
Molecular Modeling the Proteins from the exo-xis Region of Lambda and Shigatoxigenic Bacteriophages


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
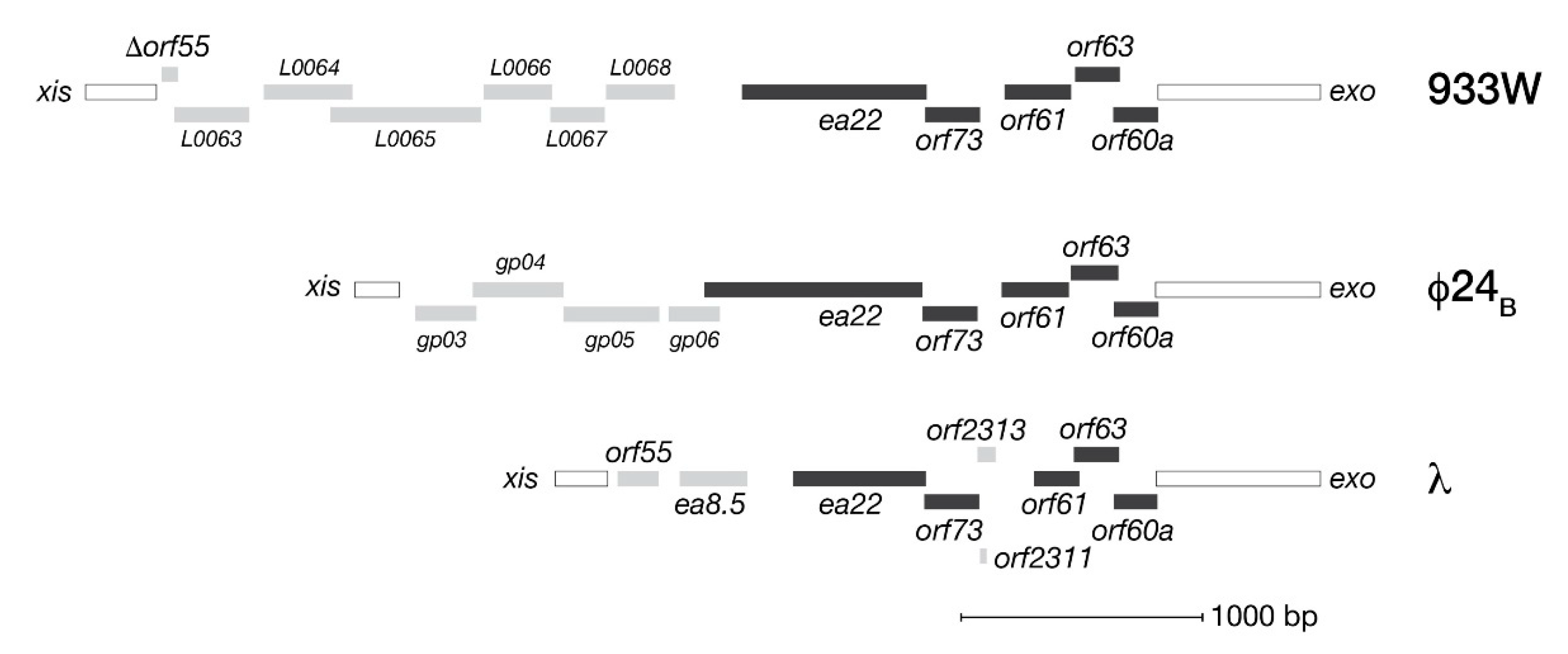
2. Conserved Genes of the exo-xis Region
3. The Interaction Landscape of exo-xis Region Proteins
3.1. Oligomerization and Phase Separation
3.2. Yeast Two-Hybrid Studies
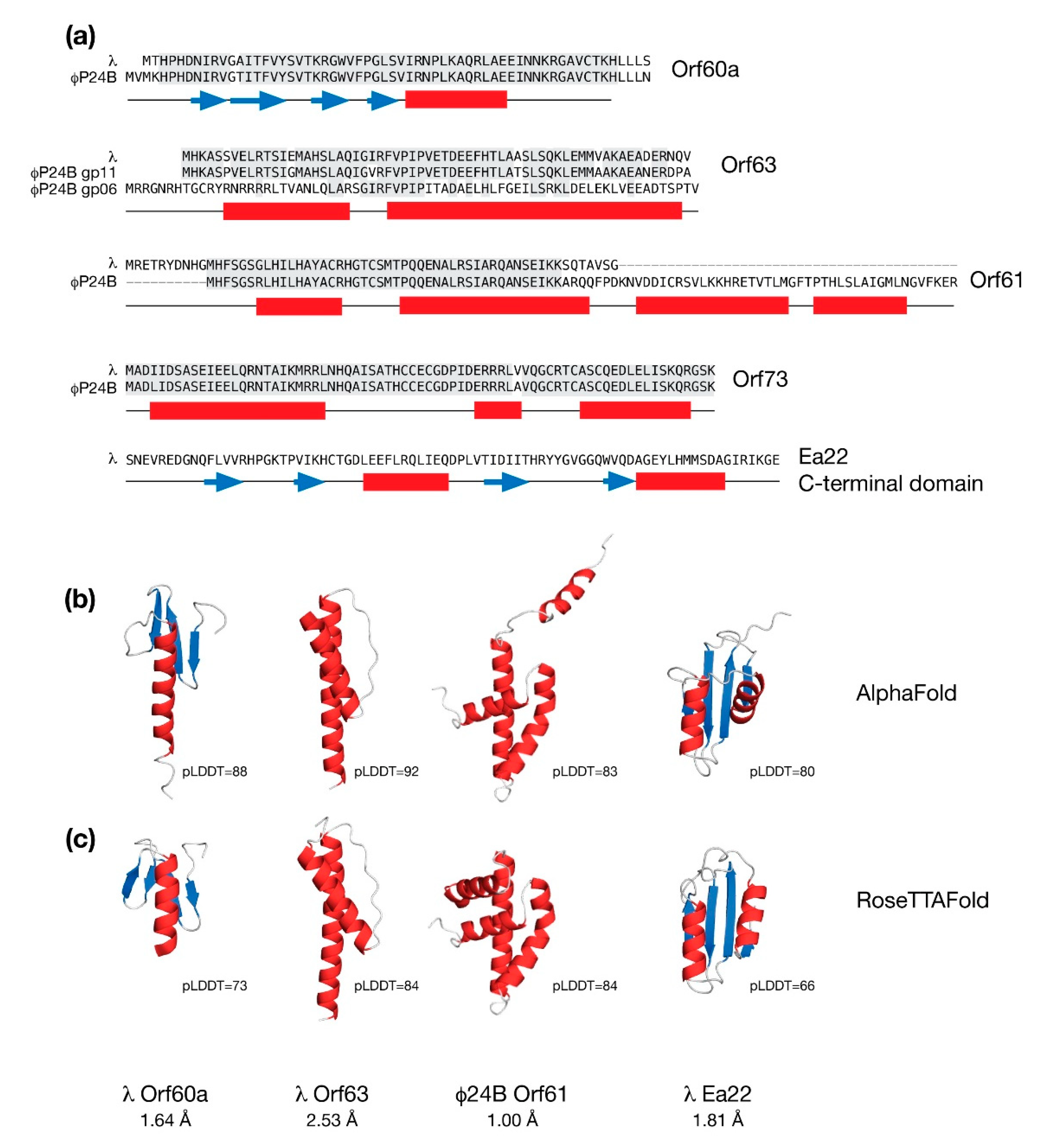
4. Structural Features of the Conserved exo-xis Region Proteins
4.1. Machine Learning Methods for Protein Structure Prediction
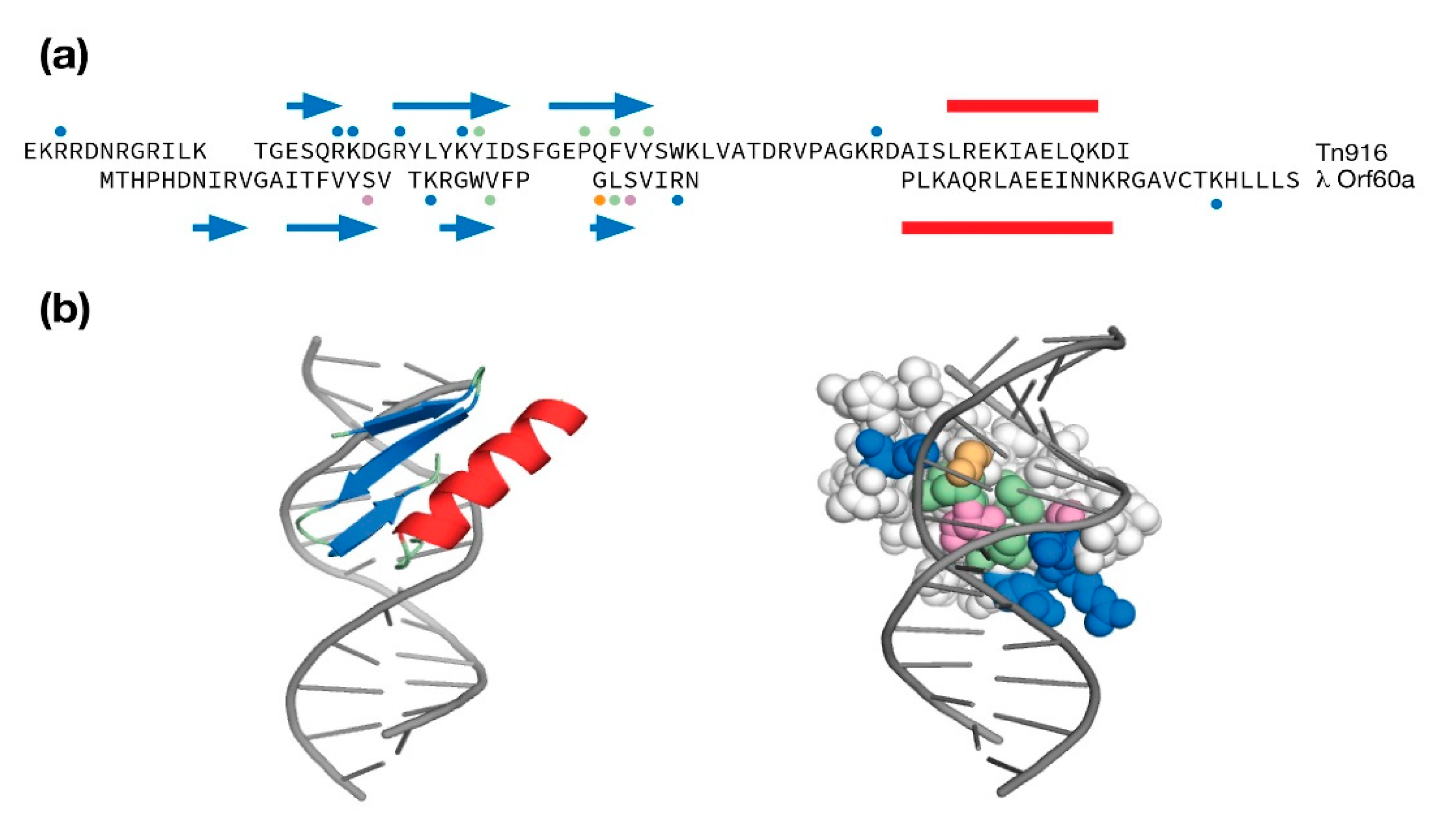
4.2. The Orf60a Gene of λ and Stx+ Phages
4.3. The Orf63 Genes of λ and Stx+ Phages
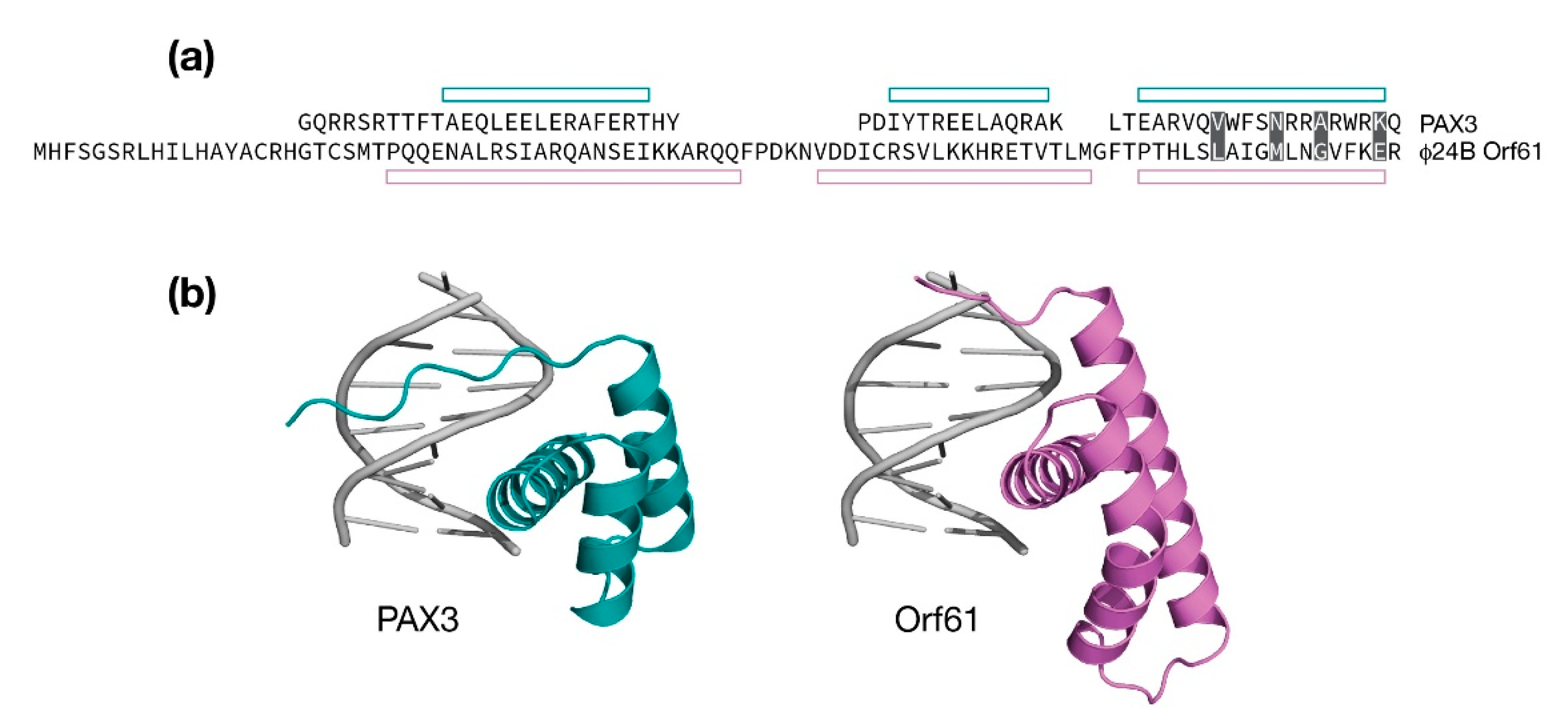
4.4. The Orf61 Gene of λ and Stx+ Phages
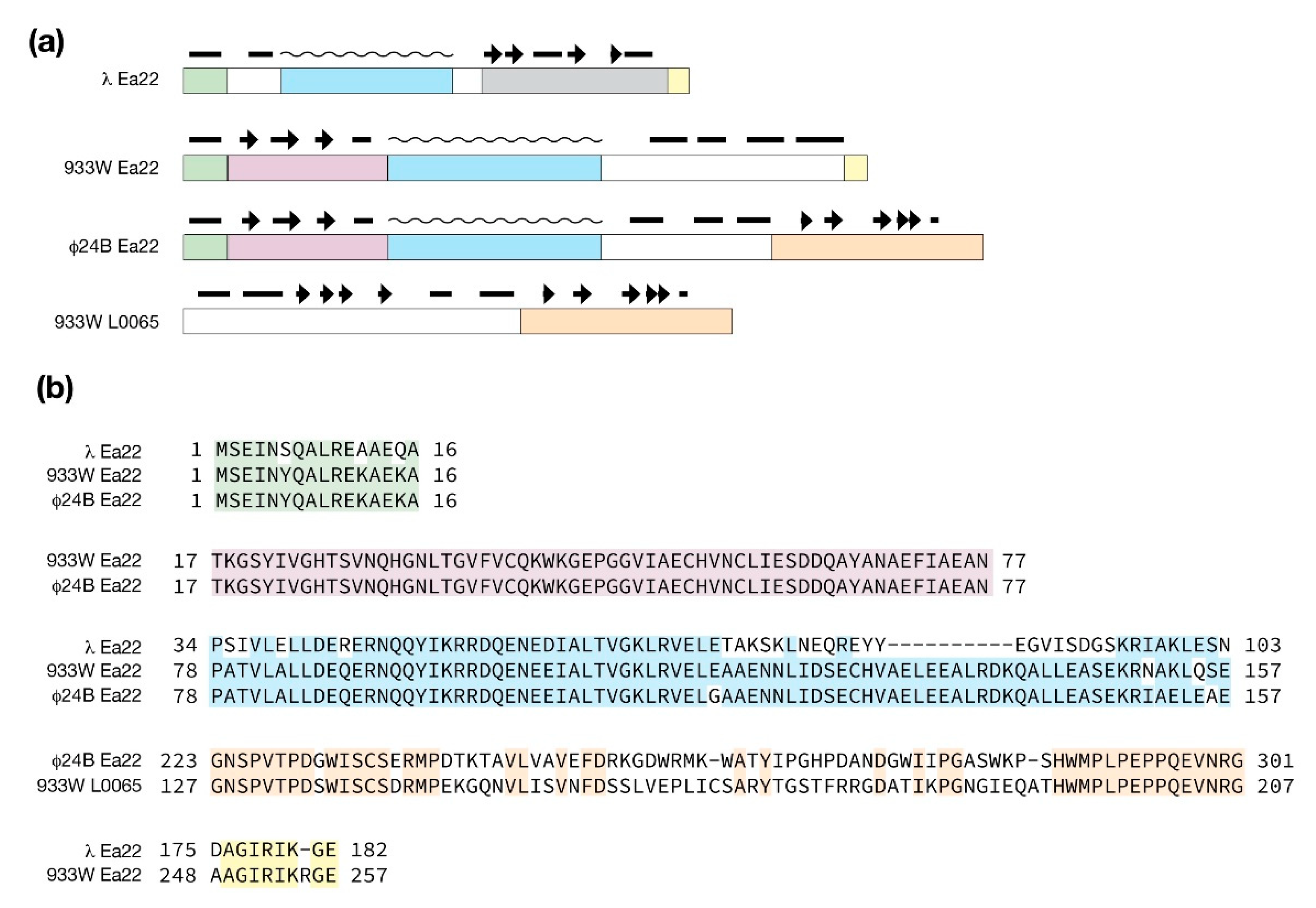
4.5. The Ea22 Genes of λ and Stx+ Phages
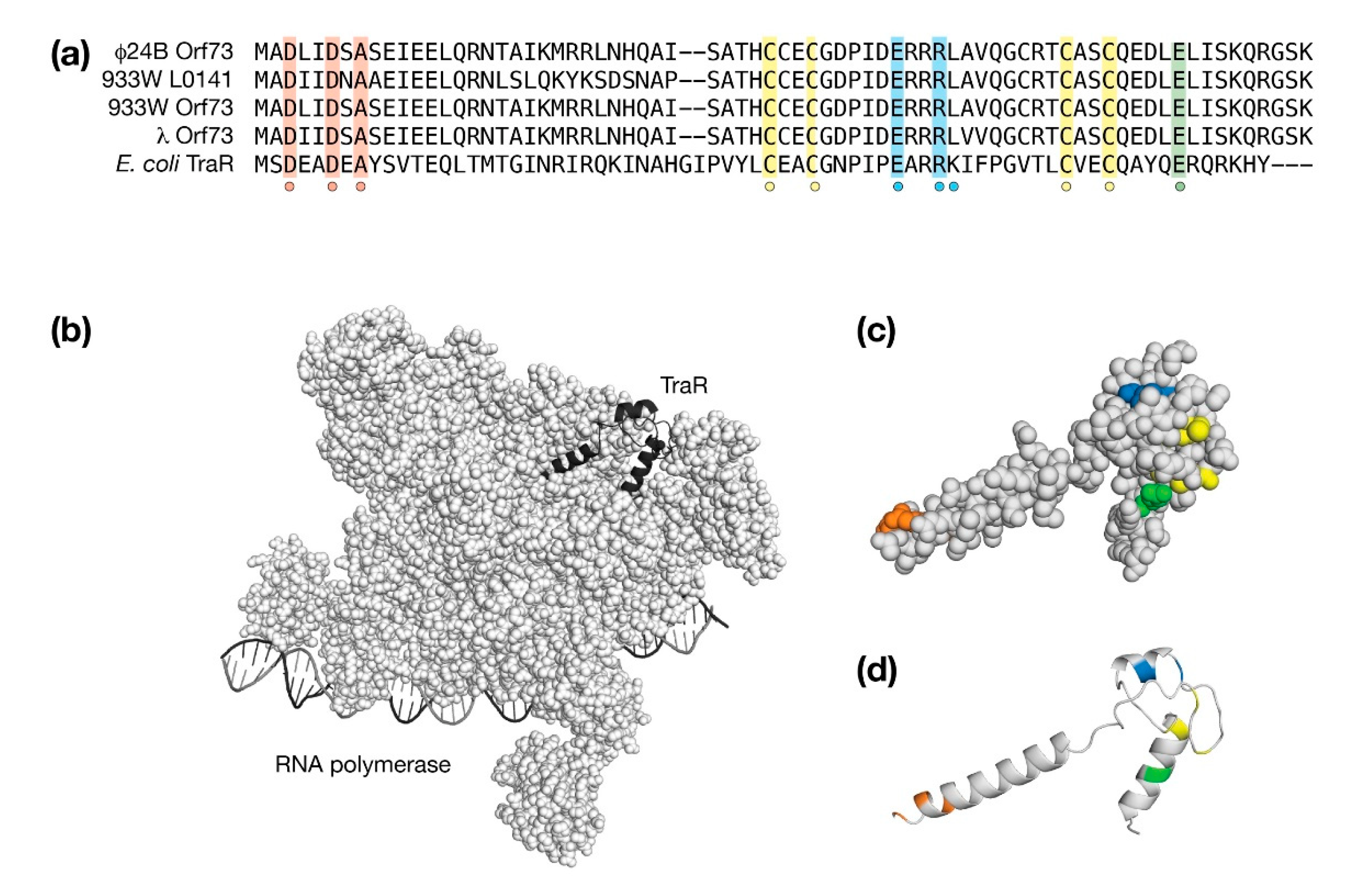
4.6. The Orf73 Genes of λ and Stx+ Phages
5. Non-Conserved exo-xis Region Proteins of Interest
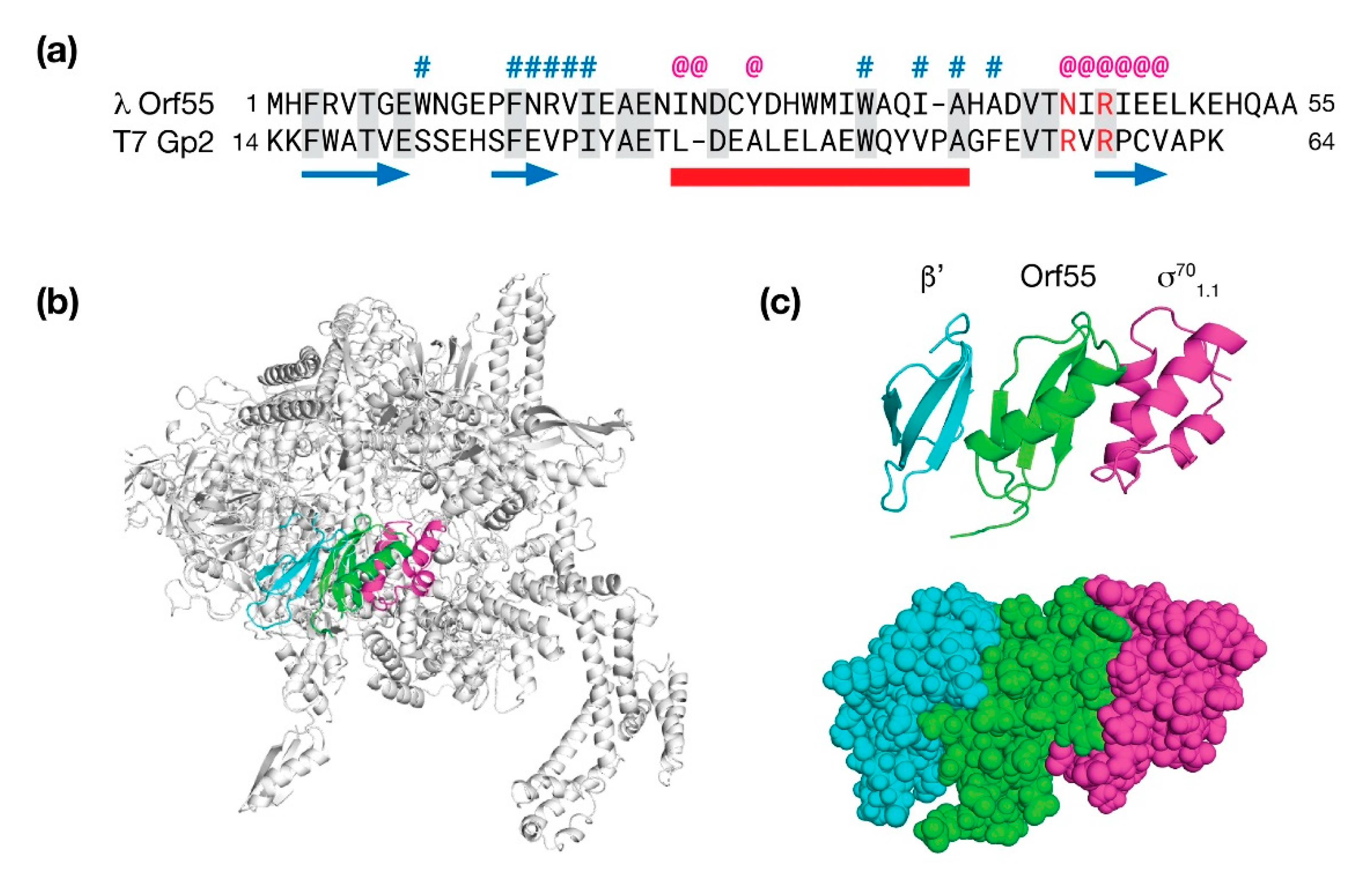
5.1. Bacteriophage λ Orf55 Is a Possible Modulator of RNA Polymerase
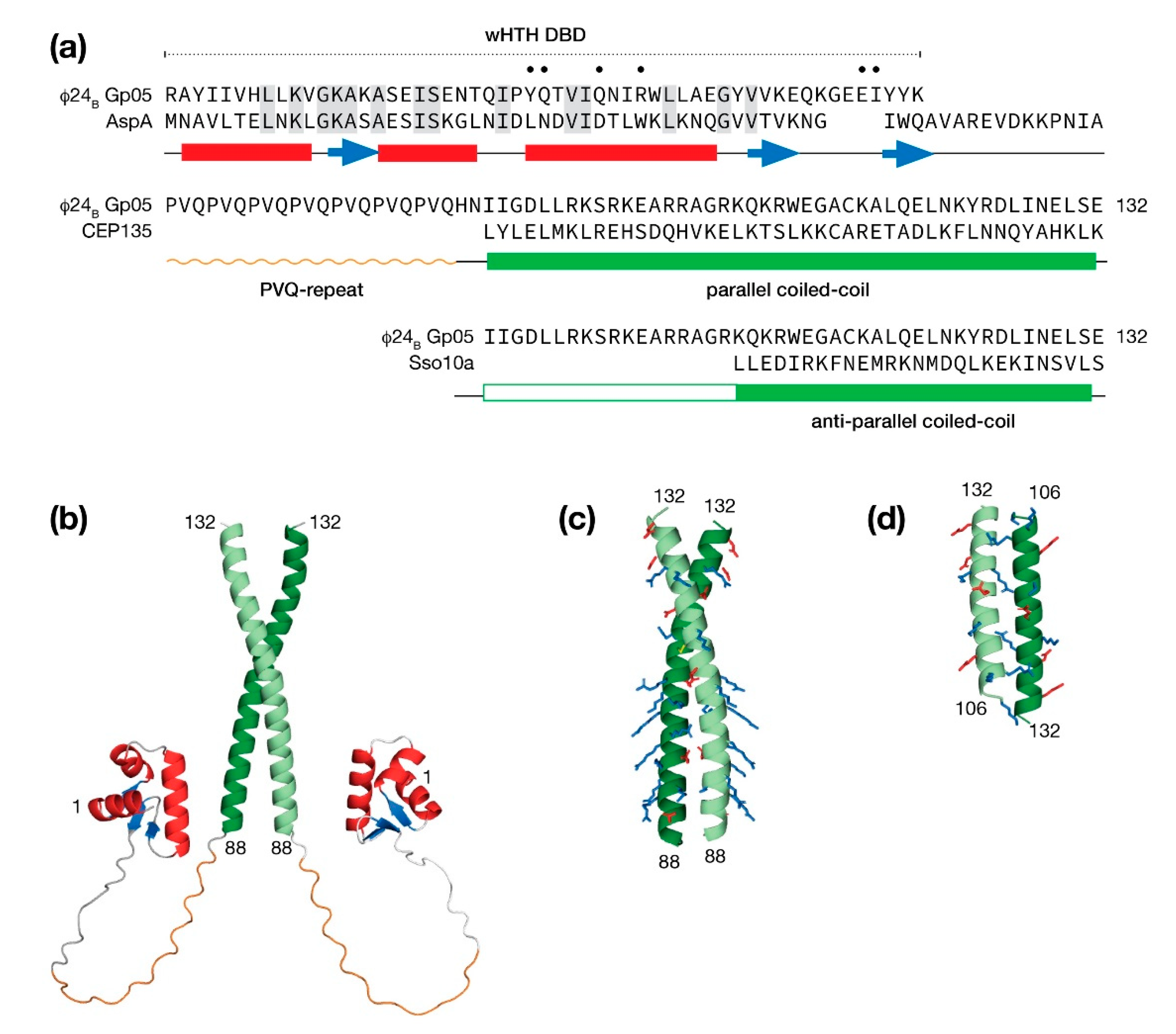
5.2. Bacteriophage φ24B Gp05 Is a Possible Transcription Factor
6. Discussion
7. Materials and Methods
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lim, J.Y.; Yoon, J.; Hovde, C.J. A Brief Overview of Escherichia Coli O157:H7 and Its Plasmid O157. J. Microbiol. Biotechnol. 2010, 20, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniesa, M.; Hammerl, J.A.; Hertwig, S.; Appel, B.; Brüssow, H. Shiga Toxin-Producing Escherichia Coli O104:H4: A New Challenge for Microbiology. Appl. Environ. Microbiol. 2012, 78, 4065–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paletta, A.C.C.; Castro, V.S.; Conte-Junior, C.A. Shiga Toxin-Producing and Enteroaggregative Escherichia Coli in Animal, Foods, and Humans: Pathogenicity Mechanisms, Detection Methods, and Epidemiology. Curr. Microbiol. 2020, 77, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Allison, H.E.; Sergeant, M.J.; James, C.E.; Saunders, J.R.; Smith, D.L.; Sharp, R.J.; Marks, T.S.; McCarthy, A.J. Immunity Profiles of Wild-Type and Recombinant Shiga-Like Toxin-Encoding Bacteriophages and Characterization of Novel Double Lysogens. Infect. Immun. 2003, 71, 3409–3418. [Google Scholar] [CrossRef] [Green Version]
