Parsek, M.R.; Wozniak, D.J. Analysis of Pseudomonas aeruginosa conditional Psl variants reveals roles for the Psl polysaccharide in adhesion and maintaining biofilm structure post attachment. J. Bacteriol. 2006, 188, 8213–8221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lequette, Y.; Rollet, E.; Delangle, A.; Greenberg, E.P.; Bohin, J.P. Linear osmoregulated periplasmic glucans are encoded by the opgGH locus of Pseudomonas aeruginosa. Microbiology 2007, 153, 3255–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulon, C.; Vinogradov, E.; Filloux, A.; Sadovskaya, I. Chemical analysis of cellular and extracellular carbohydrates of a biofilm-forming strain Pseudomonas aeruginosa PA14. PLoS ONE 2010, 5, e14220. [Google Scholar] [CrossRef]
- Bridier, A.; Dubois-Brissonnet, F.; Boubetra, A.; Thomas, V.; Briandet, R. The biofilm architecture of sixty opportunistic pathogens deciphered using a high throughput CLSM method. J. Microbiol. Meth. 2010, 82, 64–70. [Google Scholar] [CrossRef]
- Arenas, J.; Cano, S.; Nijland, R.; van Dongen, V.; Rutten, L.; van der Ende, A.; Tommassen, J. The meningococcal autotransporter AutA is implicated in autoaggregation and biofilm formation. Environ. Microbiol. 2014, 17, 1321–1337. [Google Scholar] [CrossRef] [Green Version]
- Arenas, J.; Paganelli, F.L.; Rodríguez-Castaño, P.; Cano-Crespo, S.; van der Ende, A.; van Putten, J.P.; Tommassen, J. Expression of the gene for autotransporter AutB of Neisseria meningitidis affects biofilm formation and epithelial transmigration. Front. Cell. Infect. Microbiol. 2016, 6, 162. [Google Scholar] [CrossRef] [Green Version]
- Stewart, P.S.; Peyton, B.M.; Drury, W.J.; Murga, R. Quantitative observations of heterogeneities in Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 1993, 59, 327–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Weeks, J.W.; Ntreh, A.T.; Nickels, L.M.; Wolloscheck, D. Mechanism of coupling drug transport reactions located in two different membranes. Front. Microbiol. 2015, 6, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arzanlou, M.; Chai, W.C.; Venter, H. Intrinsic, adaptive and acquired antimicrobial resistance in Gram-negative bacteria. Essays Biochem. 2017, 61, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Amado, S.; Sanz-Garcia, F.; Blanco, P.; Martinez, J.L. Fitness costs associated with the acquisition of antibiotic resistance. Essays Biochem. 2017, 61, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Breidenstein, E.B.; Hancock, R.E. Creeping baselines and adaptive resistance to antibiotics. Drug Resist. Update 2011, 14, 1–21. [Google Scholar] [CrossRef]
- El-Halfawy, O.M.; Valvano, M.A. Antimicrobial heteroresistance: An emerging field in need of clarity. Clin. Microbiol. Rev. 2015, 28, 191–207. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I.; Nicoloff, H.; Hjort, K. Mechanisms and clinical relevance of bacterial heteroresistance. Nat. Rev. Microbiol. 2019, 17, 479–496. [Google Scholar] [CrossRef]
- Band, V.I.; Crispell, E.K.; Napier, B.A.; Herrera, C.M.; Tharp, G.K.; Vavikolanu, K.; Pohl, J.; Read, T.D.; Bosinger, S.E.; Trent, M.S.; et al. Antibiotic failure mediated by a resistant subpopulation in Enterobacter cloacae. Nat. Microbiol. 2016, 1, 1–9. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, J.J.; et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019, 17, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K. Persister cellules, la dormance et les maladies infectieuses. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Wilmaerts, D.; Windels, E.M.; Verstraeten, N.; Michiels, J. General mechanisms leading to persister formation and awakening. Trends Genet. 2019, 35, 401–411. [Google Scholar] [CrossRef] [PubMed]
- De Beer, D.; Stoodley, P.; Lewandowski, Z. Liquid flow in heterogeneous biofilms. Biotechnol. Bioeng. 1994, 44, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S. Diffusion in biofilms. J. Bacteriol. 2003, 185, 1485–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadovskaya, I.; Vinogradov, E.; Li, J.; Hachani, A.; Kowalska, K.; Filloux, A. High-level antibiotic resistance in Pseudomonas aeruginosa biofilm: The ndvB gene is involved in the production of highly glycerol-phosphorylated β-(1-3)-glucans, which bind aminoglycosides. Glycobiology 2010, 20, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, K.M.; Gordon, V.D.; Murakami, K.; Borlee, B.R.; Wozniak, D.J.; Wong, G.C.; Parsek, M.R. The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011, 7, e1001264. [Google Scholar] [CrossRef]
- Wang, S.; Liu, X.; Liu, H.; Zhang, L.; Guo, Y.; Yu, S.; Wozniak, D.J.; Ma, L.Z. The exopolysaccharide Psl–eDNA interaction enables the formation of a biofilm skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Billings, N.; Millan, M.R.; Caldara, M.; Rusconi, R.; Tarasova, Y.; Stocker, R.; Ribbeck, K. The extracellular matrix component Psl provides fast-acting antibiotic defense in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2013, 9, e1003526. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [Green Version]
- Wilton, M.; Charron-Mazenod, L.; Moore, R.; Lewenza, S. Extracellular DNA acidifies biofilms and induces aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 544–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doroshenko, N.; Tseng, B.S.; Howlin, R.P.; Deacon, J.; Wharton, J.A.; Thurner, P.J.; Gilmore, B.F.; Parsek, M.R.; Stoodley, P. Extracellular DNA impedes the transport of vancomycin in Staphylococcus epidermidis biofilms preexposed to subinhibitory concentrations of vancomycin. Antimicrob. Agents Chemother. 2014, 58, 7273–7282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.; Horsman, S.R.; Charron-Mazenod, L.; Turnbull, A.L.; Mulcahy, H.; Surette, M.G.; Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2013, 13, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.D. Mechanism of bactericidal action of aminoglycosides. Microbiol. Rev. 1987, 51, 341–350. [Google Scholar] [CrossRef]
- Taber, H.W.; Mueller, J.P.; Miller, P.F.; Arrow, A.S. Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev. 1987, 51, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, R.E. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 1984, 38, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.C.; Pang, B.; King, L.B.; Tan, L.; Murrah, K.A.; Reimche, J.L.; Wren, J.T.; Richardson, S.H.; Ghandi, U.; Swords, W.E. Residence of Streptococcus pneumoniae and Moraxella catarrhalis within polymicrobial biofilm promotes antibiotic resistance and bacterial persistence in vivo. Pathog. Dis. 2014, 70, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Armbruster, C.E.; Hong, W.; Pang, B.; Weimer, K.E.; Juneau, R.A.; Turner, J.; Swords, W.E. Indirect pathogenicity of Haemophilus influenzae and Moraxella catarrhalis in polymicrobial otitis media occurs via interspecies quorum signaling. mBio 2010, 1, e00102-10. [Google Scholar] [CrossRef] [Green Version]
- Flemming, H.C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Stewart, P.S.; Franklin, M.J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Borriello, G.; Richards, L.; Ehrlich, G.D.; Stewart, P.S. Arginine or nitrate enhances antibiotic susceptibility of Pseudomonas aeruginosa in biofilms. Antimicrob. Agents Chemother. 2006, 50, 382–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haagensen, J.A.; Klausen, M.; Ernst, R.K.; Miller, S.I.; Folkesson, A.; Tolker-Nielsen, T.; Molin, S. Differentiation and distribution of colistin-and sodium dodecyl sulfate-tolerant cells in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2007, 189, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushwaha, G.S.; Oyeyemi, B.F.; Bhavesh, N.S. Stringent response protein as a potential target to intervene persistent bacterial infection. Biochimie 2019, 165, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kusser, W.; Ishiguro, E.E. Suppression of mutations conferring penicillin tolerance by interference with the stringent control mechanism of Escherichia coli. J. Bacteriol. 1987, 169, 4396–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuomanen, E.; Tomasz, A. Induction of autolysis in nongrowing Escherichia coli. J. Bacteriol. 1986, 167, 1077–1080. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; He, L.; Cui, P.; Wang, W.; Yuan, Y.; Liu, S.; Xu, T.; Zhang, S.; Wu, J.; Zhang, W.; et al. Ranking of persister genes in the same Escherichia coli genetic background demonstrates varying importance of individual persister genes in tolerance to different antibiotics. Front. Microbiol. 2015, 6, 1003. [Google Scholar] [CrossRef] [Green Version]