- Allison, H.E. Stx-Phages: Drivers and Mediators of the Evolution of STEC and STEC-like Pathogens. Future Microbiol. 2007, 2, 165–174. [Google Scholar] [CrossRef]
- Newell, D.G.; Ragione, R.M.L. Enterohaemorrhagic and Other Shiga Toxin-producing Escherichia Coli (STEC): Where Are We Now Regarding Diagnostics and Control Strategies? Transbound. Emerg. Dis. 2018, 65, 49–71. [Google Scholar] [CrossRef] [Green Version]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin Gene Expression by Shiga Toxin-Producing Escherichia Coli: The Role of Antibiotics and the Bacterial SOS Response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Gamage, S.D.; Patton, A.K.; Hanson, J.F.; Weiss, A.A. Diversity and Host Range of Shiga Toxin-Encoding Phage. Infect. Immun. 2004, 72, 7131–7139. [Google Scholar] [CrossRef] [Green Version]
- Serna, A.; Boedeker, E.C. Pathogenesis and Treatment of Shiga Toxin-Producing Escherichia Coli Infections. Curr. Opin. Gastroenterol. 2008, 24, 38–47. [Google Scholar] [CrossRef]
- Holt, G.S.; Lodge, J.K.; McCarthy, A.J.; Graham, A.K.; Young, G.; Bridge, S.H.; Brown, A.K.; Veses-Garcia, M.; Lanyon, C.V.; Sails, A.; et al. Shigatoxin Encoding Bacteriophage Φ24B Modulates Bacterial Metabolism to Raise Antimicrobial Tolerance. Sci. Rep. 2017, 7, 40424. [Google Scholar] [CrossRef] [Green Version]
- Cogliano, M.E.D.; Pinto, A.; Goldstein, J.; Zotta, E.; Ochoa, F.; Fernández-Brando, R.J.; Muniesa, M.; Ghiringhelli, P.D.; Palermo, M.S.; Bentancor, L.V. Relevance of Bacteriophage 933W in the Development of Hemolytic Uremic Syndrome (HUS). Front. Microbiol. 2018, 9, 3104. [Google Scholar] [CrossRef]
- Kozłowska, K.; Glinkowska, M.; Boss, L.; Gaffke, L.; Deptuła, J.; Węgrzyn, G. Formation of Complexes Between O Proteins and Replication Origin Regions of Shiga Toxin-Converting Bacteriophages. Front. Mol. Biosci. 2020, 7, 207. [Google Scholar] [CrossRef]
- Lawrence, D.; Baldridge, M.T.; Handley, S.A. Phages and Human Health: More Than Idle Hitchhikers. Viruses 2019, 11, 587. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xu, J.; Li, W.; Wang, S.; Li, J.; Yu, J.; Li, Y.; Wei, H. Staphylococcus Aureus Virulence Attenuation and Immune Clearance Mediated by a Phage Lysin-derived Protein. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Keen, E.C. Paradigms of Pathogenesis: Targeting the Mobile Genetic Elements of Disease. Front. Cell Infect. Microbiol. 2012, 2, 161. [Google Scholar] [CrossRef] [Green Version]
- Livny, J.; Friedman, D.I. Characterizing Spontaneous Induction of Stx Encoding Phages Using a Selectable Reporter System. Mol. Microbiol. 2004, 51, 1691–1704. [Google Scholar] [CrossRef] [Green Version]
- Bloch, S.; Nejman-Faleńczyk, B.; Dydecka, A.; Łoś, J.M.; Felczykowska, A.; Węgrzyn, A.; Węgrzyn, G. Different Expression Patterns of Genes from the Exo-Xis Region of Bacteriophage λ and Shiga Toxin-Converting Bacteriophage Φ24B Following Infection or Prophage Induction in Escherichia Coli. PLoS ONE 2014, 9, e108233. [Google Scholar] [CrossRef]
- Sergueev, K.; Court, D.; Reaves, L.; Austin, S.E. Coli Cell-Cycle Regulation by Bacteriophage Lambda. J. Mol. Biol. 2002, 324, 297–307. [Google Scholar] [CrossRef]
- Licznerska, K.; Dydecka, A.; Bloch, S.; Topka, G.; Nejman-Faleńczyk, B.; Węgrzyn, A.; Węgrzyn, G. The Role of the Exo-Xis Region in Oxidative Stress-Mediated Induction of Shiga Toxin-Converting Prophages. Oxid. Med. Cell Longev. 2016, 2016, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Baharoglu, Z.; Mazel, D. SOS, the Formidable Strategy of Bacteria against Aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, S. Trouble Is Coming: Signaling Pathways That Regulate General Stress Responses in Bacteria. J. Biol. Chem. 2019, 294, 11685–11700. [Google Scholar] [CrossRef] [Green Version]
- Łoś, J.M.; Łoś, M.; Węgrzyn, A.; Węgrzyn, G. Role of the Bacteriophage λ Exo-Xis Region in the Virus Development. Folia Microbiol. 2008, 53, 443–450. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, H.; Gu, Z.; Roberts, J.W. High-Resolution View of Bacteriophage Lambda Gene Expression by Ribosome Profiling. Proc. Natl. Acad. Sci. USA 2013, 110, 11928–11933. [Google Scholar] [CrossRef] [Green Version]