- Bernier, S.P.; Lebeaux, D.; DeFrancesco, A.S.; Valomon, A.; Soubigou, G.; Coppée, J.Y.; Ghigo, J.M.; Beloin, C. Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 2013, 9, e1003144. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [Green Version]
- Khakimova, M.; Ahlgren, H.G.; Harrison, J.J.; English, A.M.; Nguyen, D. The stringent response controls catalases in Pseudomonas aeruginosa and is required for hydrogen peroxide and antibiotic tolerance. J. Bacteriol. 2013, 195, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Martins, D.; McKay, G.; Sampathkumar, G.; Khakimova, M.; English, A.M.; Nguyen, D. Superoxide dismutase activity confers (p) ppGpp-mediated antibiotic tolerance to stationary-phase Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2018, 115, 9797–9802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viducic, D.; Ono, T.; Murakami, K.; Susilowati, H.; Kayama, S.; Hirota, K.; Miyake, Y. Functional analysis of spoT, relA and dksA genes on quinolone tolerance in Pseudomonas aeruginosa under nongrowing condition. Microbiol. Immunol. 2006, 50, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Kästle, B.; Gratani, F.L.; Goerke, C.; Wolz, C. Two small (p) ppGpp synthases in Staphylococcus aureus mediate tolerance against cell envelope stress conditions. J. Bacteriol. 2014, 196, 894–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslowska, K.H.; Makiela-Dzbenska, K.; Fijalkowska, I.J. The SOS system: A complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 2019, 60, 368–384. [Google Scholar] [CrossRef] [Green Version]
- Dörr, T.; Lewis, K.; Vulić, M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 2009, 5, e1000760. [Google Scholar] [CrossRef] [Green Version]
- Baharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, K.; Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef]
- Keren, I.; Shah, D.; Spoering, A.; Kaldalu, N.; Lewis, K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004, 186, 8172–8180. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Zhang, Z.; Khodursky, A.; Kaldalu, N.; Kurg, K.; Lewis, K. Persisters: A distinct physiological state of E. coli. BMC Microbiol. 2006, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- González Barrios, A.F.; Zuo, R.; Hashimoto, Y.; Yang, L.; Bentley, W.E.; Wood, T.K. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 2006, 188, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraikin, N.; Rousseau, C.J.; Goeders, N.; Van Melderen, L. Reassessing the role of the type II MqsRA toxin-antitoxin system in stress response and biofilm formation: mqsA is transcriptionally uncoupled from mqsR. mBio 2019, 10, e02678-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelberg-Kulka, H.; Amitai, S.; Kolodkin-Gal, I.; Hazan, R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006, 2, e135. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.A.F.; Waldor, M.K. A toxin–antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 2009, 5, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Bao, X.; Ji, L.; Chen, L.; Liu, J.; Miao, J.; Chen, D.; Bian, H.; Li, Y.; Yu, G. Resistance integrons: Class 1, 2 and 3 integrons. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillings, M.R. Integrons: Past, present, and future. Microbiol. Mol. Biol. Rev. 2014, 78, 257–277. [Google Scholar] [CrossRef] [Green Version]
- Strugeon, E.; Tilloy, V.; Ploy, M.C.; Da Re, S. The stringent response promotes antibiotic resistance dissemination by regulating integron integrase expression in biofilms. mBio 2016, 7, e00868-16. [Google Scholar] [CrossRef] [Green Version]
- Borriello, G.; Werner, E.; Roe, F.; Kim, A.M.; Ehrlich, G.D.; Stewart, P.S. Oxygen limitation contributes to antibiotic tolerance of Pseudomonas aeruginosa in biofilms. Antimicrob. Agents Chemother. 2004, 48, 2659–2664. [Google Scholar] [CrossRef] [Green Version]
- James, G.A.; Ge Zhao, A.; Usui, M.; Underwood, R.A.; Nguyen, H.; Beyenal, H.; DeLancey, E.; Hunt, A.A.; Bernstein, H.C.; Fleckman, P.; et al. Microsensor and transcriptomic signatures of oxygen depletion in biofilms associated with chronic wounds. Wound Repair Regen. 2016, 24, 373–383. [Google Scholar] [CrossRef]
- Kiamco, M.M.; Atci, E.; Mohamed, A.; Call, D.R.; Beyenal, H. Hyperosmotic agents and antibiotics affect dissolved oxygen and pH concentration gradients in Staphylococcus aureus biofilms. Appl. Environ. Microbiol. 2017, 83, e02783-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, K.R.; Pozdin, V.A.; Young, A.T.; Erb, P.D.; Wisniewski, N.A.; Magness, S.T.; Daniele, M. Integrated phosphorescence-based photonic biosensor (iPOB) for monitoring oxygen levels in 3D cell culture systems. Biosens. Bioelectron. 2019, 123, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kolpen, M.; Kühl, M.; Bjarnsholt, T.; Moser, C.; Hansen, C.R.; Liengaard, L.; Kharazmi, A.; Pressler, T.; Høiby, N.; Jensen, P.Ø. Nitrous oxide production in sputum from cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. PLoS ONE 2014, 9, e84353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tack, K.J.; Sabath, L.D. Increased minimum inhibitory concentrations with anaerobiasis for tobramycin, gentamicin, and amikacin, compared to latamoxef, piperacillin, chloramphenicol, and clindamycin. Chemotherapy 1985, 31, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J.; Williamson, K.S.; Folsom, J.P.; Boegli, L.; James, G.A. Contribution of stress responses to antibiotic tolerance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2015, 59, 3838–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, D.J.; Collins, J.J.; Walker, G.C. Unraveling the physiological complexities of antibiotic lethality. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 313–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.Ø.; Briales, A.; Brochmann, R.P.; Wang, H.; Kragh, K.N.; Kolpen, M.; Hempel, C.; Bjarnsholt, T.; Høiby, N.; Ciofu, O. Formation of hydroxyl radicals contributes to the bactericidal activity of ciprofloxacin against Pseudomonas aeruginosa biofilms. Pathog. Dis. 2014, 70, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Du, D.; van Veen, H.W.; Murakami, S.; Pos, K.M.; Luisi, B.F. Structure, mechanism and cooperation of bacterial multidrug transporters. Curr. Opin. Struc. Biol. 2015, 33, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, T.; Tsuchiya, T. Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 2009, 1794, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Kodama, K.; Shiota, S.; Mine, T.; Kataoka, A.; Mizushima, T.; Tsuchiya, T. NorM, a putative multidrug efflux protein, of Vibrio parahaemolyticus and its homolog in Escherichia coli. Antimicrob. Agents Chemother. 1998, 42, 1778–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.X.; Kuroda, T.; Mima, T.; Morita, Y.; Mizushima, T.; Tsuchiya, T. An H+-coupled multidrug efflux pump, PmpM, a member of the MATE family of transporters, from Pseudomonas aeruginosa. J. Bacteriol. 2004, 186, 262–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.Z.; Chen, J.; Mizushima, T.; Kuroda, T.; Tsuchiya, T. AbeM, an H+-coupled Acinetobacter baumannii multidrug efflux pump belonging to the MATE family of transporters. Antimicrob. Agents Chemother. 2005, 49, 4362–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.; Bay, D.C.; Turner, R.J. Few Conserved amino acids in the small multidrug resistance transporter EmrE influence drug polyselectivity. Antimicrob. Agents Chemother. 2018, 62, e00461-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, I.T.; Brown, M.H.; Dunstan, S.J.; Skurray, R.A. Molecular characterization of the staphylococcal multidrug resistance export protein QacC. J. Bacteriol. 1995, 177, 2827–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousa, J.J.; Bruner, S.D. Structural and mechanistic diversity of multidrug transporters. Nat. Prod. Rep. 2016, 33, 1255–1267. [Google Scholar] [CrossRef]
- Schindler, B.D.; Kaatz, G.W. Multidrug efflux pumps of Gram-positive bacteria. Drug Resist. Updat. 2016, 27, 1–13. [Google Scholar] [CrossRef]
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug efflux pumps in Staphylococcus aureus: An update. Open Microbiol. J. 2013, 7, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Nishino, K.; Yamaguchi, A. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J. Bacteriol. 2001, 183, 5639–5644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, K.A.; Liu, Q.; Henderson, P.J.; Paulsen, I.T. Homologs of the Acinetobacter baumannii AceI transporter represent a new family of bacterial multidrug efflux systems. mBio 2015, 6, e01982-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Chen, M.; Yu, Z.; Bell, J.M.; Wang, H.; Forrester, I.; Villareal, H.; Jakana, J.; Du, D.; Luisi, B.F.; et al. In situ structure and assembly of the multidrug efflux pump AcrAB-TolC. Nat. Commun. 2019, 10, 2635. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Yonehara, R.; Ishizaka-Ikeda, E.; Miyazaki, N.; Maeda, S.; Iwasaki, K.; Nakagawa, A.; Yamashita, E. Structures of the wild-type MexAB-OprM tripartite pump reveal its complex formation and drug efflux mechanism. Nat. Commun. 2019, 10, 1520. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, P.; Sacha, P.; Hauschild, T.; Zórawski, M.; Krawczyk, M.; Tryniszewska, E. Multidrug resistant Acinetobacter baumannii—the role of AdeABC (RND family) efflux pump in resistance to antibiotics. Folia Histochem. Cytobiol. 2008, 46, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xiao, M.; Horiyama, T.; Zhang, Y.; Li, X.; Nishino, K.; Yan, A. The multidrug efflux pump MdtEF protects against nitrosative damage during the anaerobic respiration in Escherichia coli. J. Biol. Chem. 2011, 286, 26576–26584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Woo, J.; Ahn, J. Cellular and molecular responses of Salmonella Typhimurium to antimicrobial-induced stresses during the planktonic-to-biofilm transition. Lett. Appl. Microbiol. 2012, 55, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Kvist, M.; Hancock, V.; Klemm, P. Inactivation of efflux pumps abolishes bacterial biofilm formation. Appl. Environ. Microbiol. 2008, 74, 7376–7382. [Google Scholar] [CrossRef] [Green Version]
- Ikonomidis, A.; Tsakris, A.; Kanellopoulou, M.; Maniatis, A.N.; Pournaras, S. Effect of the proton motive force inhibitor carbonyl cyanide-m-chlorophenylhydrazone (CCCP) on Pseudomonas aeruginosa biofilm development. Lett. Appl. Microbiol. 2008, 47, 298–302. [Google Scholar] [CrossRef]
- Baugh, S.; Phillips, C.R.; Ekanayaka, A.S.; Piddock, L.J.; Webber, M.A. Inhibition of multidrug efflux as a strategy to prevent biofilm formation. J. Antimicrob. Chemother. 2014, 69, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Gillis, R.J.; White, K.G.; Choi, K.H.; Wagner, V.E.; Schweizer, H.P.; Iglewski, B.H. Molecular basis of azithromycin-resistant Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2005, 49, 3858–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamp, S.J.; Gjermansen, M.; Johansen, H.K.; Tolker-Nielsen, T. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 2008, 68, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mah, T.F. Involvement of a novel efflux system in biofilm-specific resistance to antibiotics. J. Bacteriol. 2008, 190, 4447–4452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bay, D.C.; Stremick, C.A.; Slipski, C.J.; Turner, R.J. Secondary multidrug efflux pump mutants alter Escherichia coli biofilm growth in the presence of cationic antimicrobial compounds. Res. Microbiol. 2017, 168, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Winterhalter, M.; Pagès, J.M. Outer membrane porins. Subcell. Biochem. 2019, 92, 79–123. [Google Scholar] [CrossRef] [PubMed]
- Davin-Regli, A.; Bolla, J.M.; James, C.E.; Lavigne, J.P.; Chevalier, J.; Garnotel, E.; Molitor, A. Membrane permeability and regulation of drug “influx and efflux” in enterobacterial pathogens. Curr. Drug Targets 2008, 9, 750–759. [Google Scholar] [CrossRef]
- Van Boxtel, R.; Wattel, A.A.; Arenas, J.; Goessens, W.H.; Tommassen, J. Acquisition of carbapenem resistance by plasmid-encoded-AmpC-expressing Escherichia coli. Antimicrob. Agents Chemother. 2016, 61, e01413-16. [Google Scholar] [CrossRef] [Green Version]
- Gaddy, J.A.; Tomaras, A.P.; Actis, L.A. The Acinetobacter baumannii 19606 OmpA protein plays a role in biofilm formation on abiotic surfaces and in the interaction of this pathogen with eukaryotic cells. Infect. Immun. 2009, 77, 3150–3160. [Google Scholar] [CrossRef] [Green Version]
- Vuotto, C.; Longo, F.; Pascolini, C.; Donelli, G.; Balice, M.P.; Libori, M.F.; Tiracchia, V.; Salvia, A.; Varaldo, P.E. Biofilm formation and antibiotic resistance in Klebsiella pneumoniae urinary strains. J. Appl. Microbiol. 2017, 123, 1003–1018. [Google Scholar] [CrossRef]
- Edgar, R.; Bibi, E. MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J. Bacteriol. 1997, 179, 2274–2280. [Google Scholar] [CrossRef] [Green Version]
- Bohn, C.; Bouloc, P. The Escherichia coli cmlA gene encodes the multidrug efflux pump Cmr/MdfA and is responsible for isopropyl-beta-D-thiogalactopyranoside exclusion and spectinomycin sensitivity. J. Bacteriol. 1998, 180, 6072–6075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Z.; Plésiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Alberti, M.; Lynch, C.; Nikaido, H.; Hearst, J.E. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol. Microbiol. 1996, 19, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, H.; Takumi-Kobayashi, A.; Theisen, U.; Hirata, T.; Nishino, K.; Yamaguchi, A. AcrS/EnvR represses expression of the acrAB multidrug efflux genes in Escherichia coli. J. Bacteriol. 2008, 190, 6276–6279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, K.; Yamaguchi, A. Role of histone-like protein H-NS in multidrug resistance of Escherichia coli. J. Bacteriol. 2004, 186, 1423–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, Y.; Oshima, T.; Mori, H.; Aono, R.; Yamamoto, K.; Ishihama, A.; Utsumi, R. Transcriptional regulation of drug efflux genes by EvgAS, a two-component system in Escherichia coli. Microbiology 2003, 149, 2819–2828. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, S.; Yang, S.; Davidson, A.L.; Zechiedrich, E.L. Control of the AcrAB multidrug efflux pump by quorum-sensing regulator SdiA. Mol. Microbiol. 2002, 43, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Kettles, R.A.; Tschowri, N.; Lyons, K.J.; Sharma, P.; Hengge, R.; Webber, M.A.; Grainger, D.C. The Escherichia coli MarA protein regulates the ycgZ-ymgABC operon to inhibit biofilm formation. Mol. Microbiol. 2019, 112, 1609–1625. [Google Scholar] [CrossRef]
- Heacock-Kang, Y.; Sun, Z.; Zarzycki-Siek, J.; Poonsuk, K.; McMillan, I.A.; Chuanchuen, R.; Hoang, T.T. Two regulators, PA3898 and PA2100, modulate the Pseudomonas aeruginosa multidrug resistance MexAB-OprM and EmrAB efflux pumps and biofilm formation. Antimicrob. Agents Chemother. 2018, 62, e01459-18. [Google Scholar] [CrossRef] [Green Version]
- Di Cagno, R.; De Angelis, M.; Calasso, M.; Gobbetti, M. Proteomics of the bacterial cross-talk by quorum sensing. J. Proteomics. 2011, 74, 19–34. [Google Scholar] [CrossRef]
- Arevalo-Ferro, C.; Hentzer, M.; Reil, G.; Görg, A.; Kjelleberg, S.; Givskov, M.; Riedel, K.; Eberl, L. Identification of quorum-sensing regulated proteins in the opportunistic pathogen Pseudomonas aeruginosa by proteomics. Environ. Microbiol. 2003, 5, 1350–1369. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T.; Jensen, P.Ø.; Burmølle, M.; Hentzer, M.; Haagensen, J.A.; Hougen, H.P.; Calum, H.; Madsen, K.F.; Moser, C.; Molin, S.; et al. Pseudomonas aeruginosa tolerance to tobramycin, hydrogen peroxide and polymorphonuclear leukocytes is quorum-sensing dependent. Microbiology 2005, 151, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popat, R.; Crusz, S.A.; Messina, M.; Williams, P.; West, S.A.; Diggle, S.P. Quorum-sensing and cheating in bacterial biofilms. Proc. Biol. Sci. 2012, 279, 4765–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarwood, J.M.; Bartels, D.J.; Volper, E.M.; Greenberg, E.P. Quorum sensing in Staphylococcus aureus biofilms. J. Bacteriol. 2004, 186, 1838–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, J.L.; Cagnazzo, J.; Phan, C.Q.; Barnes, A.M.; Dunny, G.M. Multiple roles for Enterococcus faecalis glycosyltransferases in biofilm-associated antibiotic resistance, cell envelope integrity, and conjugative transfer. Antimicrob. Agents Chemother. 2015, 59, 4094–4105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan, R.; Que, Y.A.; Maura, D.; Strobel, B.; Majcherczyk, P.A.; Hopper, L.R.; Wilbur, D.J.; Hreha, T.N.; Barquera, B.; Rahme, L.G. Auto poisoning of the respiratory chain by a quorum-sensing-regulated molecule favors biofilm formation and antibiotic tolerance. Curr. Biol. 2016, 26, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.R.; Déziel, E.; d’Argenio, D.A.; Lépine, F.; Emerson, J.; McNamara, S.; Gibson, R.L.; Ramsey, B.W.; Miller, S.I. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2006, 103, 19890–19895. [Google Scholar] [CrossRef] [Green Version]
- Orazi, G.; O’Toole, G.A. Pseudomonas aeruginosa alters Staphylococcus aureus sensitivity to vancomycin in a biofilm model of cystic fibrosis infection. mBio 2017, 8, e00873-17. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.P.; Fouhy, Y.; Garcia, B.F.; Watt, S.A.; Niehaus, K.; Yang, L.; Tolker-Nielsen, T.; Dow, J.M. Interspecies signalling via the Stenotrophomonas maltophilia diffusible signal factor influences biofilm formation and polymyxin tolerance in Pseudomonas aeruginosa. Mol. Microbiol. 2008, 68, 75–86. [Google Scholar] [CrossRef]