- Dydecka, A.; Bloch, S.; Rizvi, A.; Perez, S.; Nejman-Falenczyk, B.; Topka, G.; Gasior, T.; Necel, A.; Wegrzyn, G.; Donaldson, L.W.; et al. Bad Phages in Good Bacteria: Role of the Mysterious Orf63 of λ and Shiga Toxin-Converting Φ24B Bacteriophages. Front. Microbiol. 2017, 8, 1618. [Google Scholar] [CrossRef]
- Zdrojewska, K.; Dydecka, A.; Nejman-Faleńczyk, B.; Topka, G.; Necel, A.; Węgrzyn, A.; Węgrzyn, G.; Bloch, S. Role of Orf73 in the Development of Lambdoid Bacteriophages during Infection of the Escherichia Coli Host. Acta Biochim. Pol. 2019. [Google Scholar] [CrossRef]
- Dydecka, A.; Nejman-Faleńczyk, B.; Bloch, S.; Topka, G.; Necel, A.; Donaldson, L.W.; Węgrzyn, G.; Węgrzyn, A. Roles of Orf60a and Orf61 in Development of Bacteriophages λ and Φ24B. Viruses 2018, 10, 553. [Google Scholar] [CrossRef] [Green Version]
- Dydecka, A.; Bloch, S.; Necel, A.; Topka, G.; Węgrzyn, A.; Tong, J.; Donaldson, L.W.; Węgrzyn, G.; Nejman-Faleńczyk, B. The Ea22 Gene of Lambdoid Phages: Preserved Prolysogenic Function despite of High Sequence Diversity. Virus Genes 2020, 56, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Azaldegui, C.A.; Vecchiarelli, A.G.; Biteen, J.S. The Emergence of Phase Separation as an Organizing Principle in Bacteria. Biophys. J. 2020, 120, 1123–1138. [Google Scholar] [CrossRef]
- Janissen, R.; Arens, M.M.A.; Vtyurina, N.N.; Rivai, Z.; Sunday, N.D.; Eslami-Mossallam, B.; Gritsenko, A.A.; Laan, L.; de Ridder, D.; Artsimovitch, I.; et al. Global DNA Compaction in Stationary-Phase Bacteria Does Not Affect Transcription. Cell 2018, 174, 1188–1199.e14. [Google Scholar] [CrossRef] [Green Version]
- Harami, G.M.; Kovács, Z.J.; Pancsa, R.; Pálinkás, J.; Baráth, V.; Tárnok, K.; Málnási-Csizmadia, A.; Kovács, M. Phase Separation by SsDNA Binding Protein Controlled via Protein−protein and Protein−DNA Interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 26206–26217. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Casjens, S.; Uetz, P. The Protein Interaction Map of Bacteriophage Lambda. BMC Microbiol. 