- Guérin, J.; Bigot, S.; Schneider, R.; Buchanan, S.K.; Jacob-Dubuisson, F. Two-partner secretion: Combining efficiency and simplicity in the secretion of large proteins for bacteria-host and bacteria-bacteria interactions. Front. Cell. Infect. Microbiol. 2017, 7, 148. [Google Scholar] [CrossRef]
- Pérez, A.; Merino, M.; Rumbo-Feal, S.; Álvarez-Fraga, L.; Vallejo, J.A.; Beceiro, A.; Ohneck, E.J.; Mateos, J.; Fernández-Puente, P.; Actis, L.A.; et al. The FhaB/FhaC two-partner secretion system is involved in adhesion of Acinetobacter baumannii AbH12O-A2 strain. Virulence 2017, 8, 959–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neil, R.B.; Apicella, M.A. Role of HrpA in biofilm formation of Neisseria meningitidis and regulation of the hrpBAS transcripts. Infect. Immun. 2009, 77, 2285–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, S.K.; Diner, E.J.; de Roodenbeke, C.T.K.; Burgess, B.R.; Poole, S.J.; Braaten, B.A.; Jones, A.M.; Webb, J.S.; Hayes, C.S.; Cotter, P.A.; et al. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 2010, 468, 439–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, J.; Schipper, K.; van Ulsen, P.; van der Ende, A.; Tommassen, J. Domain exchange at the 3’end of the gene encoding the fratricide meningococcal two-partner secretion protein A. BMC Genet. 2013, 14, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, E.C.; Perault, A.I.; Marlatt, S.A.; Cotter, P.A. Interbacterial signaling via Burkholderia contact-dependent growth inhibition system proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 8296–8301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Baltekin, Ö.; Wäneskog, M.; Elkhalifa, D.; Hammarlöf, D.L.; Elf, J.; Koskiniemi, S. Contact-dependent growth inhibition induces high levels of antibiotic-tolerant persister cells in clonal bacterial populations. EMBO J. 2018, 37, e98026. [Google Scholar] [CrossRef] [PubMed]
- Arenas, J.; de Maat, V.; Catón, L.; Krekorian, M.; Herrero, J.C.; Ferrara, F.; Tommassen, J. Fratricide activity of MafB protein of N. meningitidis strain B16B6. BMC Microbiol. 2015, 15, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, J.; Catón, L.; van den Houven, T.; de Maat, V.; Cruz herrero, J.; Tommassen, J. The outer-membrane protein MafA of Neisseria meningitidis constitutes a novel protein secretion pathway specific for the fratricide protein MafB. Virulence 2020, 11, 1701–1715. [Google Scholar] [CrossRef]
- Hausner, M.; Wuertz, S. High rates of conjugation in bacterial biofilms as determined by quantitative in situ analysis. Appl. Environ. Microbiol. 1999, 65, 3710–3713. [Google Scholar] [CrossRef] [Green Version]
- Molin, S.; Tolker-Nielsen, T. Gene transfer occurs with enhanced efficiency in biofilms and induces enhanced stabilisation of the biofilm structure. Cur. Opin. Biotechnol. 2003, 14, 255–261. [Google Scholar] [CrossRef]
- Hathroubi, S.; Mekni, M.A.; Domenico, P.; Nguyen, D.; Jacques, M. Biofilms: Microbial shelters against antibiotics. Microb. Drug Resist. 2017, 23, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Savage, V.J.; Chopra, I.; O’Neill, A.J. Staphylococcus aureus biofilms promote horizontal transfer of antibiotic resistance. Antimicrob. Agents Chemother. 2013, 57, 1968–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, W.D.; Atkinson, R.M.; Goel, R.K.; Toleman, M.A.; Benson, L.S.; Porucznik, C.A.; VanDerslice, J.A. Horizontal transfer of the blaNDM-1 gene to Pseudomonas aeruginosa and Acinetobacter baumannii in biofilms. FEMS Microbiol. Lett. 2017, 364, fnx048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, G.P.; Ben-Yehuda, S. Intercellular nanotubes mediate bacterial communication. Cell 2011, 144, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pande, S.; Shitut, S.; Freund, L.; Westermann, M.; Bertels, F.; Colesie, C.; Bischofs, I.B.; Kost, C. Metabolic cross-feeding via intercellular nanotubes among bacteria. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kouzel, N.; Oldewurtel, E.R.; Maier, B. Gene transfer efficiency in gonococcal biofilms: Role of biofilm age, architecture, and pilin antigenic variation. J. Bacteriol. 2015, 197, 2422–2431. [Google Scholar] [CrossRef] [Green Version]
- Hannan, S.; Ready, D.; Jasni, A.S.; Rogers, M.; Pratten, J.; Roberts, A.P. Transfer of antibiotic resistance by transformation with eDNA within oral biofilms. FEMS Immunol. Med. Microbiol. 2010, 59, 345–349. [Google Scholar] [CrossRef]
- Solheim, H.T.; Sekse, C.; Urdahl, A.M.; Wasteson, Y.; Nesse, L.L. Biofilm as an environment for dissemination of stx genes by transduction. Appl. Environ. Microbiol. 2013, 79, 896–900. [Google Scholar] [CrossRef] [Green Version]
- Rumbo, C.; Fernández-Moreira, E.; Merino, M.; Poza, M.; Mendez, J.A.; Soares, N.C.; Mosquera, A.; Chaves, F.; Bou, G. Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: A new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2011, 55, 3084–3090. [Google Scholar] [CrossRef] [Green Version]
- Woodford, N.; Ellington, M.J. The emergence of antibiotic resistance by mutation. Clin. Microbiol. Infect. 2007, 13, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, J.W.; Yeesin, P.; Simmons, L.A.; Wang, J.D. Sources of spontaneous mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.; Coenye, T. The role of reactive oxygen species in antibiotic-mediated killing of bacteria. Trends Microbiol. 2017, 25, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Leong, P.M.; Hsia, H.C.; Miller, J.H. Analysis of spontaneous base substitutions generated in mismatch-repair-deficient strains of Escherichia coli. J. Bacteriol. 1986, 168, 412–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaper, R.M.; Dunn, R.L. Spectra of spontaneous mutations in Escherichia coli strains defective in mismatch correction: The nature of in vivo DNA replication errors. Proc. Natl. Acad. Sci. USA 1987, 84, 6220–6224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, G.M.; Blázquez, J. Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin. Infect. Dis. 2003, 37, 1201–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciá, M.D.; Blanquer, D.; Togores, B.; Sauleda, J.; Pérez, J.L.; Oliver, A. Hypermutation is a key factor in development of multiple-antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob. Agents Chemother. 2005, 49, 3382–3386. [Google Scholar] [CrossRef] [Green Version]
- Driffield, K.; Miller, K.; Bostock, J.M.; O’Neill, A.J.; Chopra, I. Increased mutability of Pseudomonas aeruginosa in biofilms. J. Antimicrob. Chemother. 2008, 61, 1053–1056. [Google Scholar] [CrossRef] [Green Version]
- Prunier, A.L.; Malbruny, B.; Laurans, M.; Brouard, J.; Duhamel, J.F.; Leclercq, R. High rate of macrolide resistance in Staphylococcus aureus strains from patients with cystic fibrosis reveals high proportions of hypermutable strains. J. Infect. Dis. 2003, 187, 1709–1716. [Google Scholar] [CrossRef] [Green Version]
- Román, F.; Cantón, R.; Pérez-Vázquez, M.; Baquero, F.; Campos, J. Dynamics of long-term colonization of respiratory tract by Haemophilus influenzae in cystic fibrosis patients shows a marked increase in hypermutable strains. J. Clin. Microbiol. 2004, 42, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, B.; Le Gall-David, S.; Vincent, P.; Le Bars, H.; Buffet-Bataillon, S.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Is biofilm formation related to the hypermutator phenotype in clinical Enterobacteriaceae isolates? FEMS Microbiol. Lett. 2013, 347, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Ninio, J. Transient mutators: A semiquantitative analysis of the influence of translation and transcription errors on mutation rates. Genetics 1991, 129, 957–962. [Google Scholar] [PubMed]
- Springer, B.; Kidan, Y.G.; Prammananan, T.; Ellrott, K.; Böttger, E.C.; Sander, P. Mechanisms of streptomycin resistance: Selection of mutations in the 16S rRNA gene conferring resistance. Antimicrob. Agents Chemother. 2001, 45, 2877–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guénard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plésiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, S.M.; Ernst, R.K.; Miller, S.I. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 2004, 186, 575–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paltansing, S.; Kraakman, M.; van Boxtel, R.; Kors, I.; Wessels, E.; Goessens, W.; Tommassen, J.; Bernards, A. Increased expression levels of chromosomal AmpC β-lactamase in clinical Escherichia coli isolates and their effect on susceptibility to extended-spectrum cephalosporins. Microb. Drug Resist. 2015, 21, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Walther-Rasmussen, J.; Høiby, N. Class A carbapenemases. J. Antimicrob. Chemother. 2007, 60, 470–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafourian, S.; Sadeghifard, N.; Soheili, S.; Sekawi, Z. Extended spectrum beta-lactamases: Definition, classification and epidemiology. Curr. Issues Mol. Biol. 2015, 17, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Hentzer, M.; Riedel, K.; Rasmussen, T.B.; Heydorn, A.; Andersen, J.B.; Parsek, M.R.; Rice, S.A.; Eberl, L.; Molin, S.; Høiby, N.; et al. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology 2002, 148, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Manefield, M.; Rasmussen, T.B.; Henzter, M.; Andersen, J.B.; Steinberg, P.; Kjelleberg, S.; Givskov, M. Halogenated furanones inhibit quorum sensing through accelerated LuxR turnover. Microbiology 2002, 148, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Park, J.H.; Cho, H.S.; Joo, S.W.; Cho, M.H.; Lee, J. Anti-biofilm activities of quercetin and tannic acid against Staphylococcus aureus. Biofouling 2013, 29, 491–499. [Google Scholar] [CrossRef]
- Manner, S.; Skogman, M.; Goeres, D.; Vuorela, P.; Fallarero, A. Systematic exploration of natural and synthetic flavonoids for the inhibition of Staphylococcus aureus biofilms. Int. J. Mol. Sci. 2013, 14, 19434–19451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rémy, B.; Mion, S.; Plener, L.; Elias, M.; Chabrière, E.; Daudé, D. Interference in bacterial quorum sensing: A biopharmaceutical perspective. Front. Pharmacol. 2018, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.C.; Patel, S.K.; Kang, Y.C.; Lee, J.K. Quorum sensing inhibitors as antipathogens: Biotechnological applications. Biotechnol. Adv. 2019, 37, 68–90. [Google Scholar] [CrossRef] [PubMed]
- Andresen, L.; Tenson, T.; Hauryliuk, V. Cationic bactericidal peptide 1018 does not specifically target the stringent response alarmone (p) ppGpp. Sci. Rep. 2016, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef] [Green Version]
- Reffuveille, F.; de la Fuente-Núñz, C.; Mansour, S.; Hancock, R.E. A broad-spectrum antibiofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob. Agents Chemother. 2014, 58, 5363–5371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adil, M.; Singh, K.; Verma, P.K.; Khan, A.U. Eugenol-induced suppression of biofilm-forming genes in Streptococcus mutans: An approach to inhibit biofilms. J. Glob. Antimicrob. Resist. 2014, 2, 286–292. [Google Scholar] [CrossRef]
- Gopal, R.; Lee, J.H.; Kim, Y.G.; Kim, M.S.; Seo, C.H.; Park, Y. Anti-microbial, anti-biofilm activities and cell selectivity of the NRC-16 peptide derived from which flounder, Glyptocephalus cynoglossus. Mar. Drugs 2013, 11, 1836–1852. [Google Scholar] [CrossRef] [Green Version]
- Dosler, S.; Karaaslan, E.; Alev Gerceker, A. Antibacterial and anti-biofilm activities of melittin and colistin, alone and in combination with antibiotics against Gram-negative bacteria. J. Chemother. 2016, 28, 95–103. [Google Scholar] [CrossRef]
- Picoli, T.; Peter, C.M.; Zani, J.L.; Waller, S.B.; Lopes, M.G.; Boesche, K.N.; Vargas, G.D.; Hübner, S.O.; Fischer, G. Melittin and its potential in the destruction and inhibition of the biofilm formation by Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa isolated from bovine milk. Microb. Pathog. 2017, 112, 57–62. [Google Scholar] [CrossRef]
- Bardbari, A.M.; Arabestani, M.R.; Karami, M.; Keramat, F.; Aghazadeh, H.; Alikhani, M.Y.; Bagheri, K.P. Highly synergistic activity of melittin with imipenem and colistin in biofilm inhibition against multidrug-resistant strong biofilm producer strains of Acinetobacter baumannii. Eur. J. Clin. Microbiol. 2018, 37, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zai, Y.; Xi, X.; Ma, C.; Wang, L.; Zhou, M.; Shaw, C.; Chen, T. A novel membrane-disruptive antimicrobial peptide from frog skin secretion against cystic fibrosis isolates and evaluation of anti-MRSA effect using Galleria mellonella model. Biochim. Biophys. Acta 2019, 1863, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yüksel, E.; Karakeçili, A. Antibacterial activity on electrospun poly (lactide-co-glycolide) based membranes via magainin II grafting. Mater. Sci. Eng. C 2014, 45, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Kang, N.H.; Ko, S.J.; Park, J.; Park, E.; Shin, D.W.; Kim, S.H.; Lee, S.A.; Lee, J.I.; Lee, S.H.; et al. Antibacterial and antibiofilm activity and mode of action of magainin 2 against drug-resistant Acinetobacter baumannii. Int. J. Mol. Sci. 2018, 19, 3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtreimer Kandiyote, N.; Mohanraj, G.; Mao, C.; Kasher, R.; Arnusch, C.J. Synergy on surfaces: Anti-biofouling interfaces using surface-attached antimicrobial peptides PGLa and magainin-2. Langmuir 2018, 34, 11147–11155. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Sambanthamoorthy, K.; Palys, T.; Paranavitana, C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 2013, 49, 131–137. [Google Scholar] [CrossRef]
- Kang, J.; Dietz, M.J.; Li, B. Antimicrobial peptide LL-37 is bactericidal against Staphylococcus aureus biofilms. PLoS ONE 2019, 14, e0216676. [Google Scholar] [CrossRef] [Green Version]
- Maisetta, G.; Grassi, L.; Di Luca, M.; Bombardelli, S.; Medici, C.; Brancatisano, F.L.; Esin, S.; Batoni, G. Anti-biofilm properties of the antimicrobial peptide temporin 1Tb and its ability, in combination with EDTA, to eradicate Staphylococcus epidermidis biofilms on silicone catheters. Biofouling 2016, 32, 787–800. [Google Scholar] [CrossRef]
- Grassi, L.; Maisetta, G.; Maccari, G.; Esin, S.; Batoni, G. Analogs of the frog-skin antimicrobial peptide temporin 1Tb exhibit a wider spectrum of activity and a stronger antibiofilm potential as compared to the parental peptide. Front. Chem. 2017, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Luca, V.; Stringaro, A.; Colone, M.; Pini, A.; Mangoni, M.L. Esculentin(1-21), an amphibian skin membrane-active peptide with potent activity on both planktonic and biofilm cells of the bacterial pathogen Pseudomonas aeruginosa. Cell. Mol. Life Sci. 2013, 70, 2773–2786. [Google Scholar] [CrossRef] [PubMed]
- Beljantseva, J.; Kudrin, P.; Jimmy, S.; Ehn, M.; Pohl, R.; Varik, V.; Tozawa, Y.; Shingler, V.; Tenson, T.; Rejman, D.; et al. Molecular mutagenesis of ppGpp: Turning a RelA activator into an inhibitor. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, R.; Brady, K.M.; Greenberg, E.P.; Qi, F.; Yarbrough, D.K.; He, J.; McHardy, I.; Anderson, M.H.; Shi, W. Enhancement of antimicrobial activity against Pseudomonas aeruginosa by coadministration of G10KHc and tobramycin. Antimicrob. Agents Chemother. 2006, 50, 3833–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molhoek, E.M.; van Dijk, A.; Veldhuizen, E.J.; Haagsman, H.P.; Bikker, F.J. A cathelicidin-2-derived peptide effectively impairs Staphylococcus epidermidis biofilms. Int. J. Antimicrob. Agents 2011, 37, 476–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casciaro, B.; Moros, M.; Rivera-Fernández, S.; Bellelli, A.; Jesús, M.; Mangoni, M.L. Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a(1-21)NH2 as a reliable strategy for antipseudomonal drugs. Acta Biomater. 2017, 47, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisetta, G.; Grassi, L.; Esin, S.; Serra, I.; Scorciapino, M.A.; Rinaldi, A.C.; Batoni, G. The semi-synthetic peptide lin-SB056-1 in combination with EDTA exerts strong antimicrobial and antibiofilm activity against Pseudomonas aeruginosa in conditions mimicking cystic fibrosis sputum. Int. J. Mol. Sci. 2017, 18, 1994. [Google Scholar] [CrossRef]
- Khalifa, L.; Brosh, Y.; Gelman, D.; Coppenhagen-Glazer, S.; Beyth, S.; Poradosu-Cohen, R.; Que, Y.A.; Beyth, N.; Hazan, R. Targeting Enterococcus faecalis biofilms with phage therapy. Appl. Environ. Microbiol. 2015, 81, 2696–2705. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, M.M.; Frezza, D.; Romano, E.; Marmo, P.; De Angelis, L.H.; Perini, N.; Thaller, M.C.; Di Lallo, G. The lytic bacteriophage vB_EfaH_EF1TV, a new member of the Herelleviridae family, disrupts biofilm produced by Enterococcus faecalis clinical strains. J. Glob. Antimicrob. Resist. 2020, 21, 68–75. [Google Scholar] [CrossRef]
- Yuan, Y.; Qu, K.; Tan, D.; Li, X.; Wang, L.; Cong, C.; Xiu, Z.; Xu, Y. Isolation and characterization of a bacteriophage and its potential to disrupt multi-drug resistant Pseudomonas aeruginosa biofilms. Microb. Pathog. 2019, 128, 329–336. [Google Scholar] [CrossRef]