2011, 11, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasche, S.; Wuchty, S.; Rajagopala, S.V.; Uetz, P. The Protein Interaction Network of Bacteriophage Lambda with Its Host, Escherichia Coli. J. Virol. 2013, 87, 12745–12755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parma, D.H.; Snyder, M.; Sobolevski, S.; Nawroz, M.; Brody, E.; Gold, L. The Rex System of Bacteriophage Lambda: Tolerance and Altruistic Cell Death. Gene Dev. 1992, 6, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toothman, P.; Herskowitzt, I. Rex-Dependent Exclusion of Lambdoid Phages I. Prophage Requirements for Exclusion. Virology 1980, 102, 133–146. [Google Scholar] [CrossRef]
- Yin, Z.; Kaelber, J.T.; Ebright, R.H. Structural Basis of Q-Dependent Antitermination. Proc. Natl. Acad. Sci. USA 2019, 116, 18384–18390. [Google Scholar] [CrossRef] [Green Version]
- Ayed, S.H.; Cloutier, A.D.; McLeod, L.J.; Foo, A.C.Y.; Damry, A.M.; Goto, N.K. Dissecting the Role of Conformational Change and Membrane Binding by the Bacterial Cell Division Regulator MinE in the Stimulation of MinD ATPase Activity. J. Biol. Chem. 2017, 292, 20732–20743. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-T.; Villar, M.T.; Artigues, A.; Lutkenhaus, J. MinE Conformational Dynamics Regulate Membrane Binding, MinD Interaction, and Min Oscillation. Proc. Natl. Acad. Sci. USA 2017, 114, 7497–7504. [Google Scholar] [CrossRef] [Green Version]
- Kwan, J.J.; Smirnova, E.; Khazai, S.; Evanics, F.; Maxwell, K.L.; Donaldson, L.W. The Solution Structures of Two Prophage Homologues of the Bacteriophage λ Ea8.5 Protein Reveal a Newly Discovered Hybrid Homeodomain/Zinc-Finger Fold. Biochemistry 2013, 52, 3612–3614. [Google Scholar] [CrossRef]
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F.; et al. Whole Genome Comparison of a Large Collection of Mycobacteriophages Reveals a Continuum of Phage Genetic Diversity. eLife 2015, 4, e06416. [Google Scholar] [CrossRef]
- Hatfull, G.F. Dark Matter of the Biosphere: The Amazing World of Bacteriophage Diversity. J. Virol. 2015, 89, 8107–8110. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Jha, S.K.; Ramanathan, A.; Ewetz, R.; Velasquez, A.; Jha, S. Protein Folding Neural Networks Are Not Robust. arXiv 2021, arXiv:2109.04460. [Google Scholar]
- Chowdhury, R.; Bouatta, N.; Biswas, S.; Rochereau, C.; Church, G.M.; Sorger, P.K.; AlQuraishi, M. Single-Sequence Protein Structure Prediction Using Language Models from Deep Learning. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. LDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [Green Version]
- Wojciak, J.M.; Connolly, K.M.; Clubb, R.T. NMR Structure of the Tn916 Integrase–DNA Complex. Nat. Struct. Biol. 1999, 6, 366–373. [Google Scholar] [CrossRef]
- Wojciak, J.M.; Sarkar, D.; Landy, A.; Clubb, R.T. Arm-Site Binding by λ-Integrase: Solution Structure and Functional Characterization of Its Amino-Terminal Domain. Proc. Natl. Acad. Sci. USA 2002, 99, 3434–3439. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.D.; Yamasaki, K.; Ohme-Takagi, M.; Tateno, M.; Suzuki, M. A Novel Mode of DNA Recognition by a Β-sheet Revealed by the Solution Structure of the GCC-box Binding Domain in Complex with DNA. EMBO J. 1998, 17, 5484–5496. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.A.; Simpson, P.; Huyton, T.; Roper, D.; Matthews, S. Solution Structure and Interactions of the Escherichia Coli Cell Division Activator Protein CedA. Biochemistry 2005, 44, 6738–6744. [Google Scholar] [CrossRef]
- Birrane, G.; Soni, A.; Ladias, J.A.A. Structural Basis for DNA Recognition by the Human PAX3 Homeodomain. Biochemistry 2009, 48, 1148–1155. [Google Scholar] [CrossRef]
- Tong, J.; Nejman-Faleńczyk, B.; Bloch, S.; Węgrzyn, A.; Węgrzyn, G.; Donaldson, L.W. Ea22 Proteins from Lambda and Shiga Toxin-Producing Bacteriophages Balance Structural Diversity with Functional Similarity. ACS Omega 2020, 5, 12236–12244. [Google Scholar] [CrossRef]
- Gopalkrishnan, S.; Ross, W.; Chen, A.Y.; Gourse, R.L. TraR Directly Regulates Transcription Initiation by Mimicking the Combined Effects of the Global Regulators DksA and PpGpp. Proc. Natl. Acad. Sci. USA 2017, 114, E5539–E5548. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gopalkrishnan, S.; Chiu, C.; Chen, A.Y.; Campbell, E.A.; Gourse, R.L.; Ross, W.; Darst, S.A. E. Coli TraR Allosterically Regulates Transcription Initiation by Altering RNA Polymerase Conformation. eLife 2019, 8, e49375. [Google Scholar] [CrossRef] [PubMed]