- Gutiérrez, D.; Vandenheuvel, D.; Martínez, B.; Rodríguez, A.; Lavigne, R.; García, P. Two phages, phiIPLA-RODI and phiIPLA-C1C, lyse mono- and dual-species staphylococcal biofilms. Appl. Environ. Microbiol. 2015, 81, 3336–3348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xu, L.L.; Gan, D.; Zhang, X. In vitro study of bacteriophage AB3 endolysin LysAB3 activity against Acinetobacter baumannii biofilm and biofilm-bound A. baumannii. Clin. Lab. 2018, 64, 1021–1030. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Z.; Mi, L.; Huang, Y.; Li, P.; Liu, H.; Yuan, X.; Niu, W.; Jiang, N.; Bai, C.; et al. Identification and characterization of capsule depolymerase Dpo48 from Acinetobacter baumannii phage IME200. PeerJ 2019, 7, e6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedi, M.S.; Verma, V.; Chhibber, S. Amoxicillin and specific bacteriophage can be used together for eradication of biofilm of Klebsiella pneumoniae B5055. World J. Microbiol. Biot. 2009, 25, 1145. [Google Scholar] [CrossRef]
- Rahman, M.; Kim, S.; Kim, S.M.; Seol, S.Y.; Kim, J. Characterization of induced Staphylococcus aureus bacteriophage SAP-26 and its anti-biofilm activity with rifampicin. Biofouling 2011, 27, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Alves, D.R.; Gaudion, A.; Bean, J.E.; Esteban, P.P.; Arnot, T.C.; Harper, D.R.; Kot, W.; Hanser, L.H.; Enright, M.C.; Jenkins, A.T.A. Combined use of bacteriophage K and a novel bacteriophage to reduce Staphylococcus aureus biofilm formation. Appl. Environ. Microbiol. 2014, 80, 6694–6703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, D.; McAuliffe, O.; O’Mahony, J.; Coffey, A. Development of a broad-host-range phage cocktail for biocontrol. Bioeng. Bugs. 2011, 2, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.; McAuliffe, O.; Ross, R.P.; Coffey, A. Prevention of Staphylococcus aureus biofilm formation and reduction in established biofilm density using a combination of phage K and modified derivatives. Lett. Appl. Microbiol. 2012, 54, 286–291. [Google Scholar] [CrossRef]
- Fu, W.; Forster, T.; Mayer, O.; Curtin, J.J.; Lehman, S.M.; Donlan, R.M. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob. Agents Chemother. 2010, 54, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Alves, D.R.; Perez-Esteban, P.; Kot, W.; Bean, J.E.; Arnot, T.; Hansen, L.H.; Enright, M.C.; Jenkins, A.T.A. A novel bacteriophage cocktail reduces and disperses Pseudomonas aeruginosa biofilms under static and flow conditions. Microb. Biotechnol. 2016, 9, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.S.A.; Zahin, M.; Hasan, S.; Husain, F.M.; Ahmad, I. Inhibition of quorum sensing regulated bacterial functions by plant essential oils with special reference to clove oil. Lett. Appl. Microbiol. 2009, 49, 354–360. [Google Scholar] [CrossRef]
- Nuryastuti, T.; van der Mei, H.C.; Busscher, H.J.; Iravati, S.; Aman, A.T.; Krom, B.P. Effect of cinnamon oil on icaA expression and biofilm formation by Staphylococcus epidermidis. Appl. Environ. Microbiol. 2009, 75, 6850–6855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jesus Pimentel-Filho, N.; de Freitas Martins, M.C.; Nogueira, G.B.; Mantovani, H.C.; Vanetti, M.C.D. Bovicin HC5 and nisin reduce Staphylococcus aureus adhesion to polystyrene and change the hydrophobicity profile and Gibbs free energy of adhesion. Int. J. Food Microbiol. 2014, 190, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, M.; Yadav, V.K.; Singh, P.K.; Sharma, D.; Pandey, H.; Narvi, S.S.; Agarwal, V. Effect of cinnamon oil on quorum sensing-controlled virulence factors and biofilm formation in Pseudomonas aeruginosa. PLoS ONE. 2015, 10, e0135495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alibi, S.; Ben Selma, W.; Ramos-Vivas, J.; Smach, M.A.; Touati, R.; Boukadida, J.; Navas, J.; Mansour, H.B. Anti-oxidant, antibacterial, anti-biofilm, and anti-quorum sensing activities of four essential oils against multidrug-resistant bacterial clinical isolates. Curr. Res. Transl. Med. 2020, 68, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Raei, P.; Pourlak, T.; Memar, M.Y.; Alizadeh, N.; Aghamali, M.; Zeinalzadeh, E.; Asgharzadeh, M.; Kafil, H.S. Thymol and carvacrol strongly inhibit biofilm formation and growth of carbapenemase-producing Gram negative bacilli. Cell. Mol. Biol. 2017, 63, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Valliammai, A.; Selvaraj, A.; Yuvashree, U.; Aravindraja, C.; Karutha Pandian, S. sarA-dependent antibiofilm activity of thymol enhances the antibacterial efficacy of rifampicin against Staphylococcus aureus. Front. Microbiol. 2020, 11, 1744. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Z.; Liu, Z.; Meng, R.; Shi, C.; Chen, X.; Bu, X.; Guo, N. Phenotype and RNA-seq-based transcriptome profiling of Staphylococcus aureus biofilms in response to tea tree oil. Microb. Pathog. 2018, 123, 304–313. [Google Scholar] [CrossRef]
- Samoilova, Z.; Muzyka, N.; Lepekhina, E.; Oktyabrsky, O.; Smirnova, G. Medicinal plant extracts can variously modify biofilm formation in Escherichia coli. Antonie Van Leeuwenhoek 2014, 105, 709–722. [Google Scholar] [CrossRef]
- Da Costa Júnior, S.D.; de Oliveira Santos, J.V.; de Almeida Campos, L.A.; Pereira, M.A.; Magalhães, N.S.S.; Cavalcanti, I.M.F. Antibacterial and antibiofilm activities of quercetin against clinical isolates of Staphyloccocus aureus and Staphylococcus saprophyticus with resistance profile. IJEAB 2018, 3, 266213. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, J.; Sun, F.; Feng, W.; Sn, Y.; Qiu, X.; Xiong, L.; Liu, T.; Chen, Y. Quercetin is an effective inhibitor of quorum sensing, biofilm formation and virulence factors in Pseudomonas aeruginosa. J. Appl. Microbiol. 2016, 120, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Qayyum, S.; Sharma, D.; Bisht, D.; Khan, A.U. Identification of factors involved in Enterococcus faecalis biofilm under quercetin stress. Microb. Pathog. 2019, 126, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Lambert, R.J.W.; Skandamis, P.N.; Coote, P.J.; Nychas, G.J. A study of the minimum inhibitory concentration and mode of action of oregano essential oil, thymol and carvacrol. J. Appl. Microbiol. 2001, 91, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namivandi-Zangeneh, R.; Yang, Y.; Xu, S.; Wong, E.H.; Boyer, C. Antibiofilm platform based on the combination of antimicrobial polymers and essential oils. Biomacromolecules 2020, 21, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Raturi, K.; Dang, S.; Gupta, S.; Gabrani, R. Inhibitory effect of cinnamaldehyde alone and in combination with thymol, eugenol and thymoquinone against Staphylococcus epidermidis. J. Herb. Med. 2017, 9, 68–73. [Google Scholar] [CrossRef]
- Kali, A.; Bhuvaneshwar, D.; Charles, P.M.V.; Seetha, K.S. Antibacterial synergy of curcumin with antibiotics against biofilm producing clinical bacterial isolates. J. Basic Clin. Pharm. 2016, 7, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, E.; Yakandawala, N.; LoVetri, K.; Madhyastha, M.S. A microplate spectrofluorometric assay for bacterial biofilms. J. Ind. Microbiol. Biotchetnol. 2007, 34, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Izano, E.A.; Sadovskaya, I.; Wang, H.; Vinogradov, E.; Ragunath, C.; Ramasubbu, N.; Jabbouri, S.; Perru, M.B.; Kaplan, J.B. Poly-N-acetylglucosamine mediates biofilm formation and detergent resistance in Aggregatibacter actinomycetemcomitans. Microb. Pathog. 2007, 44, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Seidl, K.; Goerke, C.; Wolz, C.; Mack, D.; Berger-Bächi, B.; Bischoff, M. Staphylococcus aureus CcpA affects biofilm formation. Infect. Immun. 2008, 76, 2044–2050. [Google Scholar] [CrossRef] [Green Version]
- Tetz, G.V.; Artemenko, N.K.; Tetz, V.V. Effect of DNase and antibiotics on biofilm characteristics. Antimicrob. Agents Chemother. 2009, 53, 1204–1209. [Google Scholar] [CrossRef] [Green Version]
- Parks, Q.M.; Young, R.L.; Poch, K.R.; Malcolm, K.C.; Vasil, M.L.; Nick, J.A. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: Human F-actin and DNA as targets for therapy. J. Med. Microbiol. 2009, 58, 492–502. [Google Scholar] [CrossRef]