- Perederina, A.; Svetlov, V.; Vassylyeva, M.N.; Tahirov, T.H.; Yokoyama, S.; Artsimovitch, I.; Vassylyev, D.G. Regulation through the Secondary Channel—Structural Framework for PpGpp-DksA Synergism during Transcription. Cell 2004, 118, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Coll, L.; Potrykus, K.; Cashel, M.; Balsalobre, C. Mutational Analysis of Escherichia Coli GreA Protein Reveals New Functional Activity Independent of Antipause and Lethal When Overexpressed. Sci. Rep. 2020, 10, 16074. [Google Scholar] [CrossRef] [PubMed]
- Lamour, V.; Rutherford, S.T.; Kuznedelov, K.; Ramagopal, U.A.; Gourse, R.L.; Severinov, K.; Darst, S.A. Crystal Structure of Escherichia Coli Rnk, a New RNA Polymerase-Interacting Protein. J. Mol. Biol. 2008, 383, 367–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parshin, A.; Shiver, A.L.; Lee, J.; Ozerova, M.; Schneidman-Duhovny, D.; Gross, C.A.; Borukhov, S. DksA Regulates RNA Polymerase in Escherichia Coli through a Network of Interactions in the Secondary Channel That Includes Sequence Insertion 1. Proc. Natl. Acad. Sci. USA 2015, 112, E6862–E6871. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Heras, G.; Ruiz-Masó, J.A.; del Solar, G.; Espinosa, M.; Bravo, A.; Salas, M. Protein P56 from the Bacillus Subtilis Phage Φ29 Inhibits DNA-Binding Ability of Uracil-DNA Glycosylase. Nucleic Acids Res. 2007, 35, 5393–5401. [Google Scholar] [CrossRef]
- Keller, A.N.; Yang, X.; Wiedermannová, J.; Delumeau, O.; Krásný, L.; Lewis, P.J. ε, a New Subunit of RNA Polymerase Found in Gram-Positive Bacteria. J. Bacteriol. 2014, 196, 3622–3632. [Google Scholar] [CrossRef] [Green Version]
- Bae, B.; Davis, E.; Brown, D.; Campbell, E.A.; Wigneshweraraj, S.; Darst, S.A. Phage T7 Gp2 Inhibition of Escherichia Coli RNA Polymerase Involves Misappropriation of Σ70 Domain 1.1. Proc. Natl. Acad. Sci. USA 2013, 110, 19772–19777. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.W.; Green, T.; Zídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kraatz, S.; Guichard, P.; Obbineni, J.M.; Olieric, N.; Hatzopoulos, G.N.; Hilbert, M.; Sen, I.; Missimer, J.; Gönczy, P.; Steinmetz, M.O. The Human Centriolar Protein CEP135 Contains a Two-Stranded Coiled-Coil Domain Critical for Microtubule Binding. Structure 2016, 24, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Alber, T. Structure of the Leucine Zipper. Curr. Opin. Genet. Dev. 1992, 2, 205–210. [Google Scholar] [CrossRef]
- Mirdita, M.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making Protein Folding Accessible to All. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mirdita, M.; Ovchinnikov, S.; Steinegger, M. RoseTTAFold Jupyter Notebook. Available online: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/RoseTTAFold.ipynb (accessed on 5 September 2021).
- Mirdita, M.; Ovchinnikov, S.; Steinegger, M. AlphaFold2 Advanced Jupyter Notebook. Available online: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/AlphaFold2_advanced.ipynb (accessed on 5 September 2021).
- Krissinel, E.; Henrick, K. Secondary-Structure Matching (SSM), a New Tool for Fast Protein Structure Alignment in Three Dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.W.; Lemmon, G.H.; DeLuca, S.L.; Sheehan, J.H.; Meiler, J. Practically Useful: What the Rosetta Protein Modeling Suite Can Do for You. Biochemistry 2010, 49, 2987–2998. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donaldson, L.W. Molecular Modeling the Proteins from the exo-xis Region of Lambda and Shigatoxigenic Bacteriophages. Antibiotics 2021, 10, 1282. https://doi.org/10.3390/antibiotics10111282
Donaldson LW. Molecular Modeling the Proteins from the exo-xis Region of Lambda and Shigatoxigenic Bacteriophages. Antibiotics. 2021; 10(11):1282. https://doi.org/10.3390/antibiotics10111282
Chicago/Turabian StyleDonaldson, Logan W. 2021. "Molecular Modeling the Proteins from the exo-xis Region of Lambda and Shigatoxigenic Bacteriophages" Antibiotics 10, no. 11: 1282. https://doi.org/10.3390/antibiotics10111282
APA StyleDonaldson, L. W. (2021). Molecular Modeling the Proteins from the exo-xis Region of Lambda and Shigatoxigenic Bacteriophages. Antibiotics, 10(11), 1282. https://doi.org/10.3390/antibiotics10111282