- Ramsey, D.M.; Wozniak, D.J. Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol. Microbiol. 2005, 56, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Tomasz, A. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J. Biol. Chem. 2000, 275, 20496–20501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hukić, M.; Seljmo, D.; Ramovic, A.; Ibrišimović, M.A.; Dogan, S.; Hukic, J.; Bojic, E.F. The effect of lysozyme on reducing biofilms by Staphylococcus aureus, Pseudomonas aeruginosa, and Gardnerella vaginalis: An in vitro examination. Microb. Drug Resist. 2018, 24, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Eladawy, M.; El-Mowafy, M.; El-Sokkary, M.M.A.; Barwa, R. Effects of lysozyme, proteinase K, and cephalosporins on biofilm formation by clinical isolates of Pseudomonas aeruginosa. Interdiscip. Perspect. Infect. Dis. 2020, 2020, 6156720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, M.C.F.; Coutinho, B.G.; Coelho, M.L.V. Lysostaphin: A staphylococcal bacteriolysin with potential clinical applications. Pharmaceuticals 2010, 3, 1139–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, K.L.; Roberts, A.L.; Holder, R.C.; Reid, S.D. Dispersal of Group A streptococcal biofilms by the cysteine protease SpeB leads to increased disease severity in a murine model. PLoS ONE 2011, 6, e18984. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, J.H.; Cho, M.H.; Herzberg, M.; Lee, J. Acceleration of protease effect on Staphylococcus aureus biofilm dispersal. FEMS Microbiol. Lett. 2012, 335, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Wang, L.H.; Wang, J.; Dong, Y.H.; Hu, J.Y.; Zhang, L.H. Quorum quenching enzyme activity is widely conserved in the sera of mammalian species. FEBS Lett. 2005, 579, 3713–3717. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Kim, Y.S.; Ponnusamy, K.; Kweon, J.H. Application of quorum quenching to inhibit biofilm formation. Environm. Eng. Sci. 2009, 26, 1319–1324. [Google Scholar] [CrossRef]
- Gupta, P.; Chhibber, S.; Harjai, K. Efficacy of purified lactonase and ciprofloxacin in preventing systemic spread of Pseudomonas aeruginosa in murine burn wound model. Burns 2015, 41, 153–162. [Google Scholar] [CrossRef]
- Sambanthamoorthy, K.; Luo, C.; Pattabiraman, N.; Feng, X.; Koestler, B.; Waters, C.M.; Palys, T.J. Identification of small molecules inhibiting diguanylate cyclases to control bacterial biofilm development. Biofouling 2014, 30, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Cegelski, L.; Pinkner, J.S.; Hammer, N.D.; Cusumano, C.K.; Hung, C.S.; Chorell, E.; Åberg, V.; Walker, J.N.; Seed, P.C.; Almqvist, F.; et al. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat. Chem. Biol. 2009, 5, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, S.E.; Pinkner, J.S.; Chorell, E.; Dodson, K.W.; Shaffer, C.L.; Conover, M.S.; Livny, J.; Hadjifrangiskou, M.; Almqvist, F.; Hultgren, S.J. Pilicide ec240 disrupts virulence circuits in uropathogenic Escherichia coli. mBio 2014, 5, e02038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusumano, C.K.; Pinkner, J.S.; Han, Z.; Greene, S.E.; Ford, B.A.; Crowley, J.R.; Henderson, J.R.; Jenetka, J.W.; Hultgren, S.J. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci. Transl. Med. 2011, 3, 109ra115. [Google Scholar] [CrossRef] [Green Version]
- Debebe, T.; Krüger, M.; Huse, K.; Kacza, J.; Mühlberg, K.; König, B.; Birkenmeier, G. Ethyl pyruvate: An anti-microbial agent that selectively targets pathobionts and biofilms. PLoS ONE 2016, 11, e0162919. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Yang, L.; Qu, D.; Molin, S.; Tolker-Nielsen, T. Pseudomonas aeruginosa extracellular products inhibit staphylococcal growth, and disrupt established biofilms produced by Staphylococcus epidermidis. Microbiology 2009, 155, 2148–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donelli, G.; Francolini, I.; Romoli, D.; Guaglianone, E.; Piozzi, A.; Ragunath, C.; Kaplan, J.B. Synergistic activity of dispersin B and cefamandole nafate in inhibition of staphylococcal biofilm growth on polyurethanes. Antimicrob. Agents Chemother. 2007, 51, 2733–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, J.B. Therapeutic potential of biofilm-dispersing enzymes. Int. J. Artif. Organs. 2009, 32, 545–554. [Google Scholar] [CrossRef]
- Darouiche, R.O.; Mansouri, M.D.; Gawande, P.V.; Madhyastha, S. Antimicrobial and antibiofilm efficacy of triclosan and DispersinB® combination. J. Antimicrob. Chemother. 2009, 64, 88–93. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef]
- Jiang, P.; Li, J.; Han, F.; Duan, G.; Lu, X.; Gu, Y.; Yu, W. Antibiofilm activity of an exopolysaccharide from marine bacterium Vibrio sp. QY101. PLoS ONE 2011, 6, e18514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiq, D.M.; Darouiche, R.O. New strategies to prevent catheter-associated urinary tract infections. Nat. Rev. Urol. 2012, 9, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Galdiero, E.; Lombardi, L.; Falanga, A.; Libralato, G.; Guida, M.; Carotenuto, R. Biofilms: Novel strategies based on antimicrobial peptides. Pharmaceutics 2019, 11, 322. [Google Scholar] [CrossRef] [Green Version]
- Malmsten, M. Antimicrobial peptides. Upsala J. Med. Sci. 2014, 119, 199–204. [Google Scholar] [CrossRef]
- Memariani, H.; Memariani, M.; Shahidi-Dadras, M.; Nasiri, S.; Akhavan, M.M.; Moravvej, H. Melittin: From honeybees to superbugs. Appl. Microbiol. Biotech. 2019, 103, 3265–3276. [Google Scholar] [CrossRef]
- Etayash, H.; Pletzer, D.; Kumar, P.; Straus, S.K.; Hancock, R.E.W. A cyclic derivative of host-defence peptide IDR-1018 improves proteolytic stability, suppresses inflammation, and enhances in vivo activity. J. Med. Chem. 2020, 63, 9228–9236. [Google Scholar] [CrossRef]
- Martin-Serrano, Á.; Gómez, R.; Ortega, P.; de la Mata, F.J. Nanosystems as vehicles for the delivery of antimicrobial peptides (AMPs). Pharmaceutics 2019, 11, 448. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.P.; Melo, L.D.; Boas, D.V.; Sillankorva, S.; Azeredo, J. Phage therapy as an alternative or complementary strategy to prevent and control biofilm-related infections. Curr. Opin. Microbiol. 2017, 39, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Sillankorva, S.; Oliveira, R.; Vieira, M.J.; Sutherland, I.; Azeredo, J. Pseudomonas fluorescens infection by bacteriophage ΦS1: The influence of temperature, host growth phase and media. FEMS Microbiol. Lett. 2004, 241, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, M.; Hussain, T.; Das, C.R.; Andleeb, S. Characterization of Siphoviridae phage Z and studying its efficacy against multidrug-resistant Klebsiella pneumoniae planktonic cells and biofilm. J. Med. Microbiol. 2015, 64, 454–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, S.; Yao, X.; Lu, S.; Tan, Y.; Rao, X.; Li, M.; Jin, X.; Wang, J.; Zhao, Y.; Wu, N.C.; et al. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Sci. Rep. 2014, 4, 4738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.K.; Collins, J.J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4629–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.K.; Cheng, N.C.; Cheng, C.M. Biofilms in chronic wounds: Pathogenesis and diagnosis. Trends Biotechnol. 2019, 37, 505–517. [Google Scholar] [CrossRef]
- LuTheryn, G.; Glynne-Jones, P.; Webb, J.S.; Carugo, D. Ultrasound-mediated therapies for the treatment of biofilms in chronic wounds: A review of present knowledge. Microb. Biotechnol. 2020, 13, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Espada, R.; Wang, Y.; Goh, X.S.; Dai, T. Antimicrobial blue light inactivation of microbial isolates in biofilms. Lasers Surg. Med. 2020, 52, 472–478. [Google Scholar] [CrossRef]
- Dai, T.; Gupta, A.; Huang, Y.Y.; Yin, R.; Murray, C.K.; Vrahas, M.S.; Sherwood, M.E.; Tegos, G.P.; Hamblin, M.R. Blue light rescues mice from potentially fatal Pseudomonas aeruginosa burn infection: Efficacy, safety, and mechanism of action. Antimicrob. Agents Chemother. 2013, 57, 1238–1245. [Google Scholar] [CrossRef] [Green Version]
- Rupel, K.; Zupin, L.; Ottaviani, G.; Bertani, I.; Martinelli, V.; Porrelli, D.; Vodret, S.; Vuerich, R.; da Silva, D.P.; Bussani, R.; et al. Blue laser light inhibits biofilm formation in vitro and in vivo by inducing oxidative stress. NPJ Biofilms Microb. 2019, 5, 1–11. [Google Scholar] [CrossRef]
- Bak, J.; Ladefoged, S.D.; Tvede, M.; Begovic, T.; Gregersen, A. Disinfection of Pseudomonas aeruginosa biofilm contaminated tube lumens with ultraviolet C light emitting diodes. Biofouling 2010, 26, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Reshma, V.G.; Syama, S.; Sruthi, S.; Reshma, S.C.; Remya, N.S.; Mohanan, P.V. Engineered nanoparticles with antimicrobial property. Curr. Drug Metab. 2017, 18, 1040–1054. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.C.; Siedlecki, C.A. Submicron-textured biomaterial surface reduces staphylococcal bacterial adhesion and biofilm formation. Acta Biomater. 2012, 8, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Zhang, P.; Wang, S. Recent progress in biointerfaces with controlled bacterial adhesion by using chemical and physical methods. Chem. Asian J. 2014, 9, 2004–2016. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.C.; Wo, Y.; Meyerhoff, M.E.; Siedlecki, C.A. Inhibition of bacterial adhesion and biofilm formation by dual functional textured and nitric oxide releasing surfaces. Acta Biomater. 2017, 51, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Różańska, D.; Regulska-Ilow, B.; Choroszy-Król, I.; Ilow, R. [The role of Escherichia coli strain Nissle 1917 in the gastro-intestinal diseases]. Postepy Hig. Med. Dosw. Online 2014, 6, 1251–1256. [Google Scholar] [CrossRef]
- Fang, K.; Jin, X.; Hong, S.H. Probiotic Escherichia coli inhibits biofilm formation of pathogenic E. coli via extracellular activity of DegP. Sci. Rep. 2018, 8, 4939. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.Y.; Koh, E.; Wong, A.; March, J.C.; Bentley, W.E.; Lee, Y.S.; Chang, M.W. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun. 2017, 8, 15028. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
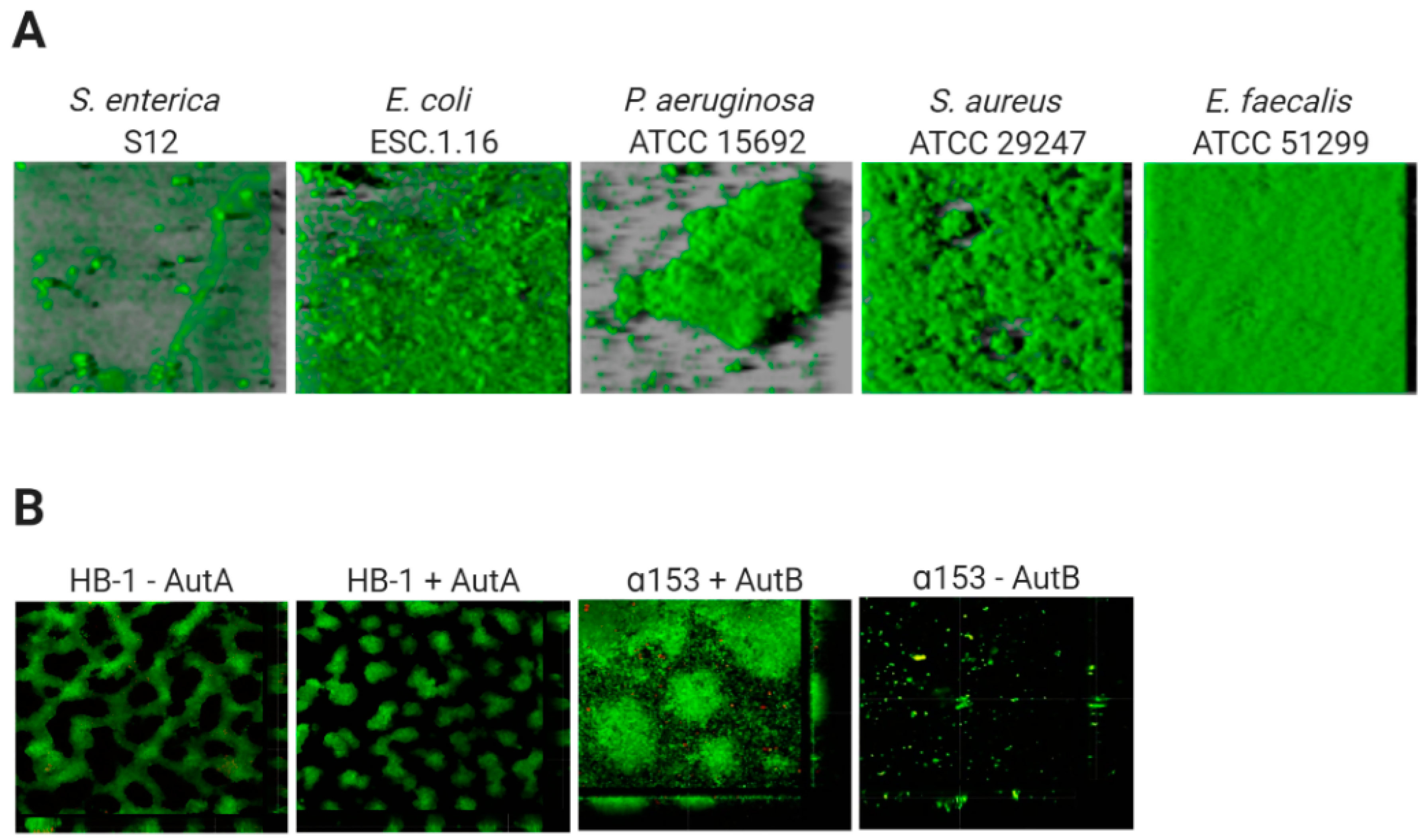{kind=link}
{kind=link}
{kind=link}
| Substance(s) | Mechanism of Action | Targets | References |
|---|---|---|---|
| Antimicrobial Peptides | |||
| Natural Antimicrobial Peptides | |||
| Melittin | Formation of short-lived pores in the membrane and increase of permeability of OM | P. aeruginosa, S. aureus, E. coli, K. pneumoniae, A. baumannii | [220,221,222,223,224] |
| Japonicin-2LF | Detergent-like activity against components of biofilm matrix; higher activity in inhibiting than in eradicating biofilms | S. aureus, MRSA, E. coli | [225] |
| Magainin 2 | Destabilizes the bacterial membrane and intracellular processes | A. baumannii, P. aeruginosa, E. coli | [226,227,228] |
| LL-37 | Membrane disruption; inhibits twitching and QS; interferes in bacterial attachment; downregulates rhlA and rhlB genes | P. aeruginosa, A. baumanni, S. aureus | [229,230,231] |
| Temporin 1Tb | Disruption of cell membrane integrity; capable of penetrating biofilm and killing bacteria; hemolytic activity | S. epidermidis, S. aureus, K. pneumoniae, P. aeruginosa, E. faecium | [232,233] |
| Synthetic Antimicrobial Peptides | |||
| 1037 | Downregulates genes of biofilm development; reduces swimming and swarming motilities | P. aeruginosa, L. monocytogenes, Burkolderia cenocepacia | [218] |
| Esculentin (1–21) | Biofilm eradication | P. aeruginosa | [234] |
| 1018 | Binds (p)ppGpp and inhibits SR; inhibits attachment, QS, and twitching motility | E. coli, S. aureus, MRSA, P. aeruginosa, A. baumannii, K. pneumoniae, A. baumannii, S. Typhimurium, E. faecium | [218,219,235] |
| STAMP G10KHc | Disrupts and permeabilizes OM and IM | P. aeruginosa | [236] |
| F2,5,12W | Reduces initial adhesion of bacteria; eliminates mature biofilms; suppresses biofilm formation | S. epidermidis | [237] |
| Combined Therapies | |||
| 1018 + antibiotics (e.g., ciprofloxacin) | Inhibition of (p)ppGpp activation; downregulation of genes that interfere with antibiotic resistance and biofilm formation | E. coli, MRSA, P. aeruginosa, K. pneumoniae, A. baumannii, S. enterica | [219] |
| Esculentin (1–21) + AuNPs (AuNPs@Esc(1–21)) | Disruption of membrane forming clusters | P. aeruginosa | [238] |
| Temporin 1Tb + EDTA | Mature biofilm eradication | S. epidermidis | [232] |
| lin-SB056-1 + EDTA | Perturbation of membrane; eradication biofilm; chelation of divalent metal ions | P. aeruginosa | [239] |
| Bacteriophages | |||
| Phages | |||
| EFDG1 | Mature biofilm eradication | E. faecium, E. faecalis | [240] |
| vB_EfaH_EF1TV | Mature biofilm eradication | E. faecalis | [241] |
| vB_PaeM_LS1 | Disrupts and avoids dispersion of biofilms; inhibits biofilm growth | P. aeruginosa | [242] |
| vB_SauM_philPLA-RODI | Penetrates biofilms; inhibits biofilm formation | S. aureus S. epidermidis | [243] |
| Phage-derived Enzymes | |||
| LysAB3 | Degradation of bacterial wall peptidoglycan, biofilm eradication | A. baumannii | [244] |
| Dpo48 | Degrades exopolysaccharide and eradicates biofilm | A. baumannii | [245] |
| Combined Phage Therapy | |||
| Phage + amoxicillin | Biofilm eradication | K. pneumoniae | [246] |
| SAP-26 + rifampicin | Hydrolysis of bacterial wall; mature biofilm eradication; reduction of biofilm growth | S. aureus | [247] |
| Phage K + DRA88 | Inhibits biofilm formation; disperses biofilms | S. aureus | [248] |
| Phage K + its derivatives (e.g., K.MS811) | Biofilm eradication | S. aureus | [249,250] |
| Phage M4 + E2005-24-39 + E2005-40-16 + W2005-24-39 + W2005-37-18-03 | Biofilm eradication | P. aeruginosa | [251] |
| DL52 + DL54 + DL60 + DL62 + DL64 + DL68 | Attachment to cell by binding to lipopolysaccharide; biofilm eradication | P. aeruginosa | [252] |
| Plant-Derived Natural Products | |||
| Essential Oils or Principal Active Compounds | |||
| Cinnamon (cinnamaldehyde) | Inhibits QS mechanism: regulates production of rhamnolipids, proteases, and alginate and swarming activity; disrupts synthesis of DNA, RNA, proteins, lipids, and polysaccharides; alters expression of genes related to biofilm formation (e.g., icaA) | E. coli, P. aeruginosa, K. pneumoniae, A. baumannii, S. epidermidis, S. aureus, MRSA, S. enteridis, S. Typhimurium | [253,254,255,256,257] |
| Clove | Disrupts QS communication: biofilm dispersal, inhibits AHL synthesis; downregulates relA gene | E. coli, P. aeruginosa, K. pneumoniae, A. baumannii, S. aureus | [253,257] |
| Thyme (thymol) | Downregulates sarA gene; increases membrane permeability; penetrates polysaccharide matrix: eradicates biofilms | E. coli, P. aeruginosa, K. pneumoniae, A. baumannii, S. aureus, S. enteridis | [257,258,259] |
| Tea tree oil | Alters expression of multiple genes related to biofilm formation (e.g., sarA, cidA, igrA, ifrB) | S. aureus | [260] |
| Oregano (carvacrol) | Increases membrane permeability; penetrates polysaccharide matrix; eradicates biofilms | K. pneumoniae, P. aeruginosa, A. baumannii | [258] |
| Halogenated furanones | QS inhibition; antagonist of LuxR | E. coli P. aeruginosa | [211,212] |
| Flavonoids (e.g., quercetin) | Represses exopolysaccharides production; inhibits rpoS gene expression; decreases swimming motility | S. aureus, E. coli, P. aeruginosa, E. faecalis | [261,262,263,264] |
| Combined Therapy | |||
| Carvacrol + eugenol | Increases membrane permeability | K. pneumoniae, P. aeruginosa, A. baumannii, S. aureus | [258,265,266] |
| Cinnamaldehyde + eugenol | Membrane permeabilization | S. epidermidis | [267] |
| Curcumin + antibiotics (e.g., ciprofloxacin) | QS inhibition | E. coli, K. pneumoniae, P. aeruginosa, S. aureus, E. faecalis | [268] |
| Enzymes | |||
| Dispersin B | Hydrolyses PNAG | S. epidermidis, S. aureus, E. coli, A. pleuropneumoniae | [38,269,270] |
| DNases | Hydrolyses DNA | A. baumannii, K. pneumoniae, E. coli, P. aeruginosa, S. aureus | [271,272,273] |
| Alginate lyase | Degrades alginate | P. aeruginosa | [274] |
| Lysozyme | Hydrolytic activity | S. pneumoniae, Gardnerella vaginalis, S. aureus, P. aeruginosa | [275,276,277] |
| Lysostaphin | Degrades cell wall | S. aureus, S. epidermidis | [278] |
| Proteases (e.g., SpeB) | Degrades cell wall | Streptococcus spp. P. aeruginosa, S. aureus | [279,280] |
| Paraoxonases (e.g., acylase I) | Inhibits QS | A. hydrophila, P. putida, P. aeruginosa | [216,281,282] |
| Lactonase | Inhibits QS | P. aeruginosa | [215,283] |
| Small molecules | |||
| Small molecules (e.g., LP 3134, LP 3145, LP 4010) | Inhibition of diguanylate cyclase | P. aeruginosa, A. baumannii | [284] |
| Pilicides (FN075, BibC6, Ec240) | Blocks synthesis of curli and Type I pili, and inhibits chaperone-usher pathway for pili biogenesis | E. coli | [285,286] |
| Mannosides | Inhibits FimH of type I pili | E. coli | [287] |
| Ethyl pyruvate | Inhibits enzymes of the glycolytic pathway | E. coli | [288] |
| Polysaccharides | |||
| Psl, Pel | Disperses biofilm | S. epidermidis | [289] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uruén, C.; Chopo-Escuin, G.; Tommassen, J.; Mainar-Jaime, R.C.; Arenas, J. Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance. Antibiotics 2021, 10, 3. https://doi.org/10.3390/antibiotics10010003
Uruén C, Chopo-Escuin G, Tommassen J, Mainar-Jaime RC, Arenas J. Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance. Antibiotics. 2021; 10(1):3. https://doi.org/10.3390/antibiotics10010003
Chicago/Turabian StyleUruén, Cristina, Gema Chopo-Escuin, Jan Tommassen, Raúl C. Mainar-Jaime, and Jesús Arenas. 2021. "Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance" Antibiotics 10, no. 1: 3. https://doi.org/10.3390/antibiotics10010003
APA StyleUruén, C., Chopo-Escuin, G., Tommassen, J., Mainar-Jaime, R. C., & Arenas, J. (2021). Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance. Antibiotics, 10(1), 3. https://doi.org/10.3390/antibiotics10